


Abstract
We present DAhunter, a web-based server that identifies homologous proteins by comparing domain architectures, the organization of protein domains. A major obstacle in comparison of domain architecture is the existence of ‘promiscuous’ domains, which carry out auxiliary functions and appear in many unrelated proteins. To distinguish these promiscuous domains from protein domains, we assigned a weight score to each domain extracted from RefSeq proteins, based on its abundance and versatility. A domain's score represents its importance in the ‘protein world’ and is used in the comparison of domain architectures. In scoring domains, DAhunter also considers domain combinations as well as single domains. To measure the similarity of two domain architectures, we developed several methods that are based on algorithms used in information retrieval (the cosine similarity, the Goodman–Kruskal γ function, and domain duplication index) and then combined these into a similarity score. Compared with other domain architecture algorithms, DAhunter is better at identifying homology. The server is available at http://www.dahunter.kr and http://localodom.kobic.re.kr/dahunter/index.htm
INTRODUCTION
There are now >600 completely sequenced genomes and >5 million unique protein sequences are available in public databases (1). A common approach for identifying protein function is to assume that proteins with similar sequences have similar functions. Thus, sequence similarity search algorithms, such as, BLAST (2) and FASTA (3), can detect sequence similarities in proteins that have not diverged greatly. However, proteins that have diverged greatly can be homologous even though they exhibit little sequence similarity (4). Thus, sequence-based homology searches can yield false negatives, especially when comparing proteins with multiple domains (5).
Domains are the building blocks of proteins and one of the most useful characteristics for determining protein function (6). The functions of the individual domains of a multi-domain protein contribute to our understanding of the properties of the protein as a whole (7). The sequential order of protein domains is known as the domain architecture. Architectures are useful for classifying evolutionarily related proteins, detecting evolutionarily distant homologs, and comparing multi-domain proteins (8,9). Several previous studies have proposed methods of domain architecture comparison for identification of protein homology. CDART (10) presents a list of proteins with similar domain architectures to a given query sequence by counting the number of shared domains. PDART (11) presents domain architectures in the leaves of a species tree. Djorklund et al. (12) proposed a ‘domain distance’, calculated as the number of domains that differ between two domain architectures.
Multi-domain proteins evolve by gene duplication and domain shuffling, causing certain domains to appear in many unrelated proteins (13). The functions of these ‘promiscuous’ domains are typically auxiliary to the primary protein function (14). Promiscuous domains also have many combinations with other domains, although the orientation of domain combination and the type of neighboring domains in proteins are generally limited. Because promiscuous domains are not directly related to homology, they should be given less importance in domain architecture comparison than non-promiscuous domains. Another important feature in domain evolution is that two- or three-domain combinations (supra-domains) are re-used in different protein contexts with different partner domains (15). These combinations should be considered, as well as a single domain, in domain architecture comparison. In this study, we define single domains and two-domain combinations within 30 amino acid residues as a ‘domain unit’.
Here we present DAhunter, a web-based server that identifies homologous proteins by comparing domain architecture. DAhunter source codes and database contents are freely available to academic users upon request.
METHODS
Calculating weight scores of domain units
The accuracy of DAhunter depends on the domain unit weight scores, which is calculated from the abundance and versatility of domain units. To obtain domain unit weight scores, we downloaded 4 234 906 protein sequences from the RefSeq Release 26 (ftp://ftp.ncbi.nih.gov/refseq/release/) (16) and classified these proteins into eukaryota, bacteria or archaea. Then we analyzed the domain content of these proteins with the Pfam (17) database. The Pfam domain annotations of all RefSeq proteins were obtained from the Similarity Matrix of Proteins (SIMAP) database (http://mips.gsf.de/simap/). This database provides a comprehensive and up-to-date dataset of the pre-calculated sequence features for all proteins in all major public sequence databases (18). For eukaryotic genes with several alternative transcripts, we kept the longest coding structure so as to retain the maximum number of domains. We filtered domain hits in proteins with a cutoff E-value of 0.01 and excluded proteins without Pfam signatures.
The Pfam-annotated proteins were converted into domain architectures, with sequences between adjacent domains divided into two types: those ≤30 residues and those >30 residues. Previous researchers have used a threshold value of 30 residues to claim that two domains have a particular functional and spatial relationship (19). We extracted domain units from the domain architectures (Table 1) and assigned weight scores according to their abundance and versatility.
Table 1.
Summary of proteins, architectures and domain units of eukaryota, bacteria and archaea (RefSeq proteins)
| Kingdom | Total proteins | Proteins with Pfam domains | Unique architectures | Domain units | |
|---|---|---|---|---|---|
| Single | Double | ||||
| Eukaryote | 1 193 766 | 750 267 | 32 737 | 4764 | 2435 |
| Bacteria | 2 781 568 | 2 170 351 | 25 913 | 4441 | 2002 |
| Archaea | 108 190a | 77 785 | 4399 | 1301 | 229 |
aDue to the small number of archaic organisms in RefSeq, the total number of archaic proteins is relatively small, compared with those of eukaryote and bacteria. The number of organisms in eukaryote, bacteria and archea in RefSeq is 1470, 1079 and 67, respectively.
To measure the abundance of a domain unit, we defined the Inverse Abundance Frequency (IAF), which is derived from the Inverse Document Frequency (IDF), a statistic commonly used in information retrieval (5). The IAF of a domain unit, d, is defined as diaf = log2 pt/(pd + α), where pt is the number of total proteins, pd is the number of proteins containing domain unit d and α is a pseudocount parameter to balance protein frequency.
To measure the versatility of a domain unit, we defined the Inverse Versatility Frequency (IVF), whose definition is also similar to that of IDF. IVF represents how many domain families are in N- and C-sides adjacent to a domain unit. The definition of IVF is divf = log2 ft/(fd + β) where ft is the number of total domain families, fd is the number of domain families adjacent to domain unit d and β is a pseudocount parameter to balance domain families.
The weight score (dws) of a domain unit is simply the product of the IAF and IVF of the domain unit: dws = diaf × divf. If a domain unit is highly promiscuous, it will have a low score. Domain unit analysis of RefSeq proteins shows that the weight score of each domain unit differ among eukaryota, bacteria and archaea. The DAhunter website provides the IAF and IVF for all domain units in the three kingdoms.
Construction of the domain architecture database
To compare domain architectures of a query protein with proteins in public databases, we built a domain architecture database, consisting of domain architectures of proteins in RefSeq, UniProKB/Swiss-Prot and UniProtKB/TrEMBL (20). The Pfam annotations of these proteins were obtained from the SIMAP database. To illustrate domain combinations in domain architectures, we represent the intervening space in domain architectures with ≤30 residues as ‘∧’ and >30 residues as ‘∼∼∼’. Thus, the three domain architectures, ‘A∧B∼∼∼C’, ‘A∼∼∼B∧C’ and ‘A∧B∧C’ (where A, B and C stand for different Pfam domains), are different even though they have the same three domains. According to the definition of a domain unit, the three architectures also have different two-domain combinations (‘A∧B∼∼∼C’ has AB; ‘A∼∼B∧C’ has BC; ‘A∧B∧C’ has AB and BC). The domain architecture database has 39 336 different domain architectures (36 634 in RefSeq; 4253 in UniProtKB/Swiss-Prot; and 10 065 in UniProtKB/TrEMBL).
Similarity between two domain architectures
To identify homologs of a query protein, DAhunter first identifies Pfam domains in the query protein using the hmmpfam program and the Pfam database. If Pfam domains are present, the server extracts domain units from the query domain architecture and selects candidate domain architectures that contain any of the query domain units from the database. Then, DAhunter compares the query domain architecture against candidate domain architectures regarding the content, order and duplication of domain units.
Domain unit content
The two sets of domain units derived from the two architectures are represented as the indices, which are built using the vector space model (VSM) (21). Domain architectures are represented by a vector in which each component corresponds to a weight score of a domain unit. The similarity of the two vectors is measured by determining their cosine similarity, a measure based on the angle between two vectors (commonly used in text mining algorithms). Thus, if x and y are vectors of two domain architectures X and Y, the cosine similarity is defined as:
| 1 |
where ‘·’ indicates the vector dot product, x·y= 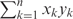 and
and 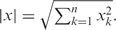 . The range of the cosine similarity is [0, 1], where 1 indicates that x and y have the same domain units and 0 indicates that they share no domain units.
. The range of the cosine similarity is [0, 1], where 1 indicates that x and y have the same domain units and 0 indicates that they share no domain units.
Domain unit order
To measure the order similarity between two domain architectures, we used the Goodman–Kruskal γ function (22), a symmetric measure based on the difference between concordant pairs (P) and discordant pairs (Q). This function is defined as:
| 2 |
where the gamma score varies from −1 to +1. We normalized the score to [0, 1] using a normalized gamma function, defined as normal_gamma(X, Y) = (1 + gamma(X, Y))/2.
Domain unit duplication
Duplication of a domain unit with a higher weight score is more important than duplication of those with a lower weight score. For example, where ‘A’ represents a high-repeat domain and ‘B’ is a low-repeat domain, ‘AB’ to ‘ABB’ is more significant than ‘AB’ to ‘AAB’. To measure duplication similarity between two domain architectures, we developed a function similar to the cosine similarity (defined above). This function uses the VSM, where each component (Cx) corresponds to the product of a weight score of a domain unit and its duplication number. The duplication similarity between two vectors is defined as:
| 3 |
where min(Cx,Cy) is a minimum index of domain unit between two vectors. The duplication score varies from 0 to 1.
The final similarity score between two domain architectures, X and Y, is obtained by combining the indices from Equations (1–3) (each normalized to [0, 1]) using a simple linear function with parameters a and b,
where a ≥ 0 and b ≥ 0. To determine the best combination of parameters a and b, we used the HomoloGene database (23), which provides information about homologies. Among the 44 481 groups present in the HomoloGene release 61, we selected 5215 groups that have more than two different domain architectures. From these groups, we obtained 8290 different domain architectures. To fix a and b, we carried out 8290 tests. In each test, one of the 8290 domain architectures was extracted and compared with the other 8289 domain architectures. The tests were performed with different combinations of a and b by allowing a and b to vary from 1.0 to 0.0 in steps of 0.1, so as to maximize the number of matched combinations in the results. Here the matched combination represents that the two groups of an extracted and the best-matched architecture in the comparison results are the same. We chose 0.8 for a and 0.3 for b, at which the number of best-matched combinations was 5431 (66%).
To evaluate the DAhunter algorithm, we compared the results of DAhunter using the HomoloGene database with those of the PDART program (using default parameters), which does not consider ‘promiscuous’ domains and domain combinations. PDART generated 4961 (59%) matched combinations of the same group. This indicates that DAhunter, which consider ‘promiscuous’ domains and domain combinations, is a better algorithm for comparing the domain architectures. The comparison results are given in DAhunter webpage.
IMPLEMENTATION
The DAhunter server consists of a web interface, a MySQL database management system (DBMS), and core programs. The web interface is implemented with static HTML and CGI scripts and MySQL DBMS is used to store the DAhunter database. The core programs were written in Perl and are divided into three main steps (Figure 1). First is the query processing step, where the server assigns Pfam domains to a query protein and extracts domain units from the Pfam annotation. Second is the comparison step, where the server selects candidate domain architectures containing domain units of a query protein and compares a query domain architecture against candidate domain architectures. Last is the sorting step, in which matched architectures are sorted according to their similarity scores.
Figure 1.

Schematic of DAhunter workflow. The DAhunter pipeline consists of three major steps: (a) query processing, (b) comparing domain architectures and (c) sorting matched domain architectures.
INPUT AND OUTPUT
Input
The query interface accepts protein sequences in the FASTA format. The user can paste the sequences directly into the input form or can upload a file from a local disk. The maximum number of input protein sequences for a single submission is 500 proteins and the length of each sequence is limited to 5000 residues. When submitting more than two protein sequences, users must input an Email address to receive DAhunter results.
Output
The output of the DAhunter service is an HTML-formatted file (Figure 2), which consists of three parts: query domain architecture with Pfam domains, matched domain architectures and domain unit information. The user can see proteins related to a domain architecture by clicking on the number of proteins in the matched domain architectures part.
Figure 2.

Screenshot of DAhunter results: (a) domain architecture of a query protein, (b) matched domain architectures and (c) domain unit information.
ACKNOWLEDGEMENTS
B.L. was supported by a grant from the KRIBB Research Initiative Program. D.L. was supported by the Ministry of Knowledge Economy, Korea, under the ITRC supervised by the IITA (IITA-2008-C1090-0801-0001). Funding to pay the Open Access publication charges for this article was provided by the Ministry of Education, Science and Technology.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 2007;8:995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 2.Ye J, McGinnis S, Madden TL. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006;34:W6–W9. doi: 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearson WR. Rapid and sensitive sequence comparison with FASTP and FASTA. Meth. Enzymol. 1990;183:63–98. doi: 10.1016/0076-6879(90)83007-v. [DOI] [PubMed] [Google Scholar]
- 4.Park J, Karplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 1998;284:1201–1210. doi: 10.1006/jmbi.1998.2221. [DOI] [PubMed] [Google Scholar]
- 5.Song N, Sedgewick RD, Durand D. Domain architecture comparison for multidomain homology identification. J. Comput. Biol. 2007;14:496–516. doi: 10.1089/cmb.2007.A009. [DOI] [PubMed] [Google Scholar]
- 6.Chothia C, Gough J, Vogel C, Teichmann SA. Evolution of the protein repertoire. Science. 2003;300:1701–1703. doi: 10.1126/science.1085371. [DOI] [PubMed] [Google Scholar]
- 7.Krishnadev O, Rekha N, Pandit SB, Abhiman S, Mohanty S, Swapna LS, Gore S, Srinivasan N. PRODOC: a resource for the comparison of tethered protein domain architectures with in-built information on remotely related domain families. Nucleic Acids Res. 2005;33:W126–W129. doi: 10.1093/nar/gki474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caetano-Anolles G, Caetano-Anolles D. An evolutionarily structured universe of protein architecture. Genome Res. 2003;13:1563–1571. doi: 10.1101/gr.1161903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukami-Kobayashi K, Minezaki Y, Tateno Y, Nishikawa K. A tree of life based on protein domain organizations. Mol. Biol. Evol. 2007;24:1181–1189. doi: 10.1093/molbev/msm034. [DOI] [PubMed] [Google Scholar]
- 10.Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: protein homology by domain architecture. Genome Res. 2002;12:1619–1623. doi: 10.1101/gr.278202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin K, Zhu L, Zhang DY. An initial strategy for comparing proteins at the domain architecture level. Bioinformatics. 2006;22:2081–2086. doi: 10.1093/bioinformatics/btl366. [DOI] [PubMed] [Google Scholar]
- 12.Bjorklund AK, Ekman D, Light S, Frey-Skott J, Elofsson A. Domain rearrangements in protein evolution. J. Mol. Biol. 2005;353:911–923. doi: 10.1016/j.jmb.2005.08.067. [DOI] [PubMed] [Google Scholar]
- 13.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 14.Patthy L. Genome evolution and the evolution of exon-shuffling—a review. Gene. 1999;238:103–114. doi: 10.1016/s0378-1119(99)00228-0. [DOI] [PubMed] [Google Scholar]
- 15.Vogel C, Berzuini C, Bashton M, Gough J, Teichmann SA. Supra-domains: evolutionary units larger than single protein domains. J. Mol. Biol. 2004;336:809–823. doi: 10.1016/j.jmb.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 16.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rattei T, Tischler P, Arnold R, Hamberger F, Krebs J, Krumsiek J, Wachinger B, Stumpflen V, Mewes W. SIMAP—structuring the network of protein similarities. Nucleic Acids Res. 2008;36:D289–D292. doi: 10.1093/nar/gkm963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apic G, Gough J, Teichmann SA. An insight into domain combinations. Bioinformatics. 2001;17(Suppl. 1):S83–S89. doi: 10.1093/bioinformatics/17.suppl_1.s83. [DOI] [PubMed] [Google Scholar]
- 20.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glenisson P, Coessens B, Van Vooren S, Mathys J, Moreau Y, De Moor B. TXTGate: profiling gene groups with text-based information. Genome Biol. 2004;5:R43. doi: 10.1186/gb-2004-5-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaroszewicz S, Simovici DA, Kuo WP, Ohno-Machado L. The Goodman-Kruskal coefficient and its applications in genetic diagnosis of cancer. IEEE Trans. Biomed. Eng. 2004;51:1095–1102. doi: 10.1109/TBME.2004.827267. [DOI] [PubMed] [Google Scholar]
- 23.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2008;36:D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]