

Abstract
Block of the channel of N-methyl-d-aspartate (NMDA) receptors by external Mg2+ (Mgo2+) has broad implications for the many physiological and pathological processes that depend on NMDA receptor activation. An essential property of channel block by Mgo2+ is its powerful voltage dependence. A widely cited explanation for the strength of the voltage dependence of block is that the Mgo2+-binding site is located deep in the channel of NMDA receptors; Mgo2+ then would sense most of the membrane potential field during block. However, recent electrophysiological and mutagenesis studies suggest that the blocking site cannot be deep enough to account for the voltage dependence of Mgo2+ block. Here we describe the basis for this discrepancy: the magnitude and voltage dependence of channel block by Mgo2+ are strongly regulated by external and internal permeant monovalent cations. Our data support a model in which access to the channel by Mgo2+ is prevented when permeant ion-binding sites at the external entrance to the channel are occupied. Mgo2+ can block the channel only when the permeant ion-binding sites are unoccupied and then can either unblock back to the external solution or permeate the channel. Unblock to the external solution is prevented if external permeant ions bind while Mg2+ blocks the channel, although permeation is still permitted. The model provides an explanation for the strength of the voltage dependence of Mgo2+ block and quantifies the interdependence of permanent and blocking ion binding to NMDA receptors.
N;;3-methyl-d-aspartate (NMDA) receptors are ligand-gated ion channels that exhibit remarkably strong voltage dependence. In contrast to the voltage-gated family of ion channels, the voltage dependence of NMDA receptor-mediated conductance changes depends on a channel-blocking ion, external Mg2+ (Mgo2+). Current flow through the channel of NMDA receptors is blocked when Mgo2+ enters and binds in the channel, and block becomes much more effective as membrane potential (Vm) is hyperpolarized. A question fundamental to NMDA receptor function is how the block by Mgo2+ achieves its powerful voltage dependence. The hypothesis first proposed to explain the voltage dependence of Mgo2+ block was that Mgo2+ binds to a site located deep in the channel (1, 2). Mgo2+ then would need to traverse part of the channel and, therefore, enter the membrane voltage field, to block; as a result, Mgo2+-blocking rate would increase with hyperpolarization (3). Mgo2+ unblocking rate similarly would decrease with hyperpolarization if Mgo2+ cannot permeate the channel. Both of these effects of Vm on blocking kinetics have been observed (4).
This direct influence of Vm on Mgo2+-blocking kinetics because of the electrical location of the Mgo2+-blocking site undoubtedly contributes to the voltage dependence of block. However, starting with the observation that the voltage dependence of the blocking rate appears anomalously high (4), data from numerous studies have suggested that other mechanisms amplify blocking voltage dependence. For the voltage dependence of channel occupation by Mgo2+ to be explained exclusively by blocking site location, the required site depth has been estimated to be about 80% through the membrane voltage field [range of estimates, 53–107% (4–12)]. However, internal Mg2+ also has been found to block the channel of NMDA receptors at a site estimated to be 55–71% through the membrane voltage field from the outside of the channel (13–16). It appears implausible that Mgo2+ on the way to its blocking site could pass by the site at which internal Mg2+ blocks. In addition, there is evidence that Mgo2+ may be able to permeate the channel of NMDA receptors under some conditions (17, 18); if this happens under physiological conditions, it would require that the blocking site be even deeper in the voltage field to explain the voltage dependence of block (4). Finally, amino acids crucial to block by Mgo2+ are located near the tip of the M2 region of NMDA receptor subunits (12, 19–22). The M2 region is a pore loop that enters and exits the membrane intracellularly and is believed to form a large portion of the channel of NMDA receptors. A physical location of the Mgo2+-blocking site near the external tip of the M2 region appears inconsistent with an electrical location near the internal extreme of the channel.
One mechanism by which the voltage dependence of block by Mgo2+ might be amplified is through a voltage-dependent influence on block of ions other than Mgo2+ (4). Changes in the concentrations of intracellular and extracellular ions other than Mg2+ have been shown to influence block by Mgo2+ (8, 17, 23) and by some organic channel blockers of NMDA receptors (24–26). Mutation of amino acids in several well-separated regions of NR2 subunits have been shown to influence Mgo2+ block (9), leaving open the possibility that block may be affected by multiple binding sites on NMDA receptors. There is evidence for Ca2+-binding sites deep in the channel (19) and on the external side of the channel (10, 27) and for permeant monovalent cation-binding sites on both sides of the channel (26). We recently demonstrated that occupation of the permeant monovalent cation-binding sites has a profound effect on the blocking kinetics of two adamantane derivatives that block the channel of NMDA receptors (26). Here, we examine how occupation of these permeant monovalent cation-binding sites influences block by Mgo2+.
Experimental Procedures
Preparation and Solutions.
Primary cultures of rat cortical neurons were prepared from 16-day embryos as described (28). Neurons were used for experiments after 15–30 days in culture.
Recordings of single-channel currents were performed by using outside-out patches (29) at room temperature (21–23°C). Pipettes were filled with one of three solutions. The control (130 mM Csi+) pipette solution contained 120 mM CsF, 10 mM CsCl, 10 mM EGTA, and 10 mM Hepes. pH was adjusted to 7.2 with CsOH. The 25 mM Csi+ solution contained 25 mM CsF, 105 mM NMDG, 10 mM EGTA, 10 mM Hepes, and 1 mM tetraethylammonium (TEA). The 8 mM Csi+ solution contained 8 mM CsF, 122 mM NMDG, 10 mM EGTA, 10 mM Hepes, and 1 mM TEA. pH of the 25 and 8 mM Csi+ solutions was adjusted to 7.2 with HCl. Osmolality of each solution was approximately 260 mosmol. Three different external solutions were used. The control (140 mM Nao+) external solution contained 140 mM NaCl, 2.8 mM KCl, 1 mM CaCl2, and 10 mM Hepes. The 105 mM Nao+ solution contained 105 mM NaCl, 2.8 mM KCl, 0.75 mM CaCl2, 10 mM Hepes, and 70 mM sucrose. The 70 mM Nao+ solution contained 70 mM NaCl, 2.8 mM KCl, 0.5 mM CaCl2, 10 mM Hepes, and 140 mM sucrose. Sucrose was used as the external NaCl substitute because all ionic substitutes considered would either block or permeate the channel of NMDA receptors. All external solutions contained 0.2 μM tetrodotoxin. pH of all external solutions was adjusted to 7.2 with NaOH. Osmolality was approximately 270 mosmol. Although external Ca2+-binding sites on the NMDA receptor have been described (10, 27), external Ca2+ does not bind significantly to the external permeant monovalent ion binding sites at the external Ca2+ to monovalent cation concentration ratios used here (26). This result was confirmed in one experiment in which Ca2+ was omitted from the external solution with the 105 mM Nao+ solution; the apparent rates of Mgo2+ block and unblock with 0 added Ca2+ was within the range of values measured in the presence of 0.75 mM Ca2+. Voltages in all experiments were corrected for junction potentials (8 mV with the 140 mM Nao+ and 130 mM Csi+ solution combination, and −2 mV with all other solution combinations).
Data Recording and Analysis.
Data recording and analysis were performed as described (26, 28). NMDA receptor-mediated, single-channel currents were activated at Vm values from −150 to −20 mV by application of 5 or 10 μM NMDA plus 10 μM glycine. Single-channel currents were low-pass-filtered and recorded on videotape. Recorded currents were played back off-line, low-pass-filtered at 3 or 4 kHz, and sampled at 40 kHz with a TL-1 interface and pclamp 6.02 (Axon Instruments). For most data the effective filter frequency resulting from cascaded filters (fc) was 3.7 kHz; events shorter than 96 μs (two times the system dead time) were deleted from histograms and omitted from histogram fits.
Open-time distributions were well-fit by a sum of two exponential components under control conditions and by one or two components in the presence of Mgo2+. When two components were present, the channel mean open time (τo) was estimated from the much larger slow component (26). The apparent blocking rate constant (k+,app) was estimated as the slope of a regression line fit to a linear plot of 1/τo vs. Mgo2+ concentration ([Mg2+]o) (30). A separate value of k+,app was estimated at each Vm in each patch by using values of τo in control conditions and at two or more Mgo2+ concentrations. Measurements of τo were not likely to be in error due to missed brief closures because correction of τo for missed closures under similar conditions did not significantly change mean open-time values (28).
Closed-time histograms in the absence and presence of Mgo2+ were well-fit by a sum of three or four exponential components. A large closed-time component appeared in the presence of Mgo2+ that was not present in the absence of Mgo2+. The mean duration of this component was used to estimate the mean duration of block (τb). Although this component generally obscured one of the control closed-time components, τb could be accurately estimated as long as the closed-time component induced by Mgo2+ was much larger than any overlapping control closed-time components (26). This was the case in all experiments except when 70 mM Nao+ was used; in this external solution, τb could be measured accurately in only one patch (used for points in Fig. 3d). A separate value of the apparent unblocking rate constant (k−,app) was estimated at each Vm in each patch by averaging the values of 1/τb measured in at least two Mgo2+ concentrations. To prevent error in measurement of τb because of missed brief openings we used [Mg2+]o values in which τo ≥ 0.4 ms (26, 28).
Figure 3.

τb and k−,app depend on [Na+]o. (a) Single-channel currents activated by 5 μM NMDA + 10 μM glycine during block by 30 μM Mgo2+ at −90 mV with [Cs+]i = 130 mM and the indicated [Na+]o. In each pair of current traces, the section of the upper single-channel record indicated by the horizontal bar is replotted below the bar at higher time resolution. (b) Closed-time histograms corresponding to the currents shown in a to the left of each histogram. Lines are four-exponential fits to the histograms. Number of events in each histogram is more than 1,000. (c) Voltage dependence of k−,app in three different [Cs+]i. Symbol meanings are as in legend to Fig. 1 c and d. (d) Voltage dependence of k−,app in three different [Na+]o. Because k−,app does not depend on [Cs+]i (c), points are averages of data at a single [Na+]o and one or more [Cs+]i. ⋄, 140 mM Nao+; ■, 105 mM Nao+; ⧫, 70 mM Nao+. Lines are drawn by using Eq. 3, with parameter values given in Table 1.
Curve fitting and simulations were performed by using sigmaplot (SPSS, Chicago). Optimal values of k+(0), b, KNa, KCs(0), and a were determined first by finding values that minimized the sum of squared errors (SSE) when fitting Eq. 2 to values of k+,app measured in 140 mM Nao+/130 mM Csi+. Small parameter adjustments then were made to provide optimal fits to all five data sets shown in Fig. 2 as judged by eye. Optimal values of k−,o(0), c, k−,i, and d were determined by first finding values that minimized the SSE when fitting Eq. 3 to values of k−,app measured in 140 mM Nao+. KNa was fixed at 34.4 mM, the value determined by fits to the data in Fig. 2. Small parameter adjustments then were made to provide optimal fits to all three data sets shown in Fig. 3d as judged by eye.
Figure 2.

k+,app depends on the concentration of both [Cs+]i and [Na+]o. (a and b) Plots of the voltage dependence of k+,app for block by Mgo2+. Symbol meanings are as in the legend to Fig. 1 c and d. Data for 140 mM Nao+/25 mM Csi+ are plotted in b as well as a for comparison with other data. Lines are drawn by using Eq. 2, with parameter values given in Table 1.
Each plotted point in the figures is the mean of measurements from n = 1–8 patches; error bars representing SD are plotted when larger than the symbol size and n > 2.
Results
Effects of Permeant Ions on Rate of Mgo2+ Block.
We investigated the influence of permeant monovalent cations on Mgo2+ block of single-channel currents through native NMDA receptors of cultured cortical neurons, which express predominantly the NR1, NR2A, and NR2B receptor subunits (26, 31). Rates of channel block and unblock by Mgo2+ were measured from the single-channel current “flicker” induced by Mgo2+ (Fig. 1a). τo (Fig. 1b) depended linearly on [Mg2+]o (Fig. 1 c and d) under all conditions, as expected for an open-channel blocker, permitting measurement of the rate constants of block (4, 6, 28, 30). Under control ionic concentrations at −50 mV, the blocking rate constant was consistent with previous measurements (4, 6). However, when the internal Cs+ concentration ([Cs+]i) was decreased from 130 to 8 mM, τo decreased more than 4-fold (Fig. 1 a and b), reflecting a large increase in the Mgo2+-blocking rate (Fig. 1c). When [Cs+]i was held at 25 mM while the external Na+ concentration ([Na+]o) was decreased, a similar increase in the blocking rate of Mgo2+ was observed (Fig. 1d). These observations indicate that Mgo2+ does not act via the classical model of open channel block (30). Because the measurable rate constant of channel block depends on the concentrations of multiple ions rather than just the blocking ion, we refer to k+,app as an “apparent” blocking rate constant.
Figure 1.

Decreasing permeant ion concentrations enhances Mgo2+-blocking rate. (a) Single-channel currents activated by 10 μM NMDA + 10 μM glycine during block by 10 μM Mgo2+ at −50 mV with [Na+]o = 140 mM and the indicated [Cs+]i. (b) Open-time histograms corresponding to the currents shown in a to the left of each histogram. Lines are double-exponential (upper two histograms) or single-exponential (lower histogram) fits to the histograms. Number of events in each histogram is more than 2,000. The principal mean open time (τo) is shown above each histogram. (c and d) Dependence of 1/τo on [Mg2+]o under the indicated conditions. ○, 140 mM Nao+/130 mM Csi+; ▵, 140 mM Nao+/25 mM Csi+; □, 140 mM Nao+/8 mM Csi+; ▴, 105 mM Nao+/25 mM Csi+; ▾, 70 mM Nao+/25 mM Csi+. Lines are regression fits to the plotted points.
To gain insight into the mechanisms by which permeant ions influence block by Mgo2+, we investigated the voltage dependence of the effect of permeant ion concentrations on k+,app. In control ionic concentrations and at Vm values from −40 to −90 mV, k+,app exhibits steep, single-exponential voltage dependence (e-fold change in 18 mV), in agreement with earlier studies (4, 6). However, the expected exponential dependence of k+,app on voltage (3, 4, 6, 30) was not observed over the entire range of Vm studied (Fig. 2); at Vm values negative of −90 mV, the voltage dependence became much weaker (e-fold change in 55 mV). The increase in k+,app induced by decreasing [Cs+]i (Fig. 1c) was observed only at depolarized Vm values (Fig. 2a). Because Cs+ efflux through the channel increases progressively with depolarization, this observation suggests that Csi+ must partially or completely permeate the channel before it can slow the rate of Mgo2+ block. In contrast, the increase in k+,app induced by decreasing [Na+]o (Fig. 1d) was largely voltage independent (Fig. 2b), suggesting that Nao+ slows the rate of Mg block by acting at an external site.
Effects of Permeant Ions on Rate of Mgo2+ Unblock.
Permeant ions also affect the rate at which Mgo2+ unblocks from the channel of NMDA receptors. Fig. 3a shows single-channel currents obtained in 140 mM Nao+ (Fig. 3a, upper traces) and 105 mM Nao+ (Fig. 3a, lower traces). At both [Na+]o values, block by Mgo2+ induced an additional component in the closed-time distributions (Fig. 3b) that was not present in the absence of Mgo2+. The time constant of the additional component corresponds to τb; the unblocking rate constant was calculated as 1/τb (4, 6, 28, 30). The block duration induced by 30 μM Mgo2+ was clearly briefer in 105 mM Nao+ than in 140 mM Nao+ (Fig. 3 a and b). Because the measurable rate constant of channel unblock depends on [Na+]o, we refer to k−,app as an “apparent” unblocking rate constant.
We next examined the voltage dependence of the effect of permeant ions on k−,app. Although changing [Cs+]i had no effect on k−,app at Vm values from −40 to −150 mV (Fig. 3c), k−,app was strongly affected by [Na+]o over nearly this entire voltage range (Fig. 3d). In 140 mM Nao+ at Vm values from −40 to −90 mV k−,app exhibited a nearly exponential voltage dependence similar to that reported previously (4, 6). However, further hyperpolarization from −90 to −140 mV did not further decrease k−,app. This could reflect permeation by Mgo2+ at very hyperpolarized Vm values, as was proposed previously based on whole-cell experiments (17). Decreasing [Na+]o to 105 mM induced an increase in k−,app at Vm values from −40 to −110 mV that was not obviously voltage-dependent. This suggests that Nao+ prevents Mgo2+ unblock to the external solution by acting at an external site. From −120 to −140 mV, where the decreased voltage dependence of k−,app suggests that Mgo2+ frequently permeates the channel, the dependence of k−,app on [Na+]o is weak (Fig. 3d). Nao+ therefore is unlikely to affect the permeation rate of Mg2+ that is blocking the channel.
Quantitative Model of the Effects of Permeant Ions on Mgo2+ Block.
Recently, we reported (26) that occupation of external permeant monovalent cation-binding sites on the channel of NMDA receptors prevents channel block and unblock by two adamantane derivatives, IEM-1754 and IEM-1857. To determine whether a similar mechanism can explain the unexpected dependence of Mgo2+ block on Vm, [Na+]o, and [Cs+]i, we developed a quantitative model of block by Mgo2+. In this model Mgo2+ can enter the channel only when external cation-binding sites are unoccupied, and so k+,app depends on [Na+]o and [Cs+]i (Fig. 2). The strong dependence of k+,app on [Na+]o (Fig. 2b) requires a model with two sites to which Nao+ can bind. The voltage independence of the effect of [Na+]o on k+,app suggests that these sites are located in a region of the outer vestibule of the channel external to the voltage field. The alternative possibility that Nao+ affects Mgo2+-blocking rate by screening surface charges on NMDA receptors seems improbable (26), because the surface charges are screened effectively by quite low concentrations of monovalent cations (23). Because the effect of [Cs+]i on k+,app exhibits relatively weak concentration dependence but strong voltage dependence (Fig. 2a), the model permits Csi+ to bind to only one of the sites after permeating the channel. The decrease in k−,app that results from increasing [Na+]o at all but the most hyperpolarized voltages (Fig. 3d) is modeled through a “lock-in” effect (32): if Nao+ binds to one or both permeant ion-binding sites while Mg2+ blocks the channel, Mg2+ cannot unblock to the external solution. [Cs+]i does not affect k−,app (Fig. 3c) because Csi+ cannot reach its external binding site while Mg2+ blocks the channel. Finally, the weakened dependence of k−,app on Vm and [Na+]o at very hyperpolarized voltages (Fig. 3d) is accounted for in the model by voltage-dependent Mg2+ permeation that is unaffected by Nao+ binding.
The resulting model is diagrammed in Fig. 4 and below in Scheme 1:
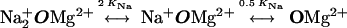 |
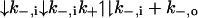 |
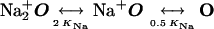 |
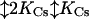 |
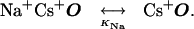 |
1 |
O represents the fully liganded NMDA receptor with its channel open and permeant ion-binding sites unoccupied. When Na+ or Cs+ is bound to O, Mgo2+ cannot enter the channel (Fig. 4a). From state O, Mgo2+ can block the channel (Fig. 4b) to create state OMg2+. Mg2+ then can either unblock to the external solution or can permeate the channel (Fig. 4c). If Na+ ion(s) occupy the external cation-binding sites while Mg2+ is blocking the channel, Mg2+ cannot unblock to the external solution, but still can permeate the channel (Fig. 4d).
Figure 4.

Schematic diagram of the NMDA receptor (purple) during block and unblock by Mgo2+. (a) When one or both of the external cation-binding sites are occupied, Mgo2+ cannot enter the channel. (b) When the external cation-binding sites are unoccupied, Mgo2+ can enter and block the channel (rate constant = k+). (c) Block by Mgo2+ can be terminated by dissociation to the external solution (rate constant = k−,o) or by permeation of the channel (rate constant = k−,i). (d) When one or both of the external cation-binding sites is occupied by Nao+, Mg2+ blocking the channel cannot dissociate to the external solution but still can permeate the channel (rate constant = k−,i).
To aid in the derivation of equations to describe Scheme 1, we made several assumptions. Because the data suggest that the cation-binding sites are located outside of the voltage field, the equilibrium dissociation constant for Na+ (KNa) was made voltage-independent. KNa was assumed to be the same whether or not Mgo2+ is bound, as was found with the IEM blockers (26). Csi+ must cross the entire membrane voltage field to bind, and so KCs must be strongly voltage-dependent. To limit adjustable parameters, we used the simplest plausible equation (Table 1) to model the voltage-dependence of Cs+ binding (26). Note that KCs is not an equilibrium constant, because it involves Cs+ permeation of the channel. The true rates (rates in the absence of permeant ions) of Mgo2+ channel block (k+), unblock to the internal solution (k−,i), and unblock to the external solution (k−,o) also were assumed to vary exponentially with Vm (3) (Table 1). Based on these assumptions, we derived the following equations for k+,app and k−,app from Scheme 1:
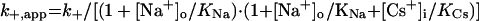 |
2 |
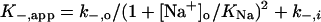 |
3 |
The model provides excellent fits to the data (Figs. 2 and 3d). Parameter values derived from fitting Scheme 1 to data are shown in Table 1. The value of KNa is in good agreement with its previous estimate (26); the small values of KCs(0) and a found here may result from an oversimplified approximation of the voltage dependence of Cs+ permeation.
Table 1.
Equations and parameter values for rate and equilibrium constants
| Parameter and equation | Value at Vm = 0 | Vm dependence |
|---|---|---|
| KNa | KNa = 34.4 mM | None |
| KCs = KCs(0) exp(Vm/a) | KCs(0) = 0.270 mM | a = −21.0 mV |
| k+ = k+(0) exp(Vm/b) | k+(0) = 1.10⋅109 M−1s−1 | b = −55.0 mV |
| k−,o = k−,o(0) exp(Vm/c) | k−,o(0) = 1.10⋅105 s−1 | c = 52.7 mV |
| k−,i = k−,i(0) exp(Vm/d) | k−,i(0) = 61.8 s−1 | d = −50.0 mV |
| KMg = KMg(0) exp(Vm/e) | KMg(0) = 101 μM | e = 26.9 mV |
The equations for k+, k−,o, k−,i, and KMg can be used to calculate the rate or equilibrium constant value in the absence of permeant monovalent cations. KMg, which equals k−,o/k+, takes into account unbinding of Mg2+ to the external solution but not permeation. Its voltage dependence therefore can be used to calculate the δ of the Mg02+ blocking site, but its value cannot be used to predict the Mgo2+ IC50.
Discussion
These results reveal that occupation of the permeant ion-binding sites has enormous effects on the rates of Mgo2+ block and unblock. At −60 mV with control ion concentrations, occupation of cation-binding site(s) prevents Mgo2+ from blocking 99.4% of the time. In the absence of permeant ion binding, the estimated Mgo2+-blocking rate (1.10 ⋅ 109 M−1s−1 at 0 mV) is extraordinarily high: it is close to the predicted diffusion limit (33). This suggests that the highest energy barrier that Mgo2+ must cross to block the channel of the NMDA receptor is very low. Unblock of Mg2+ to the external solution is prevented by Nao+ lock-in 96.1% of the time with control ion concentrations; as a result, the fraction of blocking Mg ions that permeate is 22-fold greater than it would be if lock-in did not occur. The permeant ion-binding sites might play a role in the unusual permeation properties of the NMDA receptor, such as its high Ca2+ permeability (4, 17, 34, 35) and the ability of Mgo2+ to permeate rapidly under some experimental conditions (17, 18).
It is likely that under physiological conditions occupation of the permeant ion-binding sites has a powerful effect on equilibrium block by Mgo2+ as well as on the kinetics of block. According to the model developed here, the true Mgo2+ IC50 [the IC50 in the absence of permeant ions, calculated as (k−o + k−i)/k+)] at −60 mV is 10.9 μM. With 140 mM Nao+ and 130 mM Csi+, the predicted IC50 is 95.4 μM, nearly 10-fold higher. This increase in IC50 is predicted to be predominantly caused by the lowering of k+ by Csi+; the effects of Nao+ on IC50 are predicted to be small because increasing [Na+]o lowers both k+ and k−,o. Consistent with these predictions, in whole-cell experiments and with [Mg2+]o up to 3 mM, decreasing [Cs+]i causes a large and voltage-dependent decrease in the Mgo2+ IC50 (36). Decreasing [Na+]o has much smaller and less voltage-dependent effects (37). An effect of permeant ion-binding sites on equilibrium block by Mgo2+ is supported further by two additional observations: decreasing internal [K+] strengthens inhibition of macroscopic NMDA responses by Mgo2+ (8), and the voltage dependence of the Mgo2+ IC50 becomes greater as the voltages over which measurements are made become more depolarized (9, 12, 38). The latter observation is predicted by the curvature we observed in semilog plots of k+,app (Fig. 2) and k−,app (Fig. 3) as a function of voltage. The sensitivity of the voltage dependence of block to the voltage range over which measurements are made also may contribute to the 2-fold variability in estimates of the electrical location of the Mgo2+-blocking site (4–12). Additional possible causes of this variability include differences in the permeant monovalent cation concentrations used and difference in the subunit composition of the NMDA receptors present in the cells studied. Subunit composition is important because the voltage dependence of block is NR2 subunit-specific (9) and because inaccuracies in macroscopic measurements could be introduced by potentiation of some NMDA receptor subtypes by Mgo2+ (39).
Our data suggest that the permeant ion-binding sites are in or near the single-file region of the channel and, therefore, are likely to play a role in ion permeation. If they were the only, or the highest-affinity, monovalent permeant ion-binding sites in the channel, then half-saturation of NMDA-receptor single-channel current should occur when half these sites are occupied. Previous estimates of the permeant monovalent cation concentrations at which half-maximal saturation of NMDA-receptor single-channel currents occurs range from 0.5 to 129 mM (8, 10, 23, 40). The variation in these measurements, which were made under differing conditions and with different permeant ions, prevents their use in assessing the potential role of the permeant ion-binding sites in channel permeation. It nevertheless seems likely that other ion-binding sites, such as a site near the Q/R/N sites (19) or an intracellular permeant ion-binding site (26), are involved in monovalent cation permeation.
In the model developed here, occupation of either permeant ion-binding site prevents access of Mgo2+ to the channel. This, coupled with the observation that Nao+ binding to either site is voltage-independent, appears to imply that the sites are in close proximity. However, the KNa of either permeant ion-binding site is the same whether or not the other site is occupied. This appears to imply, in contrast, that the sites are relatively distant. Previous work suggests that the sites indeed could be in close proximity: when one of two neighboring cation-binding sites in the pore of cyclic nucleotide-gated channels binds a proton, the proton affinity of the other site is unaffected (41). Occupation of a permeant ion-binding site on NMDA receptors may affect Mgo2+ more strongly than Na+ or Cs+ because of the larger charge on Mg2+, its greater hydrated radius, its slower exchange of waters of hydration, or a combination of these factors. It also is possible that occupation of one permeant ion-binding site has a moderate effect on monovalent cation binding to the second site. To test this possibility, we fit to the Fig. 2 data a modified model in which the binding affinity of the second Na+ could be equal to or lower than the binding affinity of the first Na+. The best fits still were achieved when each Nao+ bound with the same affinity.
The model of Mgo2+ block developed here permitted us to quantify the voltage dependence of the Mgo2+ blocking and unblocking rates independent from the influence of permeant ions. As modeled here, these “true” blocking and unblocking rates satisfy the assumptions of the Woodhull model (3): most importantly, that the voltage dependence of the rates depends only on the electrical locations of the barrier to Mgo2+ entry, the Mgo2+-binding site, and the barrier to Mgo2+ permeation. If the model is correct, then we can now determine accurately the electrical locations of the channel structures that govern block by Mgo2+. The voltage dependence of k+ places the peak of the barrier to Mgo2+ entry at a fractional electrical depth of 0.23 through the membrane field; the additional voltage dependence of k−,o places the Mgo2+-binding site (δ) at 0.47; the additional voltage dependence of k−,i places the peak of the barrier to permeation at 0.73. These locations are much shallower than estimated from previous data or from our data by using traditional approaches: based on the approximately exponential voltage dependence of k+,app and k−,app from −40 to −90 mV, the peak of the barrier to Mgo2+ entry is at 75%, and the Mgo2+-binding site at 89%, through the membrane voltage field. Traditional approaches provide exaggerated estimates of electrical depth because the voltage-dependent binding of Csi+ inflates the voltage dependence of k+,app at Vm values positive of −100 mV (note in Fig. 2 the low voltage dependence of k+,app at very negative Vm values, where Csi+ has little effect on k+,app).
Our conclusion that Mgo2+ traverses only 47% of the membrane voltage field to reach its blocking site appears consistent with evidence that two asparagine residues (12, 19, 20) near the tip (22) of the M2 region of NR2 subunits help form the Mgo2+-blocking site. This electrical location of the Mgo2+-blocking site also leaves room for a nearby but distinct internal Mg2+-binding site 55–71% through the membrane voltage field (13–16). The estimated depth of the internal Mg2+-binding site is somewhat discordant with our location of the permeation barrier at 73% through the field; one possible explanation is that the electrical depth of block by internal Mg2+ has been misestimated because of occupation of an internal binding site for permeant ions (26). Finally, our results are consistent with the observation (9) that structural domains outside the M2 region have a strong influence on channel block by Mgo2+, because the M2 region is unlikely to contribute to the external permeant ion-binding sites (22, 42).
In summary, the data and model presented here provide an explanation for the high voltage dependence of block by Mgo2+, give a corrected location for the Mgo2+-blocking site, demonstrate a lock-in effect of Nao+ on Mgo2+ during block, and provide a quantitative estimate of the rate of Mgo2+ permeation.
Acknowledgments
We thank P. Ascher, J. Dilmore, and A. Qian for comments on the manuscript and J. Jaumotte for skilled technical assistance. This work was supported by grants from the National Institutes of Health to J.W.J.
Abbreviations
- k+,app
apparent blocking rate constant
- k−,app
apparent unblocking rate constant
- Csi+
internal Cs+
- Mgo2+
external Mg2+
- τb
mean duration of block
- Na0+
external Na+
- NMDA
N-methyl-d-aspartate
- τo
channel mean open time
- Vm
membrane potential
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Nature (London) 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 2.Mayer M L, Westbrook G L, Guthrie P B. Nature (London) 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 3.Woodhull A M. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascher P, Nowak L. J Physiol (London) 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayer M L, Westbrook G L. J Physiol (London) 1985;361:65–90. doi: 10.1113/jphysiol.1985.sp015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jahr C E, Stevens C F. J Neurosci. 1990;10:1830–1837. doi: 10.1523/JNEUROSCI.10-06-01830.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Huang L Y. Nature (London) 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 8.Ruppersberg J P, von Kitzing E, Schoepfer R. Semin Neurosci. 1994;6:87–96. [Google Scholar]
- 9.Kuner T, Schoepfer R. J Neurosci. 1996;16:3549–3558. doi: 10.1523/JNEUROSCI.16-11-03549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Premkumar L S, Auerbach A. Neuron. 1996;16:869–880. doi: 10.1016/s0896-6273(00)80107-5. [DOI] [PubMed] [Google Scholar]
- 11.Sharma G, Stevens C F. Proc Natl Acad Sci USA. 1996;93:9259–9263. doi: 10.1073/pnas.93.17.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wollmuth L P, Kuner T, Sakmann B. J Physiol (London) 1998;506:13–32. doi: 10.1111/j.1469-7793.1998.013bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson J W, Ascher P. Biophys J. 1990;57:1085–1090. doi: 10.1016/S0006-3495(90)82626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li-Smerin Y, Johnson J W. J Physiol (London) 1996;491:121–135. doi: 10.1113/jphysiol.1996.sp021201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kupper J, Ascher P, Neyton J. J Physiol (London) 1998;507:1–12. doi: 10.1111/j.1469-7793.1998.001bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wollmuth L P, Kuner T, Sakmann B. J Physiol (London) 1998;506:33–52. doi: 10.1111/j.1469-7793.1998.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayer M L, Westbrook G L. J Physiol (London) 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stout A K, Li-Smerin Y, Johnson J W, Reynolds I J. J Physiol (London) 1996;492:641–657. doi: 10.1113/jphysiol.1996.sp021334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burnashev N, Schoepfer R, Monyer H, Ruppersberg J P, Gunther W, Seeburg P H, Sakmann B. Science. 1992;257:1415–1419. doi: 10.1126/science.1382314. [DOI] [PubMed] [Google Scholar]
- 20.Mori H, Masaki H, Yamakura T, Mishina M. Nature (London) 1992;358:673–675. doi: 10.1038/358673a0. [DOI] [PubMed] [Google Scholar]
- 21.Kupper J, Ascher P, Neyton J. Proc Natl Acad Sci USA. 1996;93:8648–8653. doi: 10.1073/pnas.93.16.8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuner T, Wollmuth L P, Karlin A, Seeburg P H, Sakmann B. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 23.Zarei M M, Dani J A. J Gen Physiol. 1994;103:231–248. doi: 10.1085/jgp.103.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald J F, Bartlett M C, Mody I, Pahapill P, Reynolds J N, Salter M W, Schneiderman J H, Pennefather P S. J Physiol (London) 1991;432:483–508. doi: 10.1113/jphysiol.1991.sp018396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen H S, Lipton S A. J Physiol (London) 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antonov S M, Gmiro V E, Johnson J W. Nat Neurosci. 1998;1:451–461. doi: 10.1038/2167. [DOI] [PubMed] [Google Scholar]
- 27.Sharma G, Stevens C F. Proc Natl Acad Sci USA. 1996;93:14170–14175. doi: 10.1073/pnas.93.24.14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonov S M, Johnson J W. J Physiol (London) 1996;493:425–445. doi: 10.1113/jphysiol.1996.sp021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 30.Neher E, Steinbach J H. J Physiol (London) 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong J, Russell S L, Pritchett D B, Molinoff P B, Williams K. Mol Pharmacol. 1994;45:846–853. [PubMed] [Google Scholar]
- 32.Neyton J, Miller C. J Gen Physiol. 1988;92:549–567. doi: 10.1085/jgp.92.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 34.MacDermott A B, Mayer M L, Westbrook G L, Smith S J, Barker J L. Nature (London) 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 35.Jahr C E, Stevens C F. Proc Natl Acad Sci USA. 1993;90:11573–11577. doi: 10.1073/pnas.90.24.11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian A, Johnson J W. Soc Neurosci Abstr. 1997;23:943. [Google Scholar]
- 37.Qian A, Johnson J W. Soc Neurosci Abstr. 1998;24:343. [Google Scholar]
- 38.Kleckner N W, Dingledine R. Brain Res Mol Brain Res. 1991;11:151–159. [PubMed] [Google Scholar]
- 39.Paoletti P, Neyton J, Ascher P. Neuron. 1995;15:1109–1120. doi: 10.1016/0896-6273(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 40.Iino M, Ciani S, Tsuzuki K, Ozawa S, Kidokoro Y. J Membr Biol. 1997;155:143–156. doi: 10.1007/s002329900166. [DOI] [PubMed] [Google Scholar]
- 41.Root M J, MacKinnon R. Science. 1994;265:1852–1856. doi: 10.1126/science.7522344. [DOI] [PubMed] [Google Scholar]
- 42.Beck C, Wollmuth L P, Seeburg P H, Sakmann B, Kuner T. Neuron. 1999;22:559–570. doi: 10.1016/s0896-6273(00)80710-2. [DOI] [PubMed] [Google Scholar]