


Abstract
The Vienna RNA Websuite is a comprehensive collection of tools for folding, design and analysis of RNA sequences. It provides a web interface to the most commonly used programs of the Vienna RNA package. Among them, we find folding of single and aligned sequences, prediction of RNA–RNA interactions, and design of sequences with a given structure. Additionally, we provide analysis of folding landscapes using the barriers program and structural RNA alignments using LocARNA. The web server together with software packages for download is freely accessible at http://rna.tbi.univie.ac.at/.
INTRODUCTION
Over the last years, the world of RNA molecules has become a focus of research in molecular biology. This is mainly due to findings that revealed a huge variety of functional RNAs that act besides or in concert with proteins to fulfill the complex biological functions inside a cell. Consequently, the need for bioinformatical tools to analyse RNA sequences and structures has risen sharply. The Vienna RNA package (1) is a comprehensive collection of tools that offers state of the art algorithms for RNA folding, comparison, and prediction of RNA–RNA interactions. In addition, there is a set of related programs that utilize core programs of the package, such as the barriers program, which gives insights into RNA secondary structure energy landscapes, or LocARNA that generates structural alignments.
The Vienna RNA secondary structure server was originally developed in 2003 (2) and offered a web interface to the most basic programs of the Vienna RNA package. In this contribution, we report on updated versions of our existing services, namely structure prediction of single sequences (RNAfold), consensus structure prediction on a set of aligned sequences (RNAalifold) and sequence design (RNAinverse). We also present new services for RNA–RNA interaction prediction (RNAcofold, RNAup), generation of structural alignments (LocARNA) and folding kinetics (barriers, treekin).
GENERAL REMARKS
The design of the servers was guided by the aim to provide a uniform interface to our programs. Each server presents an index page with a brief description of the service, input fields for entering or uploading data, as well as the ability to set various parameters affecting the computation and the output. Options are grouped into commonly used ones, which are always visible under the ‘basic options’ heading. Less frequently used options or options that require some expert knowledge can be found in the field set ‘advanced options’, which is by default hidden to keep the index page uncluttered. All services use the standard RNA energy parameters from the Turner group (3) by default. In addition, one can select a set of parameters derived by training on both thermodynamic data and known structures, as recently proposed by Andronescu et al. (4). Parameters for folding single stranded DNA sequences (5) are provided as well. Default settings have been carefully chosen so that adjustment of parameters is rarely needed to obtain good results. Once the input data is entered and the calculation has been started by hitting the ‘Proceed’ button, the job is sent to a queueing system, which automatically distributes the job to free resources. The user is informed about the status of the job via a self updating intermediate page, which automatically forwards to the results page as soon as the job is finished. An AJAX-based polling strategy is used to minimize the traffic during the updating procedure.
All servers are equipped with test input data to demonstrate their functionality. A general help page explains basic parameters and algorithms. Furthermore, a short explanation for each option and parameter is given in the form of a pop-up help. Each server offers the possibility to notify the user upon completion of the job via email.
Graphical output from the server is provided in several formats. In all cases, we generate vector graphics in encapsulated Postscript (EPS) format, since these offer the highest print quality and many of our programs embed data in an easily parseable way within an EPS file. Postscript is however not ideal for viewing within the browser. We therefore offer structure drawings as scalable vector graphics (SVG) embedded in the output page. Most modern Internet browsers can display SVG images either natively (Firefox, Opera) or with the help of a plugin (http://www.adobe.com/svg). The possibility to generate interactive SVG images by embedding Javascript makes this format particularly attractive for web applications. Since not all platforms support Postscript without the need to install additional software (such as ghostscript, http://www.ghostscript.com), we also provide files in the more commonly used portable document format (PDF) as well as the possibility to convert EPS files via an online image converter to various bitmap formats such as GIF and PNG.
To avoid overloading our server, the size of input data is limited for each server individually. Detailed restrictions for each server are stated on their index pages. If submitted calculations exceed the limits of our server, the user should consider installing the software locally. As each server returns equivalent command line calls with all necessary options and input files, one can easily get familiar with the programs and their usage.
THE RNAfold SERVER
RNAfold is one of the core programs of the Vienna RNA package. It can be used to predict the minimum free energy (MFE) secondary structure of single sequences using the dynamic programming algorithm originally proposed by Zuker and Stiegler (6). In addition to MFE folding, equilibrium base-pairing probabilities are calculated via John McCaskill's; partition function (PF) algorithm (7). Both algorithms have been recently extended to consider circular RNA sequences (8).
The input, a single RNA or DNA sequence in plain text or FASTA format, can be pasted into the text box or uploaded as a file. Additionally, one can enter structure constraints (e.g. derived from structure probing experiments) in a separate text box. By default, both the MFE and PF algorithm will be computed.
The RNAfold server output contains the predicted MFE secondary structure in the usual dot-bracket notation, additionally mfold-style Connect (ct) files (9) can be downloaded. The secondary structure together with the sequence can be passed on to the RNAeval web server, which gives a detailed thermodynamic description according to the loop-based energy model. When PF folding was selected, base-pairing probabilities were visualized in form of a Postscript dot plot. A dot-plot is a 2D graph, where each possible base-pair is marked by a box which size is proportional to the probability of the corresponding base-pair in a n × n grid. The dot-plot therefore provides information on all possible structures rather than a single optimal one.
Since RNA structure prediction is error-prone, it is important to augment predicted structures with reliability information. Various kinds of reliability annotation can be derived from the results of partition function folding. First, we list the centroid structure (10), i.e. the structure with minimal base-pair distance to all structures in the thermodynamic ensemble (which in turn can be computed simply as the structure containing all pairs with P > 0.5). A high similarity between centroid and MFE structure indicates a reliable prediction. Another global measure of reliability is the ensemble diversity, which is the average base-pair distance between all structures in the Boltzmann ensemble.
In addition, we provide structure drawings that are colour annotated using local (per base) reliability measures. Here each base is colored by the positional entropy, where the entropy of base i is given by 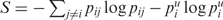 , pij is the probability of forming the pair (i, j) and
, pij is the probability of forming the pair (i, j) and 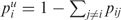 . Alternatively, bases can be colored by probability, using the pair probability for paired bases and the probability of remaining unpaired
. Alternatively, bases can be colored by probability, using the pair probability for paired bases and the probability of remaining unpaired 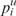 otherwise. These structure drawings are given both as EPS files as well as interactive SVG images. Finally, we produce a mountain plot, i.e. a graph that plots sequence position versus the number of base pairs that enclose that position (on average for base-pair probabilities), depicting the MFE structure, the centroid structure and pair probabilities. A sample output page of the RNAfold web server is shown in Figure 1B.
otherwise. These structure drawings are given both as EPS files as well as interactive SVG images. Finally, we produce a mountain plot, i.e. a graph that plots sequence position versus the number of base pairs that enclose that position (on average for base-pair probabilities), depicting the MFE structure, the centroid structure and pair probabilities. A sample output page of the RNAfold web server is shown in Figure 1B.
Figure 1.

(A) Screenshot of a sample output of the barriers server. We display the barrier tree and provide the transition rates matrix for download. Folding pathways between the 10 best local minima can be explored via an interactive, animated SVG file. For folding kinetic analysis, we provide an interface to the treekin program. The user can select an initial structure and a time interval. Results of the simulation will be automatically loaded into the results page. Note that simulations can be run without the need to recalculate the barrier tree. (B) Screenshot of a sample output of the RNAfold web server. The output is grouped into three sections. First, we provide results for the minimum free energy (MFE) prediction in form of the secondary structure in dot bracket notation and the free energy. Second, we list results from partition function folding, which are the ensemble free energy, the frequency of the MFE structure, the ensemble diversity and base-pairing probabilities in form of a dot plot. Additionally, we provide the centroid structure in dot bracket notation. Last, there are various graphical representation such as a secondary structure drawing and a mountain plot. An SVG file is embedded into the page to interactively explore reliability information in terms of base-pairing probabilities or positional entropy.
THE RNAalifold SERVER
RNAalifold predicts the consensus structure of a set of aligned DNA or RNA sequences (11). It extends standard dynamic programming algorithms for RNA secondary structure prediction (6) by averaging the energy contributions over all sequences and incorporating covariation terms into the energy model to reward compensatory mutations and penalize non-compatible base-pairs. Again, it supports prediction of the minimum free energy structure and base-pairing probabilities and can handle circular sequences.
The input is a single multiple sequence alignment in CLUSTAL W or FASTA format. There are only two additional parameters compared to the RNAfold server, namely ‘Weight of covariance term’ and ‘Penalty for non-compatible sequences’ which affect the covariance scoring schema and the penalization of non-compatible base-pairs of the RNAalifold algorithm. The output is similar to that of the RNAfold server, but also features a structure annotated alignment. Plots are augmented by a special colouring schema that indicates compensatory mutations. Note that the more mutations are observed that support a certain base-pair, the more evidence is given that this base-pair might be correctly predicted.
THE RNAinverse SERVER
The RNAinverse server finds sequences that fit into a pre-defined secondary structure. The input is a single secondary structure in dot-bracket notation. Additionally, one can enter an optional start sequence. The functionality and layout of this server basically equals the 2003 version (2), however, it is now capable to predict up to 100 sequences in one run. Results can be sorted online and readily submitted to the RNAfold server for detailed analysis.
THE RNAcofold SERVER
RNAcofold computes the hybridization energy and base-pairing pattern of two RNA sequences [12]. In short, this is accomplished by concatenating the sequences and treating the loop containing the concatenation point as an exterior loop. The limitation of this approach is that some common interaction motifs, such as kissing hairpins, cannot be predicted due to the knot-free condition of the Zuker algorithm. Besides providing base-pair probabilities the PF can be used to compute equilibrium constants for the dimerization reaction. This in turn can be used to predict dimer and monomer concentrations from the start concentrations of monomers.
The input consists of two sequences, which can either be pasted into two separate text fields or uploaded as two separate FASTA files. In addition to commonly used options of the Vienna RNA package, user defined initial concentrations of the monomers can be specified. We also provide a list of pre-defined initial concentrations, which are then used to generate concentration dependency plots, in the form of contour maps showing the relative concentrations of all five species, i.e. the two homodimers, the heteromer as well as the two monomers, with varying monomer start concentrations. For the special case of two identical sequences, the concentration dependency plot is reduced to an xy-graph showing the initial concentration versus the relative concentrations of the monomer and the dimer. In general, the output is similar to that of RNAfold. The two sequences are concatenated and the concatenation point is indicated by an ampersand. In addition to ΔGAB, the free energy of the heterodimer of sequence A and sequence B, ΔGbinding = Δ GAB− Δ GA−Δ GB is listed. Free energies and dot plots for all five species are provided. For detailed information and interpretation of these results we refer to ref. (12).
THE RNAup SERVER
The RNAup program (13,14) follows a different strategy for predicting RNA–RNA interactions. It models the total binding energy for the interaction at a particular site as 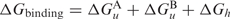 , where
, where 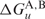 is the free energy required to make the binding region in molecule A or B accessible by removing intra-molecular structure, while Δ Gh denotes the free energy gained from forming the inter-molecular duplex. RNAup therefore first calculates the accessibility (or opening energy) for every stretch of bases up to a certain length for both molecules. In a second step, the interaction free energy is computed and combined with the opening energies to get the total binding energies.
is the free energy required to make the binding region in molecule A or B accessible by removing intra-molecular structure, while Δ Gh denotes the free energy gained from forming the inter-molecular duplex. RNAup therefore first calculates the accessibility (or opening energy) for every stretch of bases up to a certain length for both molecules. In a second step, the interaction free energy is computed and combined with the opening energies to get the total binding energies.
The input of the RNAup server consists of two sequences, which can either be pasted into two separate text fields or uploaded as two separate FASTA files. Specific options for this server are the maximal length of the unstructured region as explained earlier and a threshold for the maximum length of interaction. In the advanced options set, the user can additionally choose to calculate the accessibility individually for different types of loops, such as hairpins or interior loops. The output consists of the optimal interaction site, the secondary structure of the duplex formed and the total free energy of binding.
In addition, RNAup produces a text file containing the best binding energy for each position along the longer sequence. The same information is also provided as an x–y plot for the vicinity of the optimal-binding site.
THE BARRIERS SERVER
Rather than folding directly into their thermodynamic ground state, some RNA molecules form long-lived meta-stable structures. Thus, the equilibrium view as provided by RNAfold is not always sufficient, and one has to resort to methods that consider kinetics of RNA folding. Whether folding of a particular RNA molecule is governed by thermodynamics or folding kinetics depends on properties of the energy landscape, such as the height of energy barriers between ground state and possible folding intermediates.
The barriers program (15) performs an analysis of the energy landscape by identifying all local minima and the energy barriers separating them. This information can be conveniently depicted in the form of a barrier tree in which local minima are represented as leaves, while interior nodes represent the saddle points and the length of the edges is proportional to energy barriers. Furthermore, the barrier tree can be used as the basis of a coarse graining of the energy landscape by contracting all conformations belonging to a single local minimum into one macrostate. Since the number of such macrostates is small, the folding dynamics in this coarse grained representation can be computed by directly integrating the master equation for the underlying Markov process, as implemented in the treekin program. For further details, we refer to ref. (16).
The barriers server uses a single sequence as input, which can either be pasted or uploaded as FASTA file. Since the analysis of folding dynamics is computationally much harder than thermodynamic folding, the service is limited to fairly short sequences (currently 100 nt). Beside common options, the user can define the number of local minima as well as a minimum height for energy barriers. The output features the barrier tree and the transition rate matrix between macrostates. Moreover, we compute optimal re-folding pathways between the 10 best local minima. The user can explore these refolding pathways via interactive SVG animations, which move through the re-folding path while updating the position in the energy profile, as well the current structure in a circular and conventional structure drawing. The animation allows structure morphing similar to the RNAmovies program (17).
To obtain detailed predictions of the folding kinetics, we provide an interface to the treekin program (16), which uses macrostates and transition rates computed by the barriers program. To start a simulation with the treekin program, the user only has to choose an initial structure, as well as start and end time. Results are presented as a diagram depicting the population densities versus time. Simulations are performed on the fly and can be repeated with different initial conditions directly from the results page without the need to recompute the barriers tree. A sample output page of the barriers web server is shown in Figure 1A.
THE LocARNA SERVER
LocARNA (18) is a tool for producing structural multiple sequence alignments of RNA sequences. It is a variant of the Sankoff algorithm (19) for simultaneous folding and alignment of RNA sequences. The unrestricted Sankoff algorithm is notorious for its computational cost, scaling as 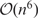 in CPU time and
in CPU time and 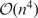 in memory. LocARNA employs a number of techniques to reduce computational complexity, most importantly by restricting the algorithm to thermodynamically likely base pairs using pair probabilities as computed by RNAfold. Multiple alignments are computed using progressive pairwise alignment along a guide tree. The current LocARNA contains several recent improvements compared to the version described in (18). Most notably, the use of stack probabilities (20), Ribosum matrices (21) instead of simple match/mismatch scores, affine gap costs, as well as optimized default parameters.
in memory. LocARNA employs a number of techniques to reduce computational complexity, most importantly by restricting the algorithm to thermodynamically likely base pairs using pair probabilities as computed by RNAfold. Multiple alignments are computed using progressive pairwise alignment along a guide tree. The current LocARNA contains several recent improvements compared to the version described in (18). Most notably, the use of stack probabilities (20), Ribosum matrices (21) instead of simple match/mismatch scores, affine gap costs, as well as optimized default parameters.
Input to the LocARNA server consists of RNA sequences in multiple FASTA format, which can either be pasted or uploaded. In the basic options field, the user can select between local and global sequence alignments. Scoring parameter, such as the choice between Ribosum or simple match/mismatch scores, gap costs, the sequence versus structure weight, can be changed in the advanced options section. Parameters affecting the speed versus accuracy trade off such as the minimal base-pair probability, can also be adjusted by the user. This probability threshold can be defined separately for the guide tree construction and the progressive alignment phase. As an output, we provide the structural alignment in CLUSTAL W format, which can either be downloaded or passed on to the RNAalifold server for consensus structure prediction and further analysis.
IMPLEMENTATION
The web server was implemented using Apache, Perl, BioPerl (22), CGI and client-side JavaScript. As of writing this article, the system makes use of two Intel Core 2 Quad machines for performing the calculations.
ACKNOWLEDGEMENTS
We thank Sebastian Will for help on the LocARNA web server, Mirela Andronescu from the group of Anne Condon for providing her revised RNA parameters and Lukas Endler for extensive testing of the web servers. This work was supported by the Austrian GEN-AU projects ‘non coding RNA’ and ‘Bioinformatics Integration Network’, as well as by the European Union as part of the FP-6 EMBIO project. Funding to pay the Open Access publication charges for this article was provided by ‘Bioinformatics Integration Network’.
Conflict of interest statement. None declared.
REFERENCES
- 1.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- 2.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 4.Andronescu M, Condon A, Hoos HH, Mathews DH, Murphy KP. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics. 2007;23:19–28. doi: 10.1093/bioinformatics/btm223. [DOI] [PubMed] [Google Scholar]
- 5.SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 8.Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006;22:1172–1176. doi: 10.1093/bioinformatics/btl023. [DOI] [PubMed] [Google Scholar]
- 9.Zuker M. Computer prediction of RNA structure. Methods Enzymol. 1989;180:262–288. doi: 10.1016/0076-6879(89)80106-5. [DOI] [PubMed] [Google Scholar]
- 10.Ding Y, Chan CY, Lawrence CE. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005;11:1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- 12.Bernhart SH, Tafer H, Mückstein U, Flamm C, Stadler PF, Hofacker IL. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol. Biol. 2006;1:3. doi: 10.1186/1748-7188-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mückstein U, Tafer H, Hackermüller J, Bernhart SH, Stadler PF, Hofacker IL. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006;22:1177–1182. doi: 10.1093/bioinformatics/btl024. [DOI] [PubMed] [Google Scholar]
- 14.Mückstein U, Tafer H, Bernhart SH, Hernandez-Rosales M, Vogel J, Stadler PF, Hofacker IL. Translational control by RNA-RNA interaction. Manuscript in preparation. [Google Scholar]
- 15.Flamm C, Hofacker IL, Stadler PF, Wolfinger MT. Barrier trees of degenerate landscapes. Z. Phys. Chem. 2002;216:1–19. [Google Scholar]
- 16.Wolfinger MT, Svrcek-Seiler AW, Flamm C, Hofacker IL, Stadler PF. Efficient computation of RNA folding dynamics. J. Phys. A: Math. Gen. 2004;37:4731–4741. [Google Scholar]
- 17.Evers D, Giegerich R. RNA movies: visualizing RNA secondary structure spaces. Bioinformatics. 1999;15:32–37. doi: 10.1093/bioinformatics/15.1.32. [DOI] [PubMed] [Google Scholar]
- 18.Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3 doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sankoff D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math. 1985;45:810–825. [Google Scholar]
- 20.Bompfünewerer AF, Backofen R, Bernhart SH, Hertel J, Hofacker IL, Stadler PF, Will S. Variations on RNA folding and alignment: lessons from Benasque. J. Math. Biol. 2008;56:129–144. doi: 10.1007/s00285-007-0107-5. [DOI] [PubMed] [Google Scholar]
- 21.Klein RJ, Eddy SR. RSEARCH: finding homologs of single structured RNA sequences. BMC Bioinform. 2003;4:44. doi: 10.1186/1471-2105-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]