

Abstract
Normally, cell cycle progression is tightly coupled to the accumulation of cell mass; however, the mechanisms whereby proliferation and cell growth are linked are poorly understood. We have identified Cyclin D2 (CycD2), a G1 cyclin implicated in mediating S phase entry, as a potential regulator of hypertrophic growth in adult post mitotic myocardium. To examine the role of CycD2 and its downstream targets, we subjected CycD2-null mice to mechanical stress. Hypertrophic growth in response to transverse aortic constriction (TAC) was attenuated in CycD2 null compared to wildtype mice. Blocking the increase in CycD2 in response to hypertrophic agonists prevented phosphorylation of CycD2-target Rb in vitro and mice deficient for Rb had potentiated hypertrophic growth. Hypertrophic growth requires new protein synthesis and transcription of tRNA genes by RNA pol III, which increases with hypertrophic signals. This load-induced increase in RNA pol III activity is augmented in Rb-deficient hearts. Rb binds and represses Brf-1 and TBP, subunits of RNA pol III-specific transcription factor B, in adult myocardium under basal conditions. However this association is disrupted in response to TAC. RNA pol III activity is unchanged in CycD2-/- myocardium after TAC, and there is no dissociation of TBP from Rb. These investigations identify an essential role for the CycD2-Rb pathway as a governor of cardiac myocyte enlargement in response to biomechanical stress and, more fundamentally, as a regulator of the load-induced activation of RNA pol III.
Keywords: Hypertrophy, Genetically altered mice, Animal models of human disease, Cell signaling/signal transduction
Introduction
During development, cell cycle progression is closely coupled to the accumulation of cell mass to maintain a uniform cell size. It has been suggested that proteins involved in cell cycle progression control both processes, thereby linking proliferation and cell growth1. In the fetal heart, the proliferative activity of cardiac myocytes is accompanied by increases in cell mass thereby maintaining myocyte size. However, in adult cardiac myocytes cell growth becomes uncoupled from proliferation after growth stimuli, resulting in hypertrophic growth2. The molecular mechanisms by which proliferation and cell growth are coupled in myocytes are poorly understood; however, cell cycle regulatory proteins typically associated with G1 exit have also been implicated in regulating cardiac myocyte cell growth3-5. CycD-Cdk4 in particular has been associated with cardiac hypertrophy, although the downstream effectors have not been identified6,7. Previous studies have indicated that inhibiting G1-Cyclin/Cdk activity in adult, post-mitotic, cardiac myocytes can attenuate hypertrophic growth8,9. Consistent with this finding, we recently reported that Myc-induced hypertrophic growth was dependent on the presence of CycD2, similar to what has been reported for Myc-induced cell cycle reentry, but was independent of Cdk2 activity10.
The association between CycD-Cdk4 represents the rate-limiting step in a complex cascade that leads to the phosphorylation of a number of proteins including the retinoblastoma gene product, or Rb8,11-13. When phosphorylated Rb becomes inactive, its association with a family of transcription factors known as E2Fs is disrupted, thereby allowing them to activate transcription14. Previous studies have determined that Rb phosphorylation by CycD-Cdk4 is essential for cell cycle progression, largely by regulating the transcriptional activities of these E2Fs15,16. However, whether this signaling pathway is also important in cardiac hypertrophy is unknown. Among the known hypertrophic signaling cascades, a direct mechanistic link to Rb is perhaps best suggested for its interaction and regulation of the RNA polymerases17,18.
Therefore, to understand the role of CycD2 and Rb proteins during cardiac hypertrophy, and to determine the mechanism by which these cell cycle regulators modulate cardiac hypertrophy in vivo, we utilized mouse models deficient for these proteins. Mice where CycD2 has been genetically ablated are viable and display normal cardiac anatomy and function19. Although normal at baseline, CycD2-deficient mice exhibited attenuated hypertrophy in response to pressure overload. To determine the importance of Rb as a downstream target, we utilized mice with a cardiac-specific deletion of Rb. These mice have normal heart size at baseline20 but hypertrophic growth in response to mechanical or pharmacological stress was increased significantly in Rb-null mice. Furthermore, activity of RNA polymerase III (pol III) was increased in Rb deficient hearts subjected to hemodynamic stress, whereas this TAC-induced increased activity was not seen in CycD2-/- myocardium. Thus, we propose that CycD and Rb work in an interdependent manner to control myocyte size in post-mitotic myocardium after mechanical stress. Our data identifies CycD2, through its phosphorylation target Rb, as a pivotal regulator of RNA pol III activation in myocardial hypertrophy.
Materials and Methods
Animal studies
Cardiac-restricted Rb-deficient mice (CRbL/L) have been described20. Cyclin D2-deficient mice were a kind gift from Dr. P. Sicinski19. For TAC, a fixed pressure overload was obtained by surgically constricting the transverse aorta, as described21. Genotypes of mice were determined by PCR as described10,20.
Isolation of cardiac myocytes and analysis
Neonatal rat ventricular myocytes (NRVM) were prepared as described10,20. Myocyte dimensions were determined and volumes calculated using a computerized morphometric system21. Details for viral propagation is available in the Supplemental Data22. For siRNA studies, NRVMs were transfected with 125nM of CycD2 siRNA or non-specific siRNA (Qiagen) with Lipofectamine 2000 (Invitrogen) according to the manufacturer's specifications.
Histology and immunostaining
For histology, hearts were either frozen in OCT compound or fixed overnight in 4% paraformaldehyde buffered with PBS and routinely processed. Details of immunostaining protocols and antibodies is available in the Supplemental Data.
RNA and Protein Analysis
Total RNA was extracted (Tri Reagent, Sigma) as per manufacturer's instructions. Northern blot and ribonucleic protection assays (RPA) were conducted as previously described20,23. Real-time quantitative PCR was conducted using the ABI PRISM 7700 Sequence Detection System; Taqman (Applied Biosystems, Foster City, CA). Primers sequences for all genes analyzed in the present study have been previously reported or are available upon request23,24.
Western blotting and immunoprecipitations was performed as we have described10,25 and details of the antibodies used are available in the Supplemental Data.
RNA pol III assays
RT–PCR analysis of ARPP P0 mRNA and primary tRNATyr or tRNALeu transcripts was carried out as previously described26,27. Gels were scanned and quantitated by a UVP image analysis system (Adobe PHOTOSHOP 4.0, Adobe Systems, Mountain View, CA).
Statistical analysis
All data are presented as mean±SEM. Results were compared by one-way analysis of variance and Fisher's PLSD or Tukey's multiple comparison post-tests, using significance at a P value <0.05.
Results
Cyclin D2-null mice display attenuated hypertrophy in response to pressure overload
Our previous work had shown that Myc's hypertrophic effects were mediated through a CycD2-dependent pathway10. In accordance with previous studies4, CycD2 protein expression increases both in vitro and in vivo in response to hypertrophic stimuli (Suppl Fig. S1). To determine if CycD2's importance was specific to Myc-induced hypertrophy or if it was more generally important in regulating hypertrophic growth, we subjected CycD2-null mice to pressure overload for 14 days. Homozygous CycD2-deficient mice are viable and appear normal but are sterile19. Although not described in the original report, CycD2-/- mice had a 22.5% reduction in body weight compared to wild-type littermates at 12 weeks (Suppl. Table S1; 23.5 ± 1.6 g CycD2-/- versus 28.8 ± 0.9 g CycD2+/+; P<0.05); however, the heart weight-to-body weight ratios were similar at baseline (4.35 ± 0.14 mg/g CycD2+/+ versus 4.40 ± 0.16 mg/g CycD2-/-; P=n.s.). As expected, TAC increased CycD2 mRNA and protein levels in wild-type but not CycD2-deficient mice (Figure 1A and 1B). CycD2 mRNA increased 75% in CycD2+/+ mice after TAC as measured by real-time PCR analyses (1.00 ± 0.18 versus 1.75 ± 0.19, P<0.05). Similarly, CycD2 protein expression increased 73% in CycD2+/+ mice after TAC as measured by Western blotting (1.62 ± 0.36 versus 2.80 ± 0.40, P<0.05). There was a 64% reduction in the hypertrophic response in CycD2-null mice compared to wildtype mice subjected to TAC (P<0.02). Two weeks of TAC induced a 25% increase in the heart weight-to-body weight ratio in CycD2 wild-type mice (Fig. 1C; HW/BW: 4.35 ± 0.14 versus 5.45 ± 0.29 mg/g, P<0.005). In contrast, CycD2-null mice displayed an attenuated hypertrophic response with only a 9% increase in HW/BW ratio (Fig. 1C; HW/BW: 4.40 ± 0.16 versus 4.81 ± 0.14 mg/g, P<0.05). Fetal cardiac genes including atrial (ANP) and B-type (BNP) natriuretic peptides as well as α-skeletal actin (α-SkA) were upregulated in response to TAC in both ventricles from mice of both genotypes despite the differences in myocardial mass suggesting not all aspects of the hypertrophic response were impaired (Fig. 1D).
Figure 1. Cyclin D2-/- mice display attenuated hypertrophy in response to pressure overload.

A, Representative RPA and quantification of CycD2 mRNA by real-time PCR normalized to GAPDH. n= 4 per group, * P<0.05 CycD2 mRNA in CycD2+/+ after TAC compared to CycD2+/+-Sham. B, Western blotting on total ventricular lysates after two weeks of TAC from the indicated genotypes and conditions. Protein quantification revealed an increase in CycD2 protein expression in CycD2+/+ hearts after TAC. Cardiac actin (α-CaA) was used to normalize total protein levels. n=4 for each group, *P<0.05 for CycD2+/+ TAC as compared to Sham. C, Two-weeks of TAC induced a significant increase in the heart weight-to-body weight ratio (HW/BW, mg/g) in CycD2+/+ but not CycD2-/- mice n=5 per group, * P<0.005 for CycD2+/+-TAC compared to CycD2+/+-Sham. An increase in the HW/BW ratio in CycD2-/- mice in response to TAC was not observed. D, Northern blot on total ventricular RNA. Expression of atrial (ANP) and B-type (BNP) natriuretic peptides, and skeletal actin (α-SkA) and 18S ribosomal RNA (18S rRNA) as a loading control is shown.
Rb regulates hypertrophic growth in vitro
Although several CycD2 phosphorylation targets are present in adult cardiac muscle, Rb is the primary target of CycD-Cdk4 kinase complexes and has been implicated, at least indirectly, in regulating protein synthesis7. Thus, we sought to determine if phosphorylation of Rb in response to hypertrophic agonists was dependent on CycD2. To knock down CycD2 expression, we transfected NRVMs with either non-specific (NS) siRNA or a specific siRNA to CycD2. As shown in Figure 2A, siRNA transfection was able to induce a 98% decrease in CycD2 protein expression in NRVMs compared to NS siRNA treatment (1.37 ± 0.29 versus 0.03 ± 0.01, P<0.05). To determine whether depleting CycD2 impaired Rb phosphorylation in response to hypertrophic agonists, NRVMs transfected with NS or CycD2 siRNA and stimulated with PE were immunostained with an antibody specific for phosphorylated serine 780 on Rb (pRbS780), a target of CycD-Cdk4. As expected, treatment of NRVMs with PE resulted in the increased expression of both CycD2 and pRbS780 in NS siRNA transfected cells (Fig. 2B, a-b). However, transfection of NRVMs with CycD2 siRNA blocked the increase in CycD2 seen with PE and diminished RbS780 phosphorylation (Fig. 2B, c-d). To determine the role of Rb in regulating hypertrophic growth, we stimulated NRVMs with PE and measured protein accumulation and myocyte size with or without Rb overexpression. Infection of NRVMs with AdRb resulted in high levels of Rb expression (Fig. 2C), which had no effect on basal amino acid incorporation in quiescent myocytes (Fig. 2C). PE increased relative protein synthesis rates by 33.4% in AdLacZ-infected cultures (1.33±0.1 versus 1.0±0.02 for vehicle-treated, LacZ-infected cells; P<0.01). This increase in protein synthesis was blocked completely by Rb (0.91±0.07 versus 0.91±0.09 for AdRb infected myocytes treated with PE versus vehicle, respectively; P=n.s., Fig. 2C & D).
Figure 2. CycD2 phosphorylates and inactivates Rb which regulates protein synthesis in vitro.

NRVM were transfected with either non-specific (NS) siRNA or specific CycD2 siRNA and treated with 100 mM phenylephrine (PE) for 24 hours. A, Western blot analysis reveals a 98% knockdown of CycD2 protein expression in CycD2 siRNA-transfected NRVM (n=3 per condition, P<0.05). B; a-b, Immunostaining of NRVMs treated with NS siRNA revealed robust staining of CycD2 and phospho-RbS780 (pRbS780) in response to PE. B; c-d, CycD2 siRNA-transfected cells revealed diminished expression of both CycD2 and pRbS780 in response to PE. Scale bar =50 μm. C and D, NRVMs cultured in serum free media for 48 hours and infected with the indicated virus. NRVM cultures were then stimulated with 100 μm PE for 24 hours. C, Relative levels of Rb expression were determined by Western blotting. C, 3H-phenylalanine incorporation was quantified and normalized to unstimulated AdLacZ cultures. The pooled results of three experiments are presented. *P<0.01 versus unstimulated AdlacZ myocytes and P<0.001 versus AdRb infected cultures.
Pressure overload results in enhanced hypertrophy in adult Rb-deficient cardiac myocytes in vivo
To determine the role of Rb in regulating the hypertrophic response in adult cardiac myocytes in vivo, we subjected cardiac-restricted Rb-deficient (CRbL/L) mice we had previously created to TAC20. These mice are phenotypically and biochemically normal at baseline. Hearts from Rb-deficient CRbL/L mice were significantly larger than those from control CRb+/+ mice after being subjected to one week of TAC (Suppl. Table S2; HW/BW: 6.40 ± 0.34 versus 5.25 ± 0.25 mg/g; P<0.05). Likewise, at two weeks (Fig. 3A), CRb+/+ mice developed a 39.3% increase in heart-to-body weight ratio (HW/BW: 5.23±0.3 versus 4.21±0.16 mg/g for Sham operated mice; P<0.05) while Rb-deficient CRbL/L mice demonstrated a 59.4% increase (HW/BW: 6.44±0.5 versus 4.41±0.19 mg/g for Sham operated mice; P<0.001). This enhancement of myocardial mass in CRbL/L mice compared to CRb+/+ animals after two weeks of banding was also significant (Fig. 3A; P<0.05). Although the role of Rb in mediating cell cycle exit in striated muscle is controversial28, increased myocyte proliferation cannot explain the increase in cardiac mass we observed as no evidence of myocyte cell cycle reentry was seen either by BrdU incorporation (Fig. 3B) or Ki-67 expression (not shown). In contrast, myocyte fiber width was significantly increased in CRbL/L mice subjected to TAC when compared to Sham-operated CRbL/L or CRb+/+ mice (12.72±1.26 versus 9.61±0.35 or 10.05±0.65 μm; P<0.05; Fig. 3C & D) suggesting the increased heart size was related to myocyte hypertrophy. This enhanced hypertrophic response could not be accounted for by extrinsic or intrinsic hemodynamic differences, since the gradient across the constriction at two weeks did not differ significantly between the genotypes (51.5±9.8 versus 60.7±4.6 mmHG; P=n.s.), and because left ventricular function was normal for both genotypes by 2D-echo or invasive hemodynamics. Measurements of contractility (+dP/dt; 5003±862 versus 4243±313 mmHg/s; P=n.s.) and relaxation (-dP/dt; -6456±1329 versus -5171±592 mmHg/s; P=n.s.) were indistinguishable between CRb+/+ and CRbL/L mice subjected to TAC for two weeks.
Figure 3. Rb deficient mice display an exaggerated hypertrophic response in vivo.
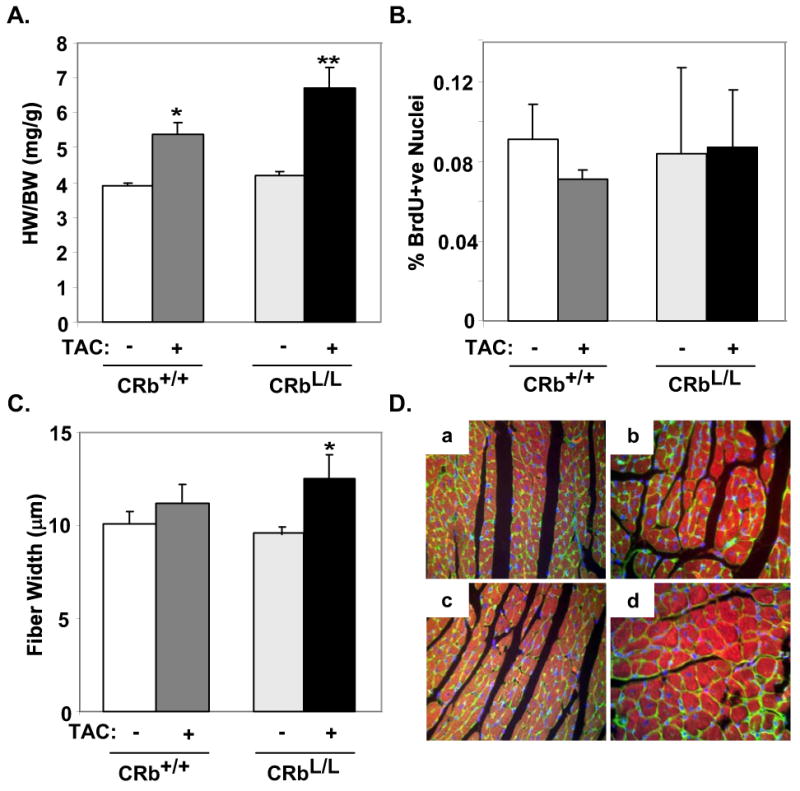
Control (CRb+/+) or Rb-deficient (CRbL/L) mice underwent Sham or TAC surgery and were followed for two weeks (n=6 for each group). A, Heart weights (mg) were normalized to body weight (g). *P<0.05 for CRb+/+-TAC versus CRb+/+-Sham and CRbL/L-Sham, **P<0.05 for CRbL/L-TAC versus CRb+/+-TAC, and P<0.0001 versus CRb+/+-Sham and CRbL/L-Sham. B, Percentage of BrdU+ve nuclei from Sham and TAC mice were quantified (n=6 per group P=n.s.). C and D, Fiber width of wheat germ agglutinin stained hearts from Sham and TAC mice were quantified (n=5 in each group). D, Representative wheat germ agglutinin stained myocardial sections from CRb+/+ (a-b) or CRbL/L (c-d) subjected to Sham (a,c) or TAC (b,d) surgery.
To determine if the exaggerated hypertrophic response to load in Rb-null myocardium was generalizable to other hypertrophic stimuli, we next subjected mice to an infusion of isoproterenol (ISO) versus the vehicle, for one week. ISO treatment caused a 26.8% increase in heart-to-body weight ratio in control CRb+/+ mice (HW/BW: 4.63±0.08 versus 5.87±0.24 mg/g; P<0.0001), but Rb-deficient CRbL/L mice demonstrated a 36.4% increase (HW/BW: 4.67±0.11 versus 6.37±0.17 mg/g; P<0.001). This induced HW/BW ratio was significantly greater in ISO-treated CRbL/L animals than in ISO-stimulated CRb+/+ mice (6.37±0.17 versus 5.87±0.24 mg/g; P<0.05). This increase in cardiac mass paralleled a corresponding increase in individual myocyte volume in the Rb-deleted background. Isolated Rb-deficient myocytes from ISO-treated CRbL/L ventricles demonstrated a 25.1% increased in myocyte volume when compared to similarly treated CRb+/+ myocytes (21962±1247 versus 17549±1891 μm3; P=0.09). Thus, in adult myocardium Rb primarily functions to constrain myocyte size in response to biomechanical stress.
Expression of E2Fs and E2F-target genes is unchanged in Rb-deficient myocardium
Although Rb associates with many proteins, classically it binds and inhibits members of the E2F family of transcription factors. Since E2Fs have been implicated in regulating cardiac hypertrophy, at least in vitro29, we examined the expression of E2F family members in CRb+/+ or CRbL/L mice at baseline and after TAC with real-time PCR. E2F-1 mRNA increased 3-fold in CRb+/+ heart in response to TAC compared to baseline (Fig. 4A; 1.00 ± 0.08 versus 3.05 ± 0.83, P<0.05); however, there were no differences in expression between CRb+/+ and CRbL/L mice (Fig. 4A). Interestingly, no significant change was noted with TAC in the expression of E2F-3 as determined by semi-quantitative PCR analysis, which was the E2F family member previously associated with hypertrophic growth in cultured NRVMs (Fig. 4A)29. No change in either E2F-4 or -5 expression was seen in response to TAC. Since E2F transcriptional activity could be altered in the absence of Rb, even if expression levels were not, we examined the levels of a panel of E2F-1 dependent target genes24. Semi-quantitative PCR revealed no significant differences in the expression of any of these E2F-target genes in CRb+/+ and CRbL/L myocardium after TAC (Fig. 4B). Thus, differences in E2F expression or activity are unlikely to account for the enhanced hypertrophic response in Rb-null myocardium.
Figure 4. Expression and activity of E2F family members is not altered in Rb-null myocardium after TAC.
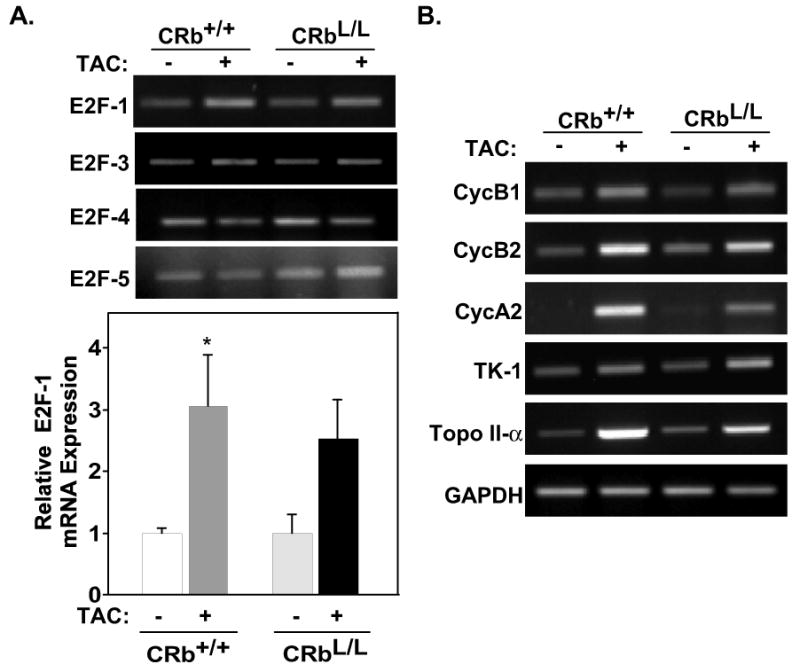
A, Representative E2F-1, -3, -4, and -5 expression obtained from semi-quantitative PCR analyses. Quantification via real-time PCR analysis (bar graph) revealed a 3-fold increase in E2F-1 mRNA expression in response to TAC in CRb+/+ mice. (n=3-4 per condition; *P<0.05). No difference was noted in the mRNA expression of E2F-3, -4, and -5. B, Semi-quantitative PCR analyses of E2F-1-target genes was examined in each genotype in response to TAC. Cyclin B1 (CycB1), Cyclin B2 (CycB2), Cyclin A2 (CycA2), thymidine kinase-1 (TK-1), topoisomerase II-α (Topo II-α).
Pressure overload induces enhanced RNA pol III activity in Rb-null hearts
RNA pol III activity is known to increase in cardiac myocytes subjected to hypertrophic signals, however, the mechanisms regulating this effect are poorly understood30. Rb is known to negatively regulate RNA pol III activity17, via binding to Brf-1 and TBP, resulting in inhibition of the pol III-specific transcription factor complex TFIIIB25,31. In order to clarify the relationship between Rb and these two main subunits of TFIIIB, in response to a hypertrophic stimulus, we performed immunoprecipitation studies using total ventricular extracts from wild-type mice exposed to TAC for 7 days. Expression of phosphorylated and total Rb, Brf-1, and TBP in Sham and TAC ventricles are displayed in Fig. 5A &B. Although total expression levels of these factors did not change with TAC, phosphorylation of Rb increased in hypertrophic ventricles (Fig. 5B). Brf-1 and TBP associated with Rb in wildtype Sham ventricles (Fig. 5C) but phosphorylation of Rb in banded ventricles disrupted this interaction and Rb was no longer associated with Brf-1 and TBP in myocardial lysates from TAC ventricles (Fig. 5C). To determine the functional significance of Rb's interactions with these factors we assayed RNA pol III activity in Rb-deficient hearts. Intron-specific primers were used to assay levels of RNA pol III–specific transcripts from tRNATyr or tRNALeu genes on RNA extracted from ventricles of mice after Sham or TAC surgery (Fig. 5D &E). RNA pol III activity was similar in Sham-operated CRb+/+ and CRbL/L myocardium. However, RNA pol III activity was increased in Rb-null hearts after TAC compared to CRb+/+ TAC animals (Fig. 5D &E). When compared to pressure-overloaded CRb+/+ mice, inducible tRNATyr transcripts in CRbL/L animals were 40% higher (1.73±0.17 versus 1.33±0.24; P<0.05), although tRNALeu transcripts were not elevated significantly (1.52±0.07 versus 1.22±0.12; P=0.12). These data are consistent with a model where Rb is bound to TFIIIB under baseline conditions but this association is disrupted by hypertrophic stimuli leading to increased RNA pol III activity.
Figure 5. Rb associates with RNA pol III subunits in vivo and regulates RNA pol III activity.
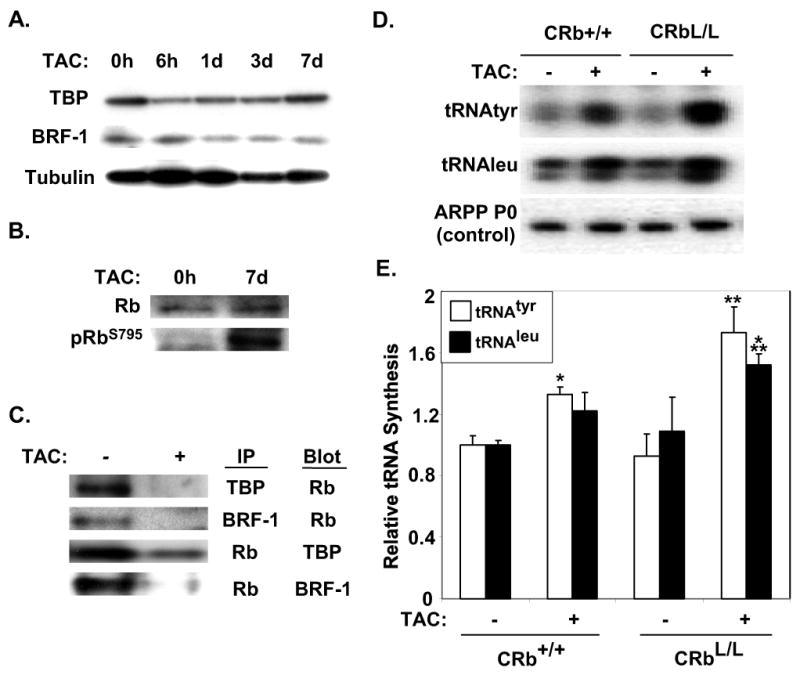
A, Representative Western blots displaying expression of Brf-1, and TBP in wild-type ventricles 0 hours (0h), 6h, 1 day (1d), 3d and 7d after TAC. B, Representative Western blots displaying expression of phospo-RbS795 (pRbS795), and total Rb 7d after TAC. C, Brf-1 and TBP subunits dissociate from Rb in response to TAC. Immunoprecipitation (IP) assays were completed with antibodies to Rb, Brf-1 and TBP as indicated using 500 μg of Sham or TAC myocardial protein lysates. Lysates were subjected to SDS-PAGE and immunoblotted using Rb, Brf-1 or TBP antibodies as indicted. D, Representative blots of RNA pol III-dependent tRNAtyr and tRNALeu transcripts. E, Quantitative results from n=5 samples are shown, *P<0.05 for tRNAtyr synthesis for CRb+/+-TAC versus CRb+/+-Sham, **P<0.05 for tRNAtyr synthesis for CRbL/L-TAC versus CRb+/+-TAC, and P<0.0001 versus CRbL/L-Sham, ***P<0.05 for tRNALeu synthesis for CRbL/L-TAC versus CRb+/+-Sham and CRbL/L-Sham.
To determine if the CycD2-Rb interaction we have proposed has functional significance in hypertrophic myocardium regulating the RNA pol III machinery, we examined the interaction of TBP and Rb in ventricles of CycD2-/- mice (Fig. 6A). Although the association of TBP and Rb is disrupted by hypertrophic stimuli in CycD2+/+ hearts as expected, TAC did not disrupt the interaction of TBP and Rb in ventricles of CycD2-/- mice (Fig. 6A). Similarly, both tRNATyr and tRNALeu transcripts were elevated in response to TAC in the CycD2+/+ animal after TAC (Fig. 6B &C; tRNATyr: 1.00 ± 0.08 versus 1.48 ± 0.07, P<0.05; tRNALeu: 1.00 ± 0.10 versus 1.54 ± 0.10, P<0.05). However, neither tRNATyr or tRNALeu transcripts were altered in CycD2-/- mice after TAC.
Figure 6. TBP does not dissociate from Rb and RNA pol III activity is not altered in CycD2-/- myocardium after TAC.

A, TBP levels remain unchanged in both CycD2+/+ and CycD2-/- mice after TAC as determined by Western blotting. A, Immunoprecipitation (IP) assays reveal that TBP does not dissociate from Rb in CycD2-/- ventricles after TAC. B, Representative blots of RNA pol III-dependent tRNAtyr and tRNALeu transcripts. C, Quantitative results from CycD2+/+ and CycD2-/- mice (Sham, n=4; TAC, n=5, respectively) are displayed; *P<0.05 for both tRNAtyr and tRNALeu transcripts in CycD2+/+-TAC versus CycD2+/+-Sham. No significant changes were noted in tRNAtyr and tRNALeu transcripts in CycD2-/- hearts.
Discussion
We have previously shown that CycD2 is critical for mediating Myc-induced hypertrophy10. In the present study we examined the role of CycD2 in mediating the hypertrophic response more generally and its possible downstream effectors. Ablation of CycD2 expression resulted in attenuated cardiac hypertrophy in response to pressure overload, conversely, the same mechanical stress led to an exaggerated hypertrophic response in an Rb-null background. This could be explained, at least in part, by the fact that the activity of RNA pol III, which is normally upregulated with hypertrophic stimuli, was further enhanced in the absence of Rb. However, RNA pol III activity remained unchanged in CycD2-/- mice after TAC as compared to wild-type counterparts. These data provide the first direct evidence for a role for the CycD-Rb pathway in regulating cardiac hypertrophy in vivo, and provide a rational mechanistic link between the observation by many investigators of the upregulation of G1 cyclins and increased protein synthesis via activation of RNA polymerase family members.
Hypertrophic was not completely abolished in CycD2-/- mice and fetal gene induction was unchanged, implying that CycD2 is not the only pathway involved in the development of hypertrophy. This conclusion is consistent with our previous data suggesting that Myc-independent hypertrophic pathways also exist10. Thus, while CycD2 may be obligate for Myc-induced hypertrophic growth, separate pathways must regulate Myc induction of fetal genes. The CycD2-Rb pathway would be expected to have a selective effect on the regulation of the translational machinery and therefore growth (increased protein synthesis) would be more affected than other transcriptional aspects of the hypertrophic response. Since we are proposing that the effects of CycD2-Rb are mediated through RNA Pol III, the transcription of genes such as ANP or BNP that are controlled by RNA Pol II would not be expected to be effected.
Our data that CycD is a critical factor in regulating cell size is seemingly at odds with existing data that CycDs are both necessary and sufficient for cardiac cell cycle progression32,33. However, under normal physiological conditions, cell cycle progression is tightly coupled to the accumulation of cell mass (cell growth)1. Thus, even in situations where CycD2 induces proliferation of cardiac myocytes, it must also be stimulating cell growth as well since the resultant new myocytes were not smaller in size and CycD2 induced “hypertrophic” growth or increased myocyte size when cell cycle progression was inhibited34. These authors concluded that cardiomyocyte hypertrophy is due to levels of cell cycle activators unable to overcome the block imposed by cell cycle inhibitors. This conclusion is consistent with studies in Drosophila, which revealed that in undifferentiated proliferating cells, CycD-Cdk4 caused accelerated cell division, while in post-mitotic cells, CycD-Cdk4 caused cell enlargement35. Thus, in the adult mouse heart where there is general agreement that Cyclin Ds are upregulated with hypertrophic stimuli, clearly the normal physiological consequence is not cell cycle reentry in mice36. This does not negate the observation that if expressed at high enough levels CycD2 can overcome the normal restraints to cell cycle reentry that are seen in adult cardiac myocytes. Our results with inducible Myc mice highlight this concept, where high level activation of Myc induced both hypertrophic growth and cell cycle reentry, although only ∼1% of the myocytes reentered the cell cycle21.
Pocket proteins, and Rb in particular, have a critical role in regulating cardiac cell cycle32,37 but little evidence exists directly implicating Rb in cell size control in mammals although several lines of evidence implicate it in this process6. Classically during cell cycle progression, Rb is inactivated in part by CycD-Cdk4 complexes freeing E2F transcription factors, which then drive cell cycle progression15,16. Although E2F-3 has been implicated in regulating the hypertrophic response, at least in vitro29, we were not able to demonstrate differences in the expression or activity of E2F-1 or -3 in Rb-null mice. This data does not rule out a role for E2F family members in regulating cell growth but does question whether they are the primary downstream effectors of the CycD-Rb pathway in cardiac hypertrophy. Given that E2F-1 levels increase with TAC and a number of E2F target genes24 linked to the control of cell size control were also regulated with TAC, E2Fs may still play a key role in regulating cardiac hypertrophy through a separate pathway.
Interestingly, deletion of CycD2 did not effect the basal cardiac phenotype in CycD2-/- mice as compared to wild-type counter-parts confirming that significant redundancy exists between CycD isoforms (D1, D2, and D3)38. However, minor differences were noted in CycD2 mice. Specifically, we noted a reduction in body weight in CycD2-/- animals as compared to wild-type mice, although the heart weight to body weight ratio was normal. Thus it is likely that other family members can substitute for many but not all functions. Likewise, cardiac-specific deletion of Rb also displayed normal basal cardiac size as compared to CRb+/+ mice. In both cases, this is consistent with the observation that basal RNA pol III activity was normal although RNA pol III activity was augmented after mechanical load elicited in Rb-deficient mice. Rb normally represses pol III transcription by binding pol III transcription factor TFIIIB and sequestering it in an inactive complex39. Our findings support a model where both Brf-1 and TBP (pivotal TFIIIB subunits) become dissociated from Rb in response to a hypertrophic stimulus in vivo. Repression of tRNA and rRNA synthesis may explain the ability of Rb to inhibit protein synthesis and hence hypertrophic growth40. Given the critical link between cell cycle progression and cell mass it not surprising that mechanisms exist that integrate these two modes of growth.
In summary, our results suggest a novel role for the CycD-Rb pathway in the regulation of hypertrophic growth in the adult heart. Future studies to better understand how this novel pathway interacts with other hypertrophic pathways to modulate the translational apparatus should provide insight into how proliferation and cell growth are linked.
Supplementary Material
Acknowledgments
We thank Dr. P. Sicinski for Cyclin D2-null mice, and K. Hayakawa for technical assistance.
Sources of Funding: This work was supported by gifts from the Laubisch Fund (WRM and KPR) as well as grants AHA EIA 0340087N, P01 HL080111 and R01 HL70748 to WRM.
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
References
- 1.Neufeld TP, Edgar BA. Connections between growth and the cell cycle. Curr Opin Cell Biol. 1998;10:784–790. doi: 10.1016/s0955-0674(98)80122-1. [DOI] [PubMed] [Google Scholar]
- 2.McGill CJ, Brooks G. Cell cycle control mechanisms and their role in cardiac growth. Cardiovasc Res. 1995;30:557–569. [PubMed] [Google Scholar]
- 3.Starksen NF, Simpson PC, Bishopric N, Coughlin SR, Lee WM, Escobedo JA, Williams LT. Cardiac myocyte hypertrophy is associated with c-myc protooncogene expression. Proc Natl Acad Sci U S A. 1986;83:8348–8350. doi: 10.1073/pnas.83.21.8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li JM, Poolman RA, Brooks G. Role of G1 phase cyclins and cyclin-dependent kinases during cardiomyocyte hypertrophic growth in rats. Am J Physiol. 1998;275:H814–H822. doi: 10.1152/ajpheart.1998.275.3.H814. [DOI] [PubMed] [Google Scholar]
- 5.Nozato T, Ito H, Tamamori M, Adachi S, Abe S, Marumo F, Hiroe M. G1 cyclins are involved in the mechanism of cardiac myocyte hypertrophy induced by angiotensin II. Jpn Circ J. 2000;64:595–601. doi: 10.1253/jcj.64.595. [DOI] [PubMed] [Google Scholar]
- 6.Tamamori-Adachi M, Ito H, Nobori K, Hayashida K, Kawauchi J, Adachi S, Ikeda MA, Kitajima S. Expression of cyclin D1 and CDK4 causes hypertrophic growth of cardiomyocytes in culture: a possible implication for cardiac hypertrophy. Biochem Biophys Res Commun. 2002;296:274–280. doi: 10.1016/s0006-291x(02)00854-9. [DOI] [PubMed] [Google Scholar]
- 7.Busk PK, Bartkova J, Strom CC, Wulf-Andersen L, Hinrichsen R, Christoffersen TE, Latella L, Bartek J, Haunso S, Sheikh SP. Involvement of cyclin D activity in left ventricle hypertrophy in vivo and in vitro. Cardiovasc Res. 2002;56:64–75. doi: 10.1016/s0008-6363(02)00510-2. [DOI] [PubMed] [Google Scholar]
- 8.Tamamori M, Ito H, Hiroe M, Terada Y, Marumo F, Ikeda MA. Essential roles for G1 cyclin-dependent kinase activity in development of cardiomyocyte hypertrophy. Am J Physiol. 1998;275:H2036–H2040. doi: 10.1152/ajpheart.1998.275.6.H2036. [DOI] [PubMed] [Google Scholar]
- 9.Nozato T, Ito H, Watanabe M, Ono Y, Adachi S, Tanaka H, Hiroe M, Sunamori M, Marum F. Overexpression of cdk Inhibitor p16INK4a by adenovirus vector inhibits cardiac hypertrophy in vitro and in vivo: a novel strategy for the gene therapy of cardiac hypertrophy. J Mol Cell Cardiol. 2001;33:1493–1504. doi: 10.1006/jmcc.2001.1412. [DOI] [PubMed] [Google Scholar]
- 10.Zhong W, Mao S, Tobis S, Angelis E, Jordan MC, Roos KP, Fishbein MC, de Alboran IM, MacLellan WR. Hypertrophic growth in cardiac myocytes is mediated by Myc through a Cyclin D2-dependent pathway. EMBO J. 2006;25:3869–3879. doi: 10.1038/sj.emboj.7601252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadoshima J, Aoki H, Izumo S. Angiotensin II and serum differentially regulate expression of cyclins, activity of cyclin-dependent kinases, and phosphorylation of retinoblastoma gene product in neonatal cardiac myocytes. Circ Res. 1997;80:228–241. doi: 10.1161/01.res.80.2.228. [DOI] [PubMed] [Google Scholar]
- 12.Kato JY, Matsuoka M, Strom DK, Sherr CJ. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Cell Biol. 1994;14:2713–2721. doi: 10.1128/mcb.14.4.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herzinger T, Reed SI. Cyclin D3 is rate-limiting for the G1/S phase transition in fibroblasts. J Biol Chem. 1998;273:14958–14961. doi: 10.1074/jbc.273.24.14958. [DOI] [PubMed] [Google Scholar]
- 14.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 16.Saavedra HI, Wu L, de Bruin A, Timmers C, Rosol TJ, Weinstein M, Robinson ML, Leone G. Specificity of E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ. 2002;13:215–225. [PubMed] [Google Scholar]
- 17.White RJ, Trouche D, Martin K, Jackson SP, Kouzarides T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature. 1996;382:88–90. doi: 10.1038/382088a0. [DOI] [PubMed] [Google Scholar]
- 18.Simone C, Bagella L, Bellan C, Giordano A. Physical interaction between pRb and cdk9/cyclinT2 complex. Oncogene. 2002;21:4158–4165. doi: 10.1038/sj.onc.1205511. [DOI] [PubMed] [Google Scholar]
- 19.Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature. 1996;384:470–474. doi: 10.1038/384470a0. [DOI] [PubMed] [Google Scholar]
- 20.MacLellan WR, Garcia A, Oh H, Frenkel P, Jordan MC, Roos KP, Schneider MD. Overlapping roles of pocket proteins in the myocardium are unmasked by germ line deletion of p130 plus heart-specific deletion of Rb. Mol Cell Biol. 2005;25:2486–2497. doi: 10.1128/MCB.25.6.2486-2497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao G, Mao S, Baumgarten G, Serrano J, Jordan MC, Roos KP, Fishbein MC, MacLellan WR. Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ Res. 2001;89:1122–1129. doi: 10.1161/hh2401.100742. [DOI] [PubMed] [Google Scholar]
- 22.Hu SX, Ji W, Zhou Y, Logothetis C, Xu HJ. Development of an adenovirus vector with tetracycline-regulatable human tumor necrosis factor alpha gene expression. Cancer Res. 1997;57:3339–3343. [PubMed] [Google Scholar]
- 23.Angelis E, Tse MY, Pang SC. Interactions between atrial natriuretic peptide and the renin-angiotensin system during salt-sensitivity exhibited by the proANP gene-disrupted mouse. Mol Cell Biochem. 2005;276:121–131. doi: 10.1007/s11010-005-3672-1. [DOI] [PubMed] [Google Scholar]
- 24.Hlaing M, Spitz P, Padmanabhan K, Cabezas B, Barker CS, Bernstein HS. E2F-1 regulates the expression of a subset of target genes during skeletal myoblast hypertrophy. J Biol Chem. 2004;279:43625–43633. doi: 10.1074/jbc.M408391200. [DOI] [PubMed] [Google Scholar]
- 25.Goodfellow SJ, Innes F, Derblay LE, MacLellan WR, Scott PH, White RJ. Regulation of RNA polymerase III transcription during hypertrophic growth. EMBO J. 2006;25:1522–1533. doi: 10.1038/sj.emboj.7601040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winter AG, Sourvinos G, Allison SJ, Tosh K, Scott PH, Spandidos DA, White RJ. RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc Natl Acad Sci U S A. 2000;97:12619–12624. doi: 10.1073/pnas.230224097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crighton D, Woiwode A, Zhang C, Mandavia N, Morton JP, Warnock LJ, Milner J, White RJ, Johnson DL. p53 represses RNA polymerase III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB. EMBO J. 2003;22:2810–2820. doi: 10.1093/emboj/cdg265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacLellan WR, Xiao G, Abdellatif M, Schneider MD. A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol. 2000;20:8903–8915. doi: 10.1128/mcb.20.23.8903-8915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vara D, Bicknell KA, Coxon CH, Brooks G. Inhibition of E2F abrogates the development of cardiac myocyte hypertrophy. J Biol Chem. 2003;278:21388–21394. doi: 10.1074/jbc.M212612200. [DOI] [PubMed] [Google Scholar]
- 30.Cutilletta AF. Muscle and nonmuscle cell RNA polymerase activities in early myocardial hypertrophy. Am J Physiol. 1981;240:H901–H907. doi: 10.1152/ajpheart.1981.240.6.H901. [DOI] [PubMed] [Google Scholar]
- 31.Larminie CG, Cairns CA, Mital R, Martin K, Kouzarides T, Jackson SP, White RJ. Mechanistic analysis of RNA polymerase III regulation by the retinoblastoma protein. EMBO J. 1997;16:2061–2071. doi: 10.1093/emboj/16.8.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, Kim KK, Field LJ. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997;99:2644–2654. doi: 10.1172/JCI119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–118. doi: 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 34.Busk PK, Hinrichsen R, Bartkova J, Hansen AH, Christoffersen TE, Bartek J, Haunso S. Cyclin D2 induces proliferation of cardiac myocytes and represses hypertrophy. Exp Cell Res. 2005;304:149–161. doi: 10.1016/j.yexcr.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 35.Datar SA, Jacobs HW, de la Cruz AF, Lehner CF, Edgar BA. The Drosophila cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 2000;19:4543–4554. doi: 10.1093/emboj/19.17.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am J Physiol. 1994;266:H1439–H1445. doi: 10.1152/ajpheart.1994.266.4.H1439. [DOI] [PubMed] [Google Scholar]
- 37.Kirshenbaum LA, Schneider MD. Adenovirus E1A represses cardiac gene transcription and reactivates DNA synthesis in ventricular myocytes, via alternative pocket protein- and p300-binding domains. J Biol Chem. 1995;270:7791–7794. doi: 10.1074/jbc.270.14.7791. [DOI] [PubMed] [Google Scholar]
- 38.Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, Akashi K, Sicinski P. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 39.Sutcliffe JE, Brown TR, Allison SJ, Scott PH, White RJ. Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription. Mol Cell Biol. 2000;20:9192–9202. doi: 10.1128/mcb.20.24.9192-9202.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larminie CG, Alzuherri HM, Cairns CA, McLees A, White RJ. Transcription by RNA polymerases I and III: a potential link between cell growth, protein synthesis and the retinoblastoma protein. J Mol Med. 1998;76:94–103. doi: 10.1007/s001090050196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.