


Abstract
There are many methods for introducing random mutations into nucleic acid sequences. Previously, we described a suite of programmes for estimating the completeness and diversity of randomized DNA libraries generated by a number of these protocols. Our programmes suggested some empirical guidelines for library design; however, no information was provided regarding library diversity at the protein (rather than DNA) level. We have now updated our web server, enabling analysis of translated libraries constructed by site-saturation mutagenesis and error-prone PCR (epPCR). We introduce GLUE-Including Translation (GLUE-IT), which finds the expected amino acid completeness of libraries in which up to six codons have been independently varied (according to any user-specified randomization scheme). We provide two tools for assisting with experimental design: CodonCalculator, for assessing amino acids corresponding to given randomized codons; and AA-Calculator, for finding degenerate codons that encode user-specified sets of amino acids. We also present PEDEL-AA, which calculates amino acid statistics for libraries generated by epPCR. Input includes the parent sequence, overall mutation rate, library size, indel rates and a nucleotide mutation matrix. Output includes amino acid completeness and diversity statistics, and the number and length distribution of sequences truncated by premature termination codons. The web interfaces are available at http://guinevere.otago.ac.nz/stats.html.
INTRODUCTION
In the past 15 years, directed evolution has developed into a broadly applicable strategy for generating new biomolecules with desirable properties, for probing protein structure and function, and for addressing fundamental questions in molecular evolution. In this approach, random mutagenesis is used to produce a large and diverse library of nucleic acid sequences, which is subsequently interrogated for rare, improved variants. Myriad protocols have been developed to produce the necessary molecular diversity (1–3). However, our ability to generate and screen randomized libraries is dwarfed by the amount of molecular diversity contained in protein sequence space. Even for a small, 100-residue protein, there are more potential amino acid sequences than there are atoms in the observable Universe (4).
Increasingly, it is recognized that high-quality libraries are critical to the success of directed evolution experiments (5,6). Previously, we argued that the likelihood of finding a variant with a desired function in a randomized library is maximized when the library is maximally diverse (7). To the experimentalist, this corresponds to a library containing as few redundant sequences (including copies of the unmutated parental gene) and as many full-length sequences (lacking premature termination codons) as possible. To aid in the design of maximally diverse libraries, we developed a suite of user-friendly programmes for estimating the completeness and diversity that they contain (4,8). These programmes were limited to estimating library diversity at the nucleic acid level, and provided no explicit information regarding the translated products of the randomized genes. In this article, we describe an expanded web server, which enables the analysis of protein diversity in randomized libraries that have been generated by site-saturation mutagenesis and error-prone PCR (epPCR). The nucleotide programmes GLUE (for randomization techniques where all DNA sequence variants are equally likely), PEDEL (Programme for Estimating Diversity in Error-prone PCR Libraries) and DRIVeR (Diversity Resulting from In Vitro Recombination) are still maintained on the website, and have been described previously (4,8).
GLUE-IT
One of our previous programmes, GLUE, is broadly applicable to any protocol where all gene variants have an equal probability of occurring in a library. The most commonly used example is site-saturation mutagenesis (also referred to as oligonucleotide-directed randomization), in which randomized bases are incorporated into one or more of the primers in a PCR, allowing the generation of diversity at specific sites in an amplified gene. Other techniques that result in equally probable daughter variants (at the DNA level) include MAX randomization (9) and versions of DNA shuffling that utilize designed oligonucleotides (10–12). GLUE is also a useful estimator of the diversity in libraries generated by incremental truncation strategies, such as Expression of Soluble Proteins by Random Incremental Truncation (ESPRIT) (13), in which variants are close to being equally probable (14).
We now introduce GLUE-Including Translation (GLUE-IT), which outputs the expected amino acid level diversity in any site-saturation mutagenesis library with up to six variable codons. The user specifies the fully or partly randomized scheme used for each of the variable codons, and the size of the library that they have constructed (or, more often, the number of clones that they plan to screen).
We provide two tools (CodonCalculator and AA-Calculator) to assist in choosing an appropriate randomization scheme for library construction. On specifying a fully or partly randomized codon, XYZ, CodonCalculator will output the possible amino acid variants and the number of times that each is encoded. AA-Calculator performs the opposite function: the user can specify a desired set of amino acids, and AA-Calculator will find the degenerate codon(s) that are optimal for encoding them. Up to 50 degenerate codons are listed, ranked according to the fraction of the XYZ-specified codons that code for the desired amino acids. AA-Calculator therefore offers a user-friendly alternative to downloading and executing the LibDesign algorithm (15), and provides users with a replacement for the Combinatorial Codons programme (16), which (as far as we are aware) is no longer available online.
On entering the randomization scheme and library size, GLUE-IT will output a summary of statistics, including the number of possible DNA and amino acid variants that are encoded by each randomized codon and the total number of possible amino acid variants in the library. The probability of a particular variant vi being present in the library is 1 − (1 − pi)L, where pi is the probability of any particular variant in the library being vi, and L is the library size. In the case of six fully randomized (NNN) codons, there are 206 = 6.4 × 107 possible variants. To quickly calculate the expected number of distinct variants in the library, 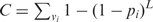 , variants are grouped according to the number of ways in which they can be encoded. Each individual amino acid can be encoded by between one and six equiprobable codons, so for six randomized codons there are at most just 66 = 46 656 different pi values to calculate. For convenience, variants that encode stop codons are assumed to be non-functional and are omitted from the final total, C.
, variants are grouped according to the number of ways in which they can be encoded. Each individual amino acid can be encoded by between one and six equiprobable codons, so for six randomized codons there are at most just 66 = 46 656 different pi values to calculate. For convenience, variants that encode stop codons are assumed to be non-functional and are omitted from the final total, C.
Consider an example in which two libraries are constructed, each containing two codons that have been targeted for randomization. This is the starting point for the Combinatorial Active site Saturation Test (CASTing), in which small, focussed libraries are produced by randomizing several sets of 2–3 amino acid positions around an enzyme active site (5,17–20). In our example, suppose that the first library contains one codon randomized according to the NNK scheme (N = G/A/T/C; K = G/T), while we use NDT (D = G/A/T) in the second position. CodonCalculator tells us that all 20 amino acids (plus one stop codon, TAG) are encoded in the NNK scheme. NDT specifies a more limited set of 12 amino acids (C, D, F, G, H, I, L, N, R, S, V and Y; one codon each). There are 32 × 12 = 384 possible codon variants in the resulting library, encoding 20 × 12 = 240 possible amino acid variants (the output of GLUE-IT includes these calculations).
Suppose that the second library is constructed using an NNB codon (B = G/T/C) and an NAY codon (Y = T/C). This library is equally diverse at the DNA level (48 × 8 = 384 variants), but less diverse when it is translated (20 × 4 = 80 amino acid variants). Because the number of DNA variants is the same in each of the two libraries, GLUE treats each library identically. The output suggests that approximately 1500 clones from each library should be screened, to ensure 98% coverage (Table 1). By considering amino acid diversity, GLUE-IT paints a rather different picture. Now it becomes clear that a much smaller screening effort would be justified for the NNB + NAY library: sampling just 500 clones would ensure that 98% of the translated library had been interrogated.
Table 1.
Completeness and diversity statistics for two hypothetical site-saturation mutagenesis libraries, in which two codons have been randomized according to different schemes (NNK + NDT or NNB + NAY)
| Library | No. of clones sampled | GLUE | GLUE-IT | ||
|---|---|---|---|---|---|
| No. of distinct DNA variants | DNA completenessa | No. of distinct amino acid variantsb | Amino acid completenessc | ||
| NNK + NDT | 100 | 88 | 0.23 | 77 | 0.32 |
| 500 | 280 | 0.73 | 196 | 0.82 | |
| 1000 | 356 | 0.93 | 229 | 0.95 | |
| 1500 | 376 | 0.98 | 237 | 0.99 | |
| NNB + NAY | 100 | 88 | 0.23 | 53 | 0.66 |
| 500 | 280 | 0.73 | 78 | 0.98 | |
| 1000 | 356 | 0.93 | 80 | 1.00 | |
| 1500 | 376 | 0.98 | 80 | 1.00 | |
aThe fraction of all possible DNA sequence variants (384 for each library) that are represented in the sample.
bNot including variants with stop codons.
cThe fraction of all possible amino acid variants (240 for Library 1; 80 for Library 2) that are sampled.
GLUE-IT also calculates the probability that a given library contains all possible amino acid variants, which is a related but distinct statistic (4). Using this, it is possible to determine empirically the number of clones to screen in order to achieve a fixed probability of sampling all variants. For example, approximately 1150 clones from the NNB + NAY library should be screened to give a 95% chance that every possible sequence variant is sampled at least once.
PEDEL AND THE PCR DISTRIBUTION
While there is a growing emphasis on targeting diversity in well-sampled, focussed libraries (5,6), epPCR remains a common means of generating random diversity at any position in a gene. Indeed, a number of commercial vendors now sell epPCR kits, usually designed to overcome the well-documented biases in nucleotide misincorporation by Taq polymerase (21). In epPCR, the potential for generating mutations at any position in a gene ensures that the number of possible variants is usually much larger than that which can be screened experimentally. Therefore, it becomes more informative to assess the number of distinct sequences that are present in the library. This is in contrast to site-saturation mutagenesis, where GLUE-IT can inform strategies for sampling all or most of the possible sequence variants.
Previously, we described PEDEL, which calculates the expected number of distinct DNA variants in an epPCR library, given the library size, the mean mutation rate and the length of the template sequence (4,8). The underlying algorithm divides the library into sub-libraries, each of which contains variants with exactly x mutations. The completeness and diversity of each sub-library is analyzed separately. The original implementation of PEDEL assumed that the number of mutations per daughter sequence (estimated by sequencing a handful of library members) follows a Poisson distribution. The web server now also includes the option to use the ‘PCR distribution’ (22), which is the preferred option provided the appropriate data from the epPCR are known. In addition to the mean mutation rate, calculations using the PCR distribution require the user to input the number of PCR thermal cycles, n, and also the PCR efficiency (i.e. the probability that any particular sequence is duplicated in a given PCR cycle; eff). In turn, estimating eff requires the experimentalist to note the initial amount of template DNA and the final yield of PCR product, so that the number of doublings in the PCR, d, can be calculated:
The PCR efficiency is then given by:
For convenience, we have included a tool for calculating eff, given d and n.
PEDEL-AA
As with site-saturation mutagenesis, it is ultimately more useful to estimate the protein sequence diversity in an epPCR library, rather than the DNA sequence diversity. Addressing this problem is not trivial, as the degeneracy of the genetic code ensures that amino acid sequence variants are not equiprobable. Many substitutions are translationally silent, and >1 substitution in a single codon is rare in epPCR, reducing the number of accessible amino acid variants.
Assessing the protein-level diversity in an epPCR library therefore requires consideration of the template sequence, the overall (DNA) mutation rate, the total size of the library and the nucleotide mutation matrix. This final parameter is an estimate of the relative frequencies of all 12 possible point mutations. It can be estimated from previous libraries [such as the one described by Shafikhani et al. (21)] or, for greater accuracy, it can be obtained by sequencing randomly chosen members of one's own library. Volles and Lansbury (23) previously described a Monte Carlo simulation programme that uses these inputs to randomly simulate every individual sequence in a library, and also produce some analytic statistics. While this powerful approach allows a variety of library statistics to be estimated, it is CPU-intensive. The programme also requires downloading and the correct formatting of input files. Therefore, we have developed and added to our web server an amino acid version of PEDEL (PEDEL-AA) that combines the sub-library analyses of PEDEL with these new inputs to determine protein sequence diversity.
A very brief description of the PEDEL-AA algorithm follows; further details are given in the notes on the website. The number of nucleotide substitutions per variant is assumed to follow the PCR distribution P(xnt). The distribution of truncated variants is then calculated and subtracted from P(xnt). Next, P(xnt) is converted to the amino acid distribution, P(xaa), by assuming that, for each xnt, the number of non-synonymous amino acid substitutions resulting from exactly xnt nucleotide substitutions follows a binomial distribution, B(xnt,f), where f is the mean number of non-synonymous amino acid substitutions per nucleotide substitution.
The input library is conceptually divided into sub-libraries Lx (x = 0, 1, 2, …) where the sub-library Lx comprises all variants in the library with exactly x amino acid substitutions. The total number of possible variants with exactly x amino acid substitutions is represented by Vx. We use two estimates for Vx – viz: Vx1 and Vx2. Vx1 is an estimate of the number of ‘easy-to-reach’ variants; that is, those variants where each substituted amino acid is accessible by just a single nucleotide substitution in the respective codon. Vx2 is the total number of variants with exactly x amino acid substitutions. Although most variants in a sub-library will be of type Vx1, variants of type Vx2 may contribute significantly to the total number of distinct variants, Cx, when the sub-library size Lx is large compared with the number of variants Vx1.
When Lx << Vx then Cx ∼ Lx (i.e. nearly all variants in Lx are distinct). For PEDEL-AA, we use this approximation when Lx < 0.1 × Vx1. This is usually the case for x ≥ 3 and almost always the case for x ≥ 4. For x = 0, 1 and 2, we calculate the expected number of distinct variants, Cx, analytically. In the rare cases where the Cx ∼ Lx approximation can not be used for all x ≥ 3, a rough approximation is used for the remaining Cx values.
The approximations used in PEDEL-AA have minimal effect for ordinary mutations rates, library sizes and epPCR template lengths. Table 2 provides an illustrative example, in which the characteristics of an α-synuclein epPCR library, as estimated by PEDEL-AA and by the Monte Carlo simulations of Volles and Lansbury (23), are compared. The results are in excellent agreement.
Table 2.
Characteristics of an α-synuclein epPCR library,a estimated by PEDEL-AA and by a previously-described Monte Carlo library diversity algorithm (23)
| Property | PEDEL-AA | Ref. (23) |
|---|---|---|
| Prematurely truncated variants (proportion of total library) | 16% | 15% |
| Number of full-length clones | 3.2 × 106 | 3.1 × 106 |
| Protein mutation frequency per amino acid | 0.016 | 0.016 |
| Mean number of mutations per protein | 2.1 | 2.1 |
| Unmutated (wild-type) sequences (proportion of total library) | 14% | 14% |
| Number of unique proteins in the library | 1.3 × 106 | 1.3 × 106 |
| Number of different point mutations in the library | 1989 | 1990 |
| Number of unique single-mutation variants in the library | 1618 | 1566 |
aThe epPCR library was constructed by Volles and Lansbury (23), and consisted of 3.77 × 106 clones with an average of 3.2 nucleotide mutations per clone. The template for randomization was 399 bp in length (coding for amino acids 8–140 of the α-synuclein protein). Table 1 of Volles and Lansbury (23) was used for the nucleotide mutation matrix.
Perhaps the most useful feature of PEDEL-AA is its ability to quickly estimate the total number of unique proteins in an epPCR library. Unlike PEDEL, PEDEL-AA is able to calculate and subtract indel-containing sequences, prematurely truncated variants and DNA sequences with synonymous substitutions from the overall estimate of useful diversity. This offers new and predictive guidelines for the process of library construction. For example, we recently performed epPCR on the 1515 bp purF gene from Escherichia coli (24). The resulting library contained 6.4 × 105 transformants and the mean mutation rate was 15.5 mutations per (DNA) sequence. PEDEL suggested that this high mutation rate was optimal; that is, at the DNA level, almost every sequence variant was unique. Reanalyzing the data with PEDEL-AA demonstrates that the very high mutation rate has produced a large number of variants with premature stop codons. In this example, the library of 6.4 × 105 unique DNA sequences only encodes 3.9 × 105 full-length, non-wild-type proteins. Comparing the PEDEL and PEDEL-AA outputs for a range of mutation rates suggests that lowering the mutation rate to approximately eight mutations per sequence would have yielded a maximally diverse library, containing 4.3 × 105 useful sequence variants (Figure 1). This could have been achieved by lowering the concentration of Mn2+ ions and/or by decreasing the number of cycles in the epPCR.
Figure 1.

The estimated numbers of unique DNA sequence variants (CDNA, dashed line) and protein sequence variants (Cprotein, solid line) in a purF epPCR library (24), plotted as a function of the DNA mutation rate, λ. The epPCR comprised 30 thermal cycles, with eff = 0.41. The library contained 6.4 × 105 clones. A total of 7549 bp of DNA sequence was obtained from randomly chosen library members, and 77 substitutions plus one single-nucleotide deletion were observed. These data were used to construct the nucleotide mutation matrix for PEDEL-AA. The library used for genetic selection experiments contained λ = 15.5 mutations per sequence; the estimated sequence diversity it contains is indicated by the vertical arrow on the right. The maximally diverse library is indicated by the vertical arrow at λ = 8. Note that, after peaking, the number of unique amino acid (but not nucleotide) variants decreases with increasing λ, due to an increasing number of truncated sequences.
In addition to a variety of overall library characteristics, a link from the PEDEL-AA output screen provides the user with an estimate of the amino acid sequence diversity contained in each sub-library (i.e. all of those variants containing exactly x mutations; 0 ≤ x ≤ 20). This data includes the fraction of the library containing no mutations (the x = 0 sub-library), and also offers an overview of the range of mutations that the experimentalist should expect to observe in their library variants. A second linked page displays two graphs. The first plots the cumulative probability that a given variant is free of stop codons, up to amino acid x. The second plots the length distribution of variants with premature stop codons. These give an instant graphical overview of regions of the template that are particularly prone to yielding truncated variants.
CONCLUSIONS
Previously, we developed GLUE, PEDEL and DRIVeR to aid in designing and analyzing randomized libraries of nucleic acid sequences (4,8). Here, we have introduced GLUE-IT and PEDEL-AA. By estimating protein-level statistics, these programmes offer new insights into the usable diversity of translated site-saturation and epPCR libraries. Our algorithms make use of some simplifying assumptions that can be avoided by using Monte Carlo simulation approaches (23). However, these assumptions have only minor effects under experimental conditions that are usually encountered in library construction. Our analytic approach is much quicker than the Monte Carlo simulation approach (especially for large library sizes), making it more amenable for use via a web server and enabling experimentalists to obtain rapid estimates of library statistics. Comprehensive mathematical notes and instructions for use can be found on the website.
ACKNOWLEDGEMENTS
We thank Dr Chris Brown (University of Otago) for his continued willingness to host our programmes on his server. Funding to pay the Open Access publication charges for this article was provided by the Institute of Molecular Biosciences at Massey University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lutz S, Patrick WM. Novel methods for directed evolution of enzymes: quality, not quantity. Curr. Opin. Biotechnol. 2004;15:291–297. doi: 10.1016/j.copbio.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Neylon C. Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: library construction methods for directed evolution. Nucleic Acids Res. 2004;32:1448–1459. doi: 10.1093/nar/gkh315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otten LG, Quax WJ. Directed evolution: selecting today's biocatalysts. Biomol. Eng. 2005;22:1–9. doi: 10.1016/j.bioeng.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Patrick WM, Firth AE, Blackburn JM. User-friendly algorithms for estimating completeness and diversity in randomized protein-encoding libraries. Protein Eng. 2003;16:451–457. doi: 10.1093/protein/gzg057. [DOI] [PubMed] [Google Scholar]
- 5.Reetz MT, Carballeira JD. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protoc. 2007;2:891–903. doi: 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- 6.Wong TS, Roccatano D, Schwaneberg U. Steering directed protein evolution: strategies to manage combinatorial complexity of mutant libraries. Environ. Microbiol. 2007;9:2645–2659. doi: 10.1111/j.1462-2920.2007.01411.x. [DOI] [PubMed] [Google Scholar]
- 7.Patrick WM, Firth AE. Strategies and computational tools for improving randomized protein libraries. Biomol. Eng. 2005;22:105–112. doi: 10.1016/j.bioeng.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Firth AE, Patrick WM. Statistics of protein library construction. Bioinformatics. 2005;21:3314–3315. doi: 10.1093/bioinformatics/bti516. [DOI] [PubMed] [Google Scholar]
- 9.Hughes MD, Nagel DA, Santos AF, Sutherland AJ, Hine AV. Removing the redundancy from randomised gene libraries. J. Mol. Biol. 2003;331:973–979. doi: 10.1016/s0022-2836(03)00833-7. [DOI] [PubMed] [Google Scholar]
- 10.Coco WM, Encell LP, Levinson WE, Crist MJ, Loomis AK, Licato LL, Arensdorf JJ, Sica N, Pienkos PT, Monticello DJ. Growth factor engineering by degenerate homoduplex gene family recombination. Nat. Biotechnol. 2002;20:1246–1250. doi: 10.1038/nbt757. [DOI] [PubMed] [Google Scholar]
- 11.Ness JE, Kim S, Gottman A, Pak R, Krebber A, Borchert TV, Govindarajan S, Mundorff EC, Minshull J. Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently. Nat. Biotechnol. 2002;20:1251–1255. doi: 10.1038/nbt754. [DOI] [PubMed] [Google Scholar]
- 12.Zha D, Eipper A, Reetz MT. Assembly of designed oligonucleotides as an efficient method for gene recombination: a new tool in directed evolution. Chem. BioChem. 2003;4:34–39. doi: 10.1002/cbic.200390011. [DOI] [PubMed] [Google Scholar]
- 13.Tarendeau F, Boudet J, Guilligay D, Mas PJ, Bougault CM, Boulo S, Baudin F, Ruigrok RW, Daigle N, Ellenberg J, et al. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 2007;14:229–233. doi: 10.1038/nsmb1212. [DOI] [PubMed] [Google Scholar]
- 14.Ostermeier M. Theoretical distribution of truncation lengths in incremental truncation libraries. Biotechnol. Bioeng. 2003;82:564–577. doi: 10.1002/bit.10604. [DOI] [PubMed] [Google Scholar]
- 15.Mena MA, Daugherty PS. Automated design of degenerate codon libraries. Protein Eng. Des. Sel. 2005;18:559–561. doi: 10.1093/protein/gzi061. [DOI] [PubMed] [Google Scholar]
- 16.Wolf E, Kim PS. Combinatorial codons: a computer program to approximate amino acid probabilities with biased nucleotide usage. Protein Sci. 1999;8:680–688. doi: 10.1110/ps.8.3.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clouthier CM, Kayser MM, Reetz MT. Designing new Baeyer-Villiger monooxygenases using restricted CASTing. J. Org. Chem. 2006;71:8431–8437. doi: 10.1021/jo0613636. [DOI] [PubMed] [Google Scholar]
- 18.Reetz MT, Bocola M, Carballeira JD, Zha D, Vogel A. Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test. Angew. Chem. Int. Ed. Engl. 2005;44:4192–4196. doi: 10.1002/anie.200500767. [DOI] [PubMed] [Google Scholar]
- 19.Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew. Chem. Int. Ed. Engl. 2006;45:7745–7751. doi: 10.1002/anie.200602795. [DOI] [PubMed] [Google Scholar]
- 20.Reetz MT, Wang LW, Bocola M. Directed evolution of enantioselective enzymes: iterative cycles of CASTing for probing protein-sequence space. Angew. Chem. Int. Ed. Engl. 2006;45:1236–1241. doi: 10.1002/anie.200502746. [DOI] [PubMed] [Google Scholar]
- 21.Shafikhani S, Siegel RA, Ferrari E, Schellenberger V. Generation of large libraries of random mutants in Bacillus subtilis by PCR-based plasmid multimerization. Biotechniques. 1997;23:304–310. doi: 10.2144/97232rr01. [DOI] [PubMed] [Google Scholar]
- 22.Drummond DA, Iverson BL, Georgiou G, Arnold FH. Why high-error-rate random mutagenesis libraries are enriched in functional and improved proteins. J. Mol. Biol. 2005;350:806–816. doi: 10.1016/j.jmb.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 23.Volles MJ, Lansbury P.T., Jr. A computer program for the estimation of protein and nucleic acid sequence diversity in random point mutagenesis libraries. Nucleic Acids Res. 2005;33:3667–3677. doi: 10.1093/nar/gki669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patrick WM, Matsumura I. A study in molecular contingency: glutamine phosphoribosylpyrophosphate amidotransferase is a promiscuous and evolvable phosphoribosylanthranilate isomerase. J. Mol. Biol. 2008;377:323–336. doi: 10.1016/j.jmb.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]