


Abstract
One major task in the post-genome era is to reconstruct proteomic and genomic interacting networks using high-throughput experiment data. To identify essential nodes/hubs in these interactomes is a way to decipher the critical keys inside biochemical pathways or complex networks. These essential nodes/hubs may serve as potential drug-targets for developing novel therapy of human diseases, such as cancer or infectious disease caused by emerging pathogens. Hub Objects Analyzer (Hubba) is a web-based service for exploring important nodes in an interactome network generated from specific small- or large-scale experimental methods based on graph theory. Two characteristic analysis algorithms, Maximum Neighborhood Component (MNC) and Density of Maximum Neighborhood Component (DMNC) are developed for exploring and identifying hubs/essential nodes from interactome networks. Users can submit their own interaction data in PSI format (Proteomics Standards Initiative, version 2.5 and 1.0), tab format and tab with weight values. User will get an email notification of the calculation complete in minutes or hours, depending on the size of submitted dataset. Hubba result includes a rank given by a composite index, a manifest graph of network to show the relationship amid these hubs, and links for retrieving output files. This proposed method (DMNC || MNC) can be applied to discover some unrecognized hubs from previous dataset. For example, most of the Hubba high-ranked hubs (80% in top 10 hub list, and >70% in top 40 hub list) from the yeast protein interactome data (Y2H experiment) are reported as essential proteins. Since the analysis methods of Hubba are based on topology, it can also be used on other kinds of networks to explore the essential nodes, like networks in yeast, rat, mouse and human. The website of Hubba is freely available at http://hub.iis.sinica.edu.tw/Hubba.
INTRODUCTION
Proteins control and mediate many biological activities via interactions with other protein partners. Information of protein networks derived from protein interactions can serve as a good starting point for understanding the molecular machinery. Besides, elucidating protein interacting partnerships may help annotate unknown proteins and provide further insight into biological networks. Various experimental strategies are available for identifying protein interactions. While the conducive for high-throughput technology on the yeast two-hybrid system, performed in bacteria, yeast, worms, flies and more recently, mice and humans (1–4), enable us to characterize physical protein–protein interactions in the genome-wide scale (5,6). Many interactomes derived from such approaches were collected by different databases, for example, Biomolecular Interaction Network Database (BIND) (7), the Database of Interacting Proteins (DIP) (8), IntAct (9), the Munich Information Center for Protein Sequences (MIPS) (10), STRING (11), REACTOME (12) and some other databases with similar purpose. Besides, some interesting interactomes of host–pathogens (4,13,14) and carcinogenesis (2), were also published recently.
A protein interaction network is naturally complicate and far from a random network. Using the network characters, such as the degree distribution, clustering, diameter and relative graphlet frequency distribution, information can be extracted from a protein–protein network (15). To identify essential nodes/hubs the protein networks is a way to decipher the critical key controllers inside biochemical pathways or complex networks. Combining the gene-expression data with a high-quality yeast protein–protein interaction dataset, Han et al. (16) deliberated on the network dynamics in protein–protein interaction networks and revealed two types of hubs. One of them is more likely to be the module organizers and the other to be the module connectors (17). These essential nodes/hubs may serve as candidates of drug-targets for developing novel therapy of human diseases, such as cancer or infectious disease caused by emerging pathogens.
There are several approaches trying to identify motif/functional modules, while few approaches were attempted to decipher the hub/essential proteins directly. For example, CFinder is a tool for predicting the function of a single protein and for discovering novel protein modules (18). Other similar tools like mfinder (19), FANMOD (20) and MAVisto (21) are designed for network motifs detection. Idowu et al. (22) use degree and BottleNeck methods to identify the possible-essential proteins in the PPI network of Bacillus Subtilis.
Here, we proposed a framework combined with self-developed algorithms and integrated platform named as Hub Objects Analyzer (Hubba) to decipher hub/essential proteins from the user-defined protein interaction networks in graphic mode. Hubba is a web-based service for exploring important nodes in an interactome network generated from specific small- or large-scale experimental methods based on graph theory. In this website, we explore the essential nodes by six characteristic analysis methods on protein–protein interaction network, including Degree, BottleNeck (BN), Edge Percolation Component (EPC), Subgraph Centrality (SC) and two characteristic analysis algorithms developed by us: Maximum Neighborhood Component (MNC) and Density of Maximum Neighborhood Component (DMNC). A double screening scheme (DSS) for exploring and identifying hubs/essential nodes from interactome networks is proposed. Hubba result includes a rank given by a composite index in DSS, a manifest graph of network to show the relationship amid these hubs via SVG viewer (http://www.adobe.com/svg/), and links of results calculated by all algorithms mentioned above. Analyzing the yeast protein interactome data (Y2H experiment) with list of essential proteins from Saccharomyces Genome Database (SGD, http://www.yeastgenome.org/), most of the Hubba high-ranked hubs (80% in top 10 hub list, and >70% in top 40 hub list) from are reported as essential proteins. Since the analysis methods of Hubba are based on topology, it can also be used on other kinds of networks to explore the essential nodes, like networks in yeast, rat, mouse and human. The clues revealed from network topological analysis will provide a new sight to experimental biologists.
SYSTEM IMPLEMENTATION
The Hubba system is built in an open-source structure: Linux (Mandriva 2007, operating system), Apache (web server), PHP (html-embedded scripting language), PostgreSQL (relational database), XMLMakerFlattener (translate data format), Graphviz (graph generator), BGL,ã LAPACK and LAPACK++ (topology calculation). The framework of whole system is depicted in Figure 1. Interaction network among hub/essential proteins can be visualized in PNG format. More annotations of biological functions related with identified hubs can be shown in SVG viewer when input file is fitting the PSI-MI format.
Figure 1.

Inside Hubba. In ‘user mode’ (top), user submits PPI dataset in PSI format, tabulated format with/without weight values and fill the note for this submission on the input page. After the calculation, user will receive the notification to views the result on the result display page. The result will be included general properties of submitted network, list of hub proteins with the graph of interaction among them, Cytoscape's format output and lists of protein ranking scores by several algorithms. In ‘system mode’ (bottom), HubbaD controls the entire process and calls programs to process input data, send notification to users and generate output files.
Algorithms used in Hubba
Hubba explores the possibly essential proteins in the interaction network by six topology-based scoring methods and a DSS.
- Topology-based scoring methods
- Degree (23): in this method, the score of a node v is assigned as the degree of v, D(v), the number of links incident to this node.
- BottleNeck (BN) (15,24): for each node v in an interaction network, a tree of shortest paths starting from v is constructed. Taking v as the root of the tree Tv, the weight of a node w in the tree Tv is the number of descendants of w, that is to say, equal to the number of shortest paths starting from v passing through w. A node w is called a bottle-neck node in Tv if the weight of w is no less than n/4, where n is the number of nodes in Tv. The score of node w, BN(v), is defined to be the number of node v such that w is a bottle-neck node in Tv.
- Edge percolated component (EPC) (25): for an interaction network G, assign a removing probability p to every edge. Let G′ be a realization of the random edge removing from G. If nodes v and w are connected in G′, set δvw be 1, otherwise set δvw be 0. The percolated connectivity of v and w, cvw, is defined to be the average of δvw over realizations. The size of percolated component containing node v, sv, is defined to be the sum of cvw over nodes w. The score of node v, EPC(v), is defined to be sv.
- Subgraph centrality (SC) (26): for a node v, the number of close walks of v of length k is denoted as μk(v). The subgraph centrality of v, SC(v), is defined to be
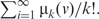
- MNC: the neighborhood of a node v, nodes adjacent to v, induce a subnetwork N(v). The score of node v, MNC(v), is defined to be the size of the maximum connected component of N(v). The neighborhood N(v) is the set of nodes adjacent to v and does not contain node v.
- DMNC: for a node v, let N be the node number and E be the edge number of MNC(v), respectively. The score of node v, DMNC(v), is defined to be E/Nε for some 1 ≤ ε ≤ 2. We may assume that the MNC has a strong community structure, such as a clique percolation in a random network. In our system, ε is set to be 1.7, which is close to 1.67, the ε-value as we assume the neighborhood sub-network has a four-community.
The double screening scheme (DSS)
Each scoring method catches certain postulated topological characteristic of essential proteins. Therefore, a DSS is proposed. That is to say, two scoring methods A and B, are used to extract mixed characteristic of essential proteins. For n, most possible essential proteins are expected in the output, the 2n top ranked proteins by method A are selected firstly. The selected 2n proteins are further ranked by method B and the n top ranked proteins are output. The number 2n is an empirical value for this double screening method. The list of yeast essential protein was integrated with the dataset from functional characterization of the Saccharomyces cerevisiae genome by gene deletion (27) and updated information from SGD (http://www.yeastgenome.org/).
Job processing and result display
The Hubba system separates a job into two modes, ‘user mode’ and ‘system mode’ (Figure 1). In ‘user mode’, protein interaction dataset can be uploaded for network analysis. Three types of data format are accepted: PSI format (Proteomics Standards Initiative, version 2.5 and 1.0), tab format and tab with weight values. The dataset may be submitted by pasting the interaction data in the query form directly, or uploading a file from the local computer. An email address is suggested to provide for those jobs may be time consuming; the Hubba daemon will notify the job completion by email. Once users verify all the parameters and submit their jobs, the process enters ‘system mode’.
All input data in a query are parsed and stored in a temporary database for the following analysis. Hubba will conduct six topological methods and the double screening scheme to submitted dataset and acquire ranking score for each node in the submitted network. The ranking score in Hubba is a composite index calculated by the DSS (DMNC || MNC) as described in the algorithm sections. After all calculations were completed, the process will be directed back to ‘user mode’ for outcome display.
There are three major options in the result page, ‘Hub Selector and Topology Moderator’, ‘Local Network Graph with Hub List’ and ‘Download Area’. In ‘Hub Selector and Topology Moderator’, users can select the top of hubs or search for particular nodes to browse the relationship among these nodes in the submitted network. Users also can manipulate on the advanced options, ‘Check the first-stage nodes’ to show the neighbors of the top/particular nodes, and ‘Display the shortest path’ to mark the shortest path distance between nodes, respectively. In this way, the connectivity among hubs can be easy identified.
An output graph in PNG format is generated by Graphviz and is shown directly in the result page of ‘Local Network Graph with Hub List’. For those query starting from the standard PSI-MI format, the biological functions related to those identified hubs can be shown in SVG viewer. All the output results, including network images and the ranking scores by the DSS and six scoring methods, can be retrieved from the ‘Download Area’. We also provide the output in gml and EPS format, which can be open in Cytoscape (http://www.cytoscape.org/) and edited with standard linux tools for further analysis.
Normally, an analysis job is completed within a few minutes and the result is pushed back to the same web browser window automatically. If a job takes longer than expected, the user can save the link as a bookmark and revisits Hubba later, or follows the link provided in the notice mail to retrieve the analysis results.
RESULTS AND CONCLUSION
The main ideas of the double screening scheme are to select methods catching diverse characters and to include most essential proteins. Firstly, the overlapping of n top lists from different methods is studied. For all the six methods applied to the protein–protein interaction dataset yeast20070107.lst (http://dip.doe-mbi.ucla.edu/), the overlaps in the top 100 ranked proteins of any two scoring methods are expressed in percentage (Supplementary Table S1). Among all methods, DMNC are found to be the one that shares the least proteins with the others. Accordingly, the topological characters extracted by DMNC may differ from those by the other methods. Second, we evaluate the performance of the six scoring method by the coverage of yeast essential proteins. As shown in Table 1, DMNC has the highest hit rate on the essential protein list. Therefore, we choose DMNC as the first method in the DSS. The second method of the double screen scheme is chosen on the same criteria. Among the five methods, MNC is the best mate of DMNC. The scheme improves the hit rate (Table 1, last column).
Table 1.
The percentage of top n ranked proteins, identified from the yeast protein interaction dataset (yeast20070107.lst) by six scoring methods and by the double screening scheme (DMNC || MNC, Hubba algorithm), to be yeast essential proteins
| Scoring methods | DSS | ||||||
|---|---|---|---|---|---|---|---|
| Degree (%) | BN (%) | EPC (%) | SC (%) | MNC (%) | DMNC (%) | DMNC || MNC (%) | |
| Top10 | 60.00 | 50.00 | 30.00 | 30.00 | 40.00 | 80.00 | 100.00 |
| Top20 | 35.00 | 40.00 | 30.00 | 25.00 | 50.00 | 80.00 | 95.00 |
| Top30 | 43.33 | 46.67 | 30.00 | 23.33 | 46.67 | 76.67 | 83.33 |
| Top40 | 45.00 | 47.50 | 30.00 | 20.00 | 45.00 | 70.00 | 72.50 |
| Top50 | 50.00 | 46.00 | 26.00 | 18.00 | 48.00 | 66.00 | 76.00 |
| Top60 | 50.00 | 48.33 | 31.67 | 15.00 | 46.67 | 61.67 | 70.00 |
| Top70 | 48.57 | 47.14 | 34.29 | 14.29 | 48.57 | 57.14 | 67.14 |
| Top80 | 47.50 | 46.25 | 35.00 | 12.50 | 48.75 | 55.50 | 65.50 |
| Top90 | 47.78 | 46.67 | 37.78 | 12.22 | 47.78 | 56.67 | 63.33 |
| Top100 | 48.00 | 45.00 | 41.00 | 14.00 | 48.00 | 58.00 | 63.00 |
For example, 8 of the top 10 proteins found by DMNC has been identified as yeast essential proteins [% = (8/10) × 100%].
Hubba is constructed as a user-friendly interface for dataset uploading and result displaying. After the analysis process is completed, Hubba provides a community graph of the top n ranked (n ≤ 100) hub/essential proteins with the identifier provided in the input dataset (Figure 2, a graph of top 10 list). We utilize a coloring scheme, from red to green, as a cue of the ranking score and a line pattern to discriminate direct interaction (solid line) from indirect interaction (dotted line). Furthermore, the advanced options of browsing the neighborhood of these hubs and the shortest path distance between hub nodes. Hubba has been applied to discover hubs/essential proteins from the PPI dataset (downloaded from IntAct website) of five model organisms. The more precalculated results are available in our help page (http://hub.iis.sinica.edu.tw/Hubba/help.htm).
Figure 2.

The top10 nodes of the dataset yeast20070107.lst explored in Hubba. A coloring scheme is used to display the ranking score of each node. When the node is redder, the ranking of it will be higher. Solid lines indicate the connected nodes interact to each other (direct connections), while dotted lines with a number indicate the shortest path (distance) between two linking nodes (indirect connections).
Identifying hubs or fragile motifs are very important in network biology. For example, based on the overview of the interaction among human proteins and proteins from 190 pathogen stains is revealed that both viral and bacterial pathogens tend to interact with hub and bottlenecks in the human PPI network (28). Chuang et al. (29) applied a protein-network-based approach to analyze the expression profiles of the two cohorts of breast cancer patients. They found several notorious cancer markers, such as P53, KRAS, HRAS, HER-2/neu and PIK3CA, are located on the interconnecting bottleneck of many expression-responsive genes, while these markers could not serve as indicators of the disease state using gene-expression data alone. Feldman and his co-workers (30) conclude some network properties of human inheritable diseases. They found that genes and proteins harboring variation causing the same disease phenotype tend to form directly connected clusters. A similar purpose for identifying disease-associated proteins can be found in Hubba, which accepts a query of an interested list on a user-defined network and provides output for the shortest path among them. In this way, nodes in the paths may serve as candidates related to the disorder the query list involved.
The topological analysis like Hubba is dependent on the completion and accuracy of the input interactome dataset. While this platform provides a chance to build a network related to the scenario the customized interaction dataset derived. Therefore, the secrets hidden inside the networks with specific spatiotemporal scenarios will be deciphered and sketched. We hope this approach can lead to a new strategy for exploring the mechanism of cancer formation and pathogens infection. And it may lead to new therapies and novel insights in understanding basic mechanisms controlling normal cellular processes and disease pathologies.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank National Science Council (NSC)/National Research Program of Genomic Medicine (NRPGM), Taiwan, for financially supporting this research through NSC 96-3112-B-001-002 to C-.Y.L. and NSC 95-2221-E-008 -055 to C-.W.H. Funding to pay the Open Access publication charges for this article was provided by NSC 96-3112-B-001-002 to C-.Y. L.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. PNAS. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonsson PF, Bates PA. Global topological features of cancer proteins in the human interactome. Bioinformatics. 2006;22:2291–2297. doi: 10.1093/bioinformatics/btl390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 4.Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, et al. Herpesviral protein networks and their interaction with the human proteome. Science. 2006;311:239–242. doi: 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]
- 5.Lin CY, Chen CL, Cho CS, Wang LM, Chang CM, Chen PY, Lo CZ, Hsiung CA. hp-DPI: Helicobacter pylori database of protein interactomes—embracing experimental and inferred interactions. Bioinformatics. 2005;21:1288–1290. doi: 10.1093/bioinformatics/bti101. [DOI] [PubMed] [Google Scholar]
- 6.Lin C-Y, Chen S-H, Cho C-S, Chen C-L, Lin F-K, Lin C-H, Chen P-Y, Lo C-Z, Hsiung CA. Fly-DPI: database of protein interactomes for D. melanogaster in the approach of systems biology. BMC Bioinform. 2006;7:S18. doi: 10.1186/1471-2105-7-S5-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bader GD, Betel D, Hogue CW. BIND: the biomolecular interaction network database. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deane CM, Salwinski L, Xenarios I, Eisenberg D. Protien interactions: two methods for assessment of the reliability of high throughput observations. Mol. Cell Proteomics. 2002;5:349–356. doi: 10.1074/mcp.m100037-mcp200. [DOI] [PubMed] [Google Scholar]
- 9.Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, Dimmer E, Feuermann M, Friedrichsen A, Huntley R, et al. IntAct—open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoof H, Spannagl M, Yang L, Ernst R, Gundlach H, Haase D, Haberer G, Mayer KF. Munich information center for protein sequences plant genome resources: a framework for integrative and comparative analyses 1(W) Plant Physiol. 2005;138:1301–1309. doi: 10.1104/pp.104.059188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Kruger B, Snel B, Bork P. STRING 7—recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35:D358–D362. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vastrik I, D’Eustachio P, Schmidt E, Joshi-Tope G, Gopinath G, Croft D, de Bono B, Gillespie M, Jassal B, Lewis S, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39. doi: 10.1186/gb-2007-8-3-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dyer MD, Murali TM, Sobral BW. Computational prediction of host-pathogen protein protein interactions. Bioinformatics. 2007;23:i159–i166. doi: 10.1093/bioinformatics/btm208. [DOI] [PubMed] [Google Scholar]
- 14.Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, et al. Epstein-Barr virus and virus human protein interaction maps. Proc. Natl Acad. Sci. USA. 2007;104:7606–7611. doi: 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Przulj N, Wigle DA, Jurisica I. Functional topology in a network of protein interactions. Bioinformatics. 2004;20:340–348. doi: 10.1093/bioinformatics/btg415. [DOI] [PubMed] [Google Scholar]
- 16.Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP, et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 17.Ekman D, Sara L, Åsa KB, Arne E. What properties characterize the hub protein of the protein protein interaction network of Saccharomyces cerevisiae. Genome Biol. 2006;7:R45. doi: 10.1186/gb-2006-7-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adamcsek B, Palla G, Farkas IJ, Derenyi I, Vicsek T. CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics. 2006;22:1021–1023. doi: 10.1093/bioinformatics/btl039. [DOI] [PubMed] [Google Scholar]
- 19.Kashtan N, Itzkovitz S, Milo R, Alon U. Efficient sampling algorithm for estimating subgraph concentrations and detecting network motifs. Bioinformatics. 2004;20:1746–1758. doi: 10.1093/bioinformatics/bth163. [DOI] [PubMed] [Google Scholar]
- 20.Wernicke S, Rasche F. FANMOD: a tool for fast network motif detection. Bioinformatics. 2006;22:1152–1153. doi: 10.1093/bioinformatics/btl038. [DOI] [PubMed] [Google Scholar]
- 21.Schreiber F, Schwobbermeyer H. MAVisto: a tool for the exploration of network motifs. Bioinformatics. 2005;21:3572–3574. doi: 10.1093/bioinformatics/bti556. [DOI] [PubMed] [Google Scholar]
- 22.Idowu OC, Lynden SJ, Young MP, Andras P. Bacillus Subtilis protein interaction network analysis. In 2004 IEEE Computational Systems Bioinformatics Conference (CSB'04), 2004:623–625. [Google Scholar]
- 23.Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 24.Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007;3:e59. doi: 10.1371/journal.pcbi.0030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chin C-S, Manoj PS. Global snapshot of a protein interaction network—a percolation based approach. Bioinformatics. 2003;19:2413–2419. doi: 10.1093/bioinformatics/btg339. [DOI] [PubMed] [Google Scholar]
- 26.Estrada E, Rodríguez-Velázquez JA. Subgraph centrality in complex network. Phys. Rev. 2005;71:056103. doi: 10.1103/PhysRevE.71.056103. [DOI] [PubMed] [Google Scholar]
- 27.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 28.Dyer MD, Murali TM, Sobral BW. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008;4:e32. doi: 10.1371/journal.ppat.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feldman I, Rzhetsky A, Vitkup D. Network properties of genes harboring inherited disease mutations. Proc. Natl Acad. Sci. USA. 2008;105:4323–4328. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.