

Abstract
Mutations leading to aberrant cytoplasmic localization of nucleophosmin (NPM) are the most frequent genetic alteration in acute myelogenous leukemia (AML). NPM binds the Arf tumor suppressor and protects it from degradation. The AML-associated NPM mutant (NPMmut) also binds p19Arf but is unable to protect it from degradation, which suggests that inactivation of p19Arf contributes to leukemogenesis in AMLs. We report here that NPM regulates turnover of the c-Myc oncoprotein by acting on the F-box protein Fbw7γ, a component of the E3 ligase complex involved in the ubiquitination and proteasome degradation of c-Myc. NPM was required for nucleolar localization and stabilization of Fbw7γ. As a consequence, c-Myc was stabilized in cells lacking NPM. Expression of NPMmut also led to c-Myc stabilization because of its ability to interact with Fbw7γ and delocalize it to the cytoplasm, where it is degraded. Because Fbw7 induces degradation of other growth-promoting proteins, the NPM–Fbw7 interaction emerges as a central tumor suppressor mechanism in human cancer.
Introduction
Mutations of the nucleophosmin (NPM) gene occur in ∼35% of acute myelogenous leukemias (AMLs) and are mutually exclusive with the major AML-associated genetic abnormalities, which suggests that they represent an initiating event in myeloid leukemogenesis (Falini et al., 2005). The underlying mechanisms, however, remain unknown.
NPM is a ubiquitously expressed nucleolar protein that functions as a molecular chaperone (Okuwaki et al., 2001) and shuttles between the nucleus and cytoplasm (Borer et al., 1989). It is part of a high–molecular weight complex and physically interacts with many cellular proteins including p53 (Colombo et al., 2002), Mdm2 (Kurki et al., 2004), and Arf (Itahana et al., 2003). NPM binds Arf and protects it from degradation (Kuo et al., 2004). In NPM−/− cells, the Arf protein looses its nucleolar localization and becomes markedly unstable, which suggests that NPM is required for correct localization and stability of Arf (Colombo et al., 2005). This function of NPM is lost in mutant NPM (NPMmut), which contains a de novo nuclear export signal and mainly localizes in the cytoplasm (Mariano et al., 2006). NPMmut competes with wild-type (WT) NPM for Arf binding and targets Arf to the cytoplasm, where it becomes more susceptible to degradation (Colombo et al., 2006).
Preliminary evidence suggests, however, that NPM controls other intracellular pathways that negatively regulate cell proliferation. In NPM−/− embryos, cells proliferate more actively, accumulate DNA damage, and undergo p53-dependent apoptosis (Colombo et al., 2005), a picture that is reminiscent of the DNA damage response induced by oncogene expression in primary cells (Bartkova et al., 2005). NPM−/− cells also show aberrant mitotic figures with multiple centrosomes (Grisendi et al., 2005) and are more susceptible to transformation by activated oncogenes such as Myc and Ras (Colombo et al., 2005). Consistently, NPM± mice show accelerated Myc-induced lymphomagenesis (Grisendi et al., 2005). We report here that NPM and its AML-associated mutant regulate the stability of the c-Myc protein.
Results and discussion
We initially analyzed levels of c-Myc in lysates from NPM−/− and WT whole embryos. Western blotting (WB) revealed a marked increase of the c-Myc protein in the NPM−/− samples (Fig. 1 a, left). Levels of c-myc mRNA, instead, were equivalent in the two samples (Fig. 1 a, right), which suggests that the increased levels of c-Myc protein in the NPM−/− embryos were caused by enhanced protein stability. Accordingly, we observed a moderate increase in mRNA expression for several c-Myc target genes in NPM−/− embryos (Fig. 1 b).
Figure 1.

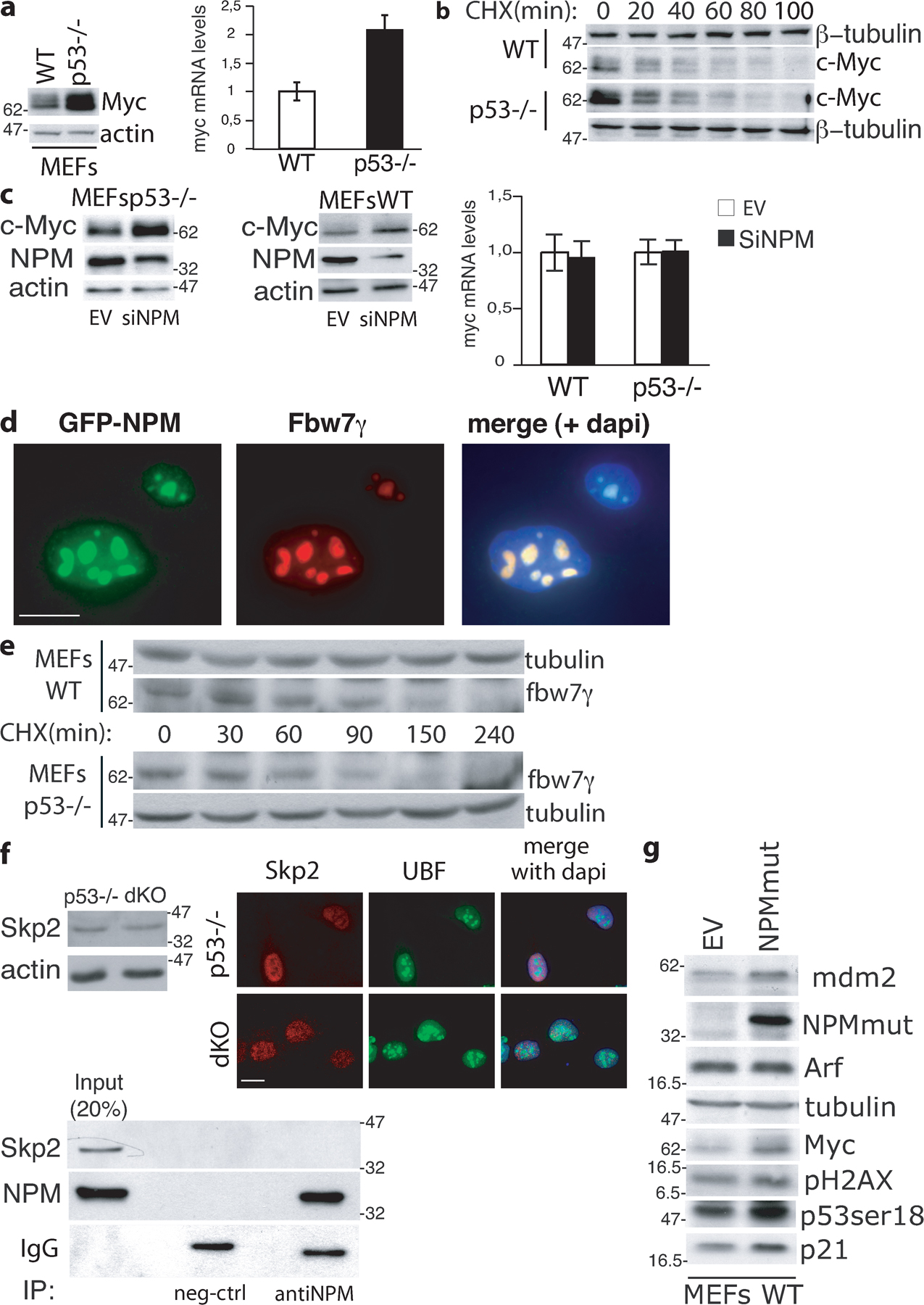
NPM regulates c-Myc protein stability. (a, left) WB analysis of two NPM WT (Nos. 1 and 2) and knockout (KO; Nos. 2 and 3) embryos at 10.5 d post coitum (Colombo et al., 2005). (right) c-Myc mRNA levels in the same embryos evaluated by QPCR (results are normalized against WT samples). (b) Expression of c-Myc target genes analyzed by QPCR using mRNA from WT and NPM KO embryos. Expression was standardized with ubiquitin and normalized against the control WT RNAs. (c) c-Myc protein levels (left) and c-Myc mRNA levels (right) in p53−/− and dKO MEFs. (d) QPCR of anti c-Myc ChIP. The percentage of DNA bound to c-Myc was calculated as described previously (Frank et al., 2001). (e) QPCR analysis of c-Myc target genes at 8 h after serum treatment. Results analyzed as in panel b. (f) c-Myc protein level in p53−/− and dKO MEFs. Cells were treated with CHX and harvested at the indicated time points. (g, left) c-Myc protein levels in p53−/− and dKO MEFs infected with retroviruses expressing the GFP-NPM1 protein (+NPM) or control retroviruses. (middle) WB analysis of the same cells during serum starvation and upon serum release. (right) QPCR analysis of c-Myc target gene expression after serum treatment (8 h) as in panel d. (h) c-Myc protein level in p53−/−, dKO, and dKO + GFP-NPM MEFs as described in panel f. Data represent the mean of three determinations ± SEM.
We then examined c-Myc levels in cultured fibroblasts (mouse embryonic fibroblasts [MEFs]) from double knockout NPM−/−;p53−/− embryos (dKO) and, as controls, MEFs from p53−/− embryos. NPM−/− MEFs, in fact, do not grow in culture due to the accumulation of DNA damage and rapid acquisition of a p53-dependent senescence-like phenotype (Colombo et al., 2005). c-Myc protein was elevated in dKO MEFs as compared with the p53−/− cells (Fig. 1 c, left) in the absence of significant variations of the amount of c-myc transcripts (Fig. 1 c, right). dKO MEFs also showed increased binding of c-Myc to target promoters, as assayed by chromatin immunoprecipitation (ChIP; Fig. 1 d), and an increased induction of c-Myc target genes after serum stimulation of starved cells (Fig. 1 e). To measure the half-life of the c-Myc protein, dKO and control cells were treated with cycloheximide (CHX; an inhibitor of de novo protein synthesis widely used to measure c-Myc protein half-life; Popov et al., 2007) and lysed at different time points to follow c-Myc protein degradation. As an internal control, we monitored expression of γ-tubulin (Colombo et al., 2005). WB in the p53−/− MEFs confirmed that c-Myc is an unstable protein with a half-life of ∼20 min (Gregory and Hann, 2000). In the dKO cells, instead, the c-Myc protein appeared to be more stable, with a calculated half-life of ∼40 min (Fig. 1 f). It appears, therefore, that NPM regulates stability of the c-Myc protein. To confirm the NPM dependency, we reconstituted NPM expression in dKO cells and measured levels of c-Myc protein, c-Myc protein stability, and c-Myc target gene expression. Notably, NPM reexpression in NPM−/− MEFs reduced the half-life of the c-Myc protein (Fig. 1 g, left; and Fig. 1 h), its steady-state and serum-stimulated levels (Fig. 1 g, middle), and expression of c-Myc target genes after serum stimulation (Fig. 1 g, right). Finally, to investigate the effects of p53 on c-Myc expression, we evaluated levels of c-myc mRNA/protein and c-Myc protein stability in WT and p53-null MEFs. As shown in Fig. S1 a (available at http://www.jcb.org/cgi/content/full/jcb.200711040/DC1), a lack of p53 led to increased levels of c-Myc mRNA and protein, as previously described (Ho et al., 2005). Notably, however, the stability of the c-Myc protein was the same in the two cell types (Fig. S1 b). Down-regulation of NPM expression by siRNA induced up-regulation of c-Myc protein levels, but not of the corresponding mRNA, in both the WT and p53−/− MEFs (Fig. S2 c), thus indicating that the effect of NPM silencing on c-Myc protein is p53 independent.
c-Myc is degraded through the ubiquitin–proteasome pathway (Salghetti et al., 1999), and its ubiquitination is controlled by two different F-box proteins, Fbw7 and Skp2 (Hann, 2006). Three isoforms of Fbw7 have been described that differ in their intracellular localization: Fbw7α (nuclear diffuse), Fbw7β (cytoplasmic), and Fbw7γ (nucleolar). Although all three Fbw7 isoforms have the potential to bind it, preliminary evidence suggests that c-Myc is prevalently degraded by Fbw7γ (Popov et al., 2007). Because NPM localizes to the nucleolus (where it colocalizes with Fbw7γ; Fig. S1 d), we investigated whether the Fbw7γ isoform is involved in the regulation of c-Myc stability by NPM. The HA-tagged version of Fbw7γ was overexpressed in dKO or p53−/− control cells previously engineered with a Myc–estrogen receptor fusion protein (Myc-ER) whose activity is induced by treatment with 4-hydroxytamoxifen (4-OHT). Treatment of p53−/− control cells with OHT rapidly induced nuclear translocation of Myc-ER (not depicted), its stabilization (Fig. 2 a), and binding to target promoters (Fig. 2 b). Overexpression of Fbw7γ reduced Myc-ER protein stabilization and promoter occupancy in p53−/− cells (Fig. 2, a and b) but had no effects in dKO cells (Fig. 2, a and b). The same experiments were repeated in the dKO and p53−/− parental cells. Overexpression of Fbw7γ slightly yet consistently reduced levels of endogenous c-Myc protein and c-Myc occupancy at target promoters in the p53−/− cells but not in dKO cells (Fig. 2, c and d). It appears, therefore, that NPM expression is critical for the ability of Fbw7γ to regulate c-Myc protein levels.
Figure 2.

NPM is required for Fbw7γ activity. (a) WB analysis in p53−/− and dKO MEFs infected with Myc-ER and, where indicated (+), HA-Fbw7γ–expressing retroviruses. Cells were treated with 4-OHT for the indicated hours or left untreated (0 h). (b) QPCR of anti–c-Myc ChIP on target promoters (as indicated) after 4-OHT treatment of the same cells as in panel a. (c) WB analysis in p53−/− and dKO MEFs infected with retroviruses expressing HA-Fbw7γ (+) or control retroviruses (−) as indicated. c-Myc levels have been calculated by densimetric analysis, normalizing band intensity to actin and control p53−/− cells. (d) QPCR analysis of anti c-Myc ChIP on target promoters (as indicated) of the same cells as in panel c. (e) dKO MEFs were cotransfected with plasmids expressing NPM1 and flag-Fbw7γ (left), flag-Fbw7α (middle), and flag-Fbw7β (right). Total lysates and immunoprecipitates were blotted with antibodies against NPM (NPMa) or anti-flag. (f) GST pull-down assay using in vitro translated 35S-labeled Fbw7γ or Fbw7α and equal amounts of GST or GST-NPM proteins. Data represent the mean of three determinations ± SEM.
We than analyzed whether NPM interacts physically with Fbw7γ and affects the localization and expression levels of Fbw7γ. Coimmuprecipitation experiments revealed that NPM forms a complex in vivo with ectopically expressed tagged Fbw7γ (Fig. 2 e, left). The interaction is likely to be direct, as recombinant GST-NPM bound in vitro translated Fbw7γ (Fig. 2 f). No interactions were detected between NPM and Fbw7α (Fig. 2, e and f) or β (Fig. 2 e).
Immunofluorescence (IF) analysis (Fig. 3 a, top) showed that Fbw7γ was concentrated in the nucleoli of p53−/− cells, as expected. In dKO MEFs, the nucleolar localization of Fbw7γ was lost, replaced by a diffuse nuclear staining. These alterations in localization were specific for the Fbw7γ isoform because Fbw7α and Fbw7β showed unchanged localization patterns in dKO and control cells (Fig. 3 a, bottom left and bottom right). Fbw7γ levels in the dKO MEFs were also consistently lower than in the control p53−/− MEFs (Fig. 3 b, left), whereas mRNAs were comparable (Fig. 3 b, right). CHX treatment revealed that the half-life of Fbw7γ in p53−/− MEFs was ∼80 min but was reduced in the dKO to ∼30 min (Fig. 3 c). The half-life of Fbw7γ protein was not influenced by expression of p53 (Fig. S1 e). Accordingly, Fbw7γ−dependent ubiquitination of c-Myc was reduced in NPM-null cells (Fig. 3 d). Finally, we analyzed the ability of Fbw7γ to inhibit c-Myc–dependent transformation. Overexpression of Fbw7γ induced a significant attenuation of the transformed phenotype in OHT-treated p53−/−;Myc-ER cells, as shown by their reduced ability to form colonies in semisolid medium (Fig. 3 e). Overexpressed Fbw7γ, instead, had no effect on OHT-treated dKO;Myc-ER cells (Fig. 3 e). In summary, NPM positively regulates the nucleolar localization and stability of the Fbw7γ protein and is essential for the ability of Fbw7 to promote c-Myc degradation and limit cell transformation. The effect of NPM on c-Myc protein stability appears to be specifically exerted through Fbw7γ, as we found no effects of NPM on expression and localization of Skp2 (Fig. S1 f).
Figure 3.

NPM is required Fbw7γ nucleolar localization and stability. (a) IF analysis of Fbw7γ (top), Fbw7α (bottom left), and Fbw7β (bottom right) localization in p53−/− and dKO MEFs transiently transfected with the corresponding flag-Fbw7 constructs (green staining). Anti-fibrillarin staining (red) is shown as a marker for nucleoli. Bars, 10 μm. (b, left) WB analysis in p53−/− and dKO MEFs infected with retroviruses expressing HA-Fbw7γ (+) or control retroviruses (−) as indicated. (right) Fbw7γ mRNA levels in p53−/− and dKO MEFs. (c) HA-Fbw7γ protein stability in p53−/− and dKO MEFs. Cells were treated with CHX and harvested at the indicated time points. (d) Effects of Fbw7γ expression on Myc ubiquitination in p53−/− and dKO cells. p53−/− (lanes 1–3) and dKO (lanes 4–6) MEFs were transfected with expression vectors for HA-tagged ubiquitin and flag-tagged Fbw7γ, as indicated. Cell lysates were IPed with an anti-Myc antibody and blotted with anti-HA (to identify ubiquitinated myc; UB-myc) and anti-Myc (as control) antibodies. Levels of flag-Fbw7γ in the input were analyzed with an anti-flag antibody. (e) Methylcellulose colony assay of p53−/− and dKO MEFs expressing Myc-ER protein and infected with control (EV) or HA-Fbw7γ–expressing retroviruses. 4-OHT was added every 3 d; colonies were counted after 15 d. Data represent the mean of three determinations ± SEM. *, P < 0.045.
c-Myc is frequently overexpressed in human cancers (Adhikary and Eilers, 2005). c-Myc protein and/or RNA are also frequently overexpressed in AMLs, though the underlying molecular mechanisms are unknown (Hoffman et al., 2002). We thus investigated whether expression of the AML-associated mutant of NPM leads to increased c-Myc expression. Ectopic expression of NPMmut in WT MEFs increased the steady-state levels of the c-Myc protein in the absence of significant variations of the amount of c-Myc transcripts (Fig. 4 a) and the expression of several Myc-target genes (Fig. 4 b). These effects were maintained in Arf−/− MEFs (Fig. 4 e) and were thus independent on the presence of Arf in the nucleolus or its interaction with Myc (Qi et al., 2004).
Figure 4.

Mutant NPM stabilizes the c-Myc protein. (a, left) WB analysis in WT and NPM± MEFs infected with retroviruses expressing NPMmut (Mut) or control (EV) retroviruses. (right) c-Myc mRNA levels in the same cell. (b) QPCR analysis of the indicated c-Myc target genes. (c, left) Growth curves of NPM± MEFs infected with retroviruses expressing NPMmut or control (EV) retroviruses. 104 cells were plated in presence of 10% serum (high serum) or 0.5% serum (low serum) as indicated. (right) The percentage of apoptosis in the same cells maintained in low serum culture conditions for 24 h. (d) QPCR analysis of the indicated c-Myc target genes in NPM± MEFs infected with EV or NPMmut retroviruses. (e, left) WB analysis in ARF−/−, p53−/−, and dKO MEFs infected with retroviruses expressing NPMmut (mut) or control (EV) retroviruses. (right) c-Myc mRNA levels in the same samples. (f) QPCR analysis of the indicated c-Myc target genes in the same cells as in panel e. Data represent the mean of three determinations ± SEM.
To gain evidence that this effect of NPMmut on c-Myc is biologically relevant, we analyzed growth of fibroblasts ectopically expressing NPMmut. Overexpression of c-Myc induces DNA damage (Dominguez-Sola et al., 2007) and, in primary fibroblasts, a p53-dependent cellular checkpoint mainly characterized by a reduction of cell proliferation; when cells are grown in low serum, it also induces apoptosis (Zindy et al., 1998). Ectopic expression of the NPMmut in MEFs induced no modifications of Arf protein levels (Fig. S1 g), it increased the levels of γH2AX (a marker of DNA damage) and phosphorylated p53 (Fig. S1 g), decreased the proliferation rate, and, in low serum conditions, induced massive apoptosis (Fig. 4 c). Together, these results suggest that apoptosis induced by NPMmut expression follows c-Myc stabilization, induction of DNA damage, and activation of p53.
We then investigated whether the effect of NPMmut on c-Myc stability is mediated by Fbw7γ. The ability of overexpressed Fbw7γ to down-regulate Myc-ER levels was completely lost in the presence of NPMmut expression (Fig. 5 a). Moreover, in the presence of NPMmut, levels of Fbw7γ protein were almost undetectable without changes of its mRNA (Fig. 5 a). Treatment of the same cells with the MG132 proteasome inhibitor for 2 h was sufficient to reconstitute Fbw7γ expression at levels similar to those observed in control cells without NPMmut (Fig. 5 b). It appears, therefore, that NPMmut induces destabilization of Fbw7γ and, as a consequence, stabilization of c-Myc.
Figure 5.

Mutant NPM delocalizes and destabilizes Fbw7γ. (a, left) WB analysis in p53−/−;Myc-ER cells infected with empty (No. 2) or HA-Fbw7γ-expressing (No. 4) retroviruses. HA-Fbw7γ–infected cells were reinfected with empty or NPMmut-expressing lentiviruses (also expressing GFP as marker). Cells were treated for 2 h with 4-OHT (Nos. 2–4). (right) QPCR analysis of Fbw7γ mRNA levels in samples Nos. 3 and 4. Data represent the mean of three determinations ± SEM. (b) WB analysis in the same cells as in panel a. Cells were or were not treated with MG132 for 2 h, as indicated. (c) dKO MEFs were cotransfected with plasmids expressing flag-Fbw7γ and NPMmut or the empty vector, as indicated. Total lysates and IPs were blotted with antibodies against NPMmut or the flag epitope. (d) IF analysis of p53−/− MEFs infected with GFP-NPMmut– and flag-Fbw7γ (red staining)-expressing constructs. A merge of the NPMmut, Fbw7γ, and DAPI staining is also shown. Bar, 10 μm. (e) Nucleus–cytoplasm fractionation in p53−/− MEFs expressing flag-Fbw7γ infected with GFP-NPMmut or control (EV) retroviruses. Total cell lysates (T) or cellular fractions (C, cytoplasm; N, nucleus) were analyzed by WB as indicated. (f) The same cells as in panel e were or were not treated with 1 μM LMB or MG132 for 3 h, as indicated. Expression of Fbw7γ was analyzed by WB on total cells lysates using anti-flag antibodies.
Finally, we investigated the mechanism through which NPMmut affects Fbw7γ protein stability. Coimmunoprecipitation experiments in NPM-deficient MEFs revealed that NPMmut is able to form a complex with Fbw7γ both in the absence (Fig. 5 c) or presence (not depicted) of WT NPM. IF analysis of the intracellular localization of Fbw7γ in p53−/− and dKO showed that expression of NPMmut causes, in both cell types, a partial displacement of Fbw7γ from the nucleus to the cytoplasm (Fig. 5 d and Fig. S2 a, available at http://www.jcb.org/cgi/content/full/jcb.200711040/DC1). These results were confirmed by WB analysis of the levels of Fbw7γ in nuclear and cytoplasmic fractions of p53−/− and dKO cells in the presence or absence of NPMmut expression (Fig. 5 e and Fig. S2 b). The ability of NPMmut to delocalize Fbw7γ and to form a complex in cells also in the presence of the WT NPM suggest that NPMmut competes with the WT protein to bind Fbw7γ. Consistently, we observed that up-regulation of c-Myc protein levels (Fig. 4 a) and Myc-target gene expression (Fig. 4 d) by NPMmut were facilitated in NPM+/− MEFs, which carry reduced levels of NPM (Grisendi et al., 2005).
To investigate if the cytoplasmic displacement of Fbw7γ correlates with its degradation, we analyzed levels of the Fbw7γ protein upon treatment of p53−/− and dKO cells with the nuclear-export inhibitor leptomycin B (LMB) as compared with treatment with the proteasome inhibitor MG132. As shown in Fig. 5 f and Fig. S2 c, LMB and MG132 increased the levels of Fbw7γ to the same extent in both cell types. However, despite the fact that NPMmut induced degradation of Fbw7γ in dKO cells (Fig. 4 e), it did not increase c-Myc protein levels in the same cells, which is consistent with the observation that, in the absence of WT NPM, the function of Fbw7γ is lost, regardless of its nuclear or cytoplasmic localization (Fig. 2, a–c). These data are consistent with the reported chaperone activity of WT NPM in maintaining the proper folding and activity of its interactors (Szebeni and Olson, 1999). In summary, we observed that expression of NPMmut induces delocalization and accelerated degradation of Fbw7γ also in the absence of the WT NPM protein, thus demonstrating that the effect of the mutant NPM on Fbw7γ is direct. Furthermore, because LMB treatment induced nuclear relocalization of NPMmut and Fbw7γ and restoration of physiological levels of Fbw7γ protein, aberrant cytoplasmic localization of the NPMmut appears critical for the degradation of Fbw7γ.
In conclusion, our findings demonstrate that NPM regulates c-Myc protein stability through its effect on the γ-isoform of the F-box E3 ubiquitin ligase Fbw7. They are consistent with a model whereby NPM binds directly to Fbw7γ and serves as a molecular chaperone to ensure its proper folding, nucleolar localization, and to prevent its degradation. This effect of NPM on Fbw7γ is relevant for the regulation of one of its substrates, c-Myc, which, in fact, accumulates in the absence of NPM.
The effect of NPM on Fbw7 regulation, however, might be more complex. Other intracellular proteins are also Fbw7-substrates, including Notch, cyclin E, and c-Jun (Nakayama and Nakayama, 2005). Because NPM does not regulate the Fbw7α and Fbw7β isoforms, it might allow regulation of Fbw7γ-specific substrates or the nucleolar site of degradation of Fbw7-common substrates. Close inspection of the regulation of Notch, cyclin E, and c-Jun protein stability and function in NPM−/− cells might help to elucidate this question.
The leukemia-associated mutant of NPM interacts with Fbw7γ and delocalizes it to the cytoplasm, thus favoring its degradation. As a consequence, c-Myc protein levels are increased in cells expressing the NPMmut. Notably, c-Myc protein is frequently overexpressed in AMLs and stabilized in several leukemia cell lines (Hoffman et al., 2002), and c-Myc overexpression favors myeloid leukemogenesis in mouse models (Luo et al., 2005). Thus, the pathway of c-Myc degradation is relevant for human cancer, which suggests that elevated c-Myc protein expression might contribute to leukemogenesis in the NPMmut AMLs.
Oncogene-induced hyperproliferation activates an Arf- and p53-dependent intracellular checkpoint that leads to cell cycle arrest or apoptosis (Di Micco et al., 2006). Remarkably, the NPMmut also favors Arf degradation, which suggests that in AMLs, a single genetic mutation might have the double effect of activating proliferation and attenuating the resulting checkpoint response. This would probably result in accelerated leukemogenesis and reduced pressure for accumulation of further genetic abnormalities. Interestingly, AMLs with NPMmut usually have normal karyotypes (Falini et al., 2005).
Materials and methods
qChIP and quantitative PCR (QPCR) analysis
MEFs were grown and processed for qChIP analysis as described previously (Frank et al., 2001) using anti-Myc (N-262; Santa Cruz Biotechnology, Inc.) antibodies. Immunoprecipitated DNA from ∼1 × 107 cell equivalents was resuspended in 300 μl of 10 mM Tris at pH 8.0. Real-time PCR was performed with 6 μl of DNA per reaction and 200 nM of primers, diluted in a final volume of 20 μl of SYBR Green reaction mix (PerkinElmer). Data represent the mean of three determinations ± SEM. A list of the used primers is shown in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200711040/DC1).
Total RNA was extracted from MEFs with an RNAeasy MiniKit (QIAGEN), including a DNase treatment before elution from the column, and processed as described previously (Frank et al., 2001). Each PCR reaction contained 10 ng of cDNA template and primers at a concentration of 800 nM in a final volume of 20 μl of SYBR Green reaction mix. Data represent the mean ± SEM of three determinations for each gene. A list of the used primers is shown in Table S1.
Cell culture, transfection, and infection
MEFs, Phoenix, and 293T packaging cell lines were cultured at 37°C and 5% CO2 in DME supplemented with 10% fetal bovine serum. Empty or recombinant retroviral or lentivirus vectors were transfected into phoenix or 293T packaging cell lines, respectively, and after 48 h, the supernatants were used to infect target cells. Transient transfections were performed using the standard calcium phosphate precipitate method for Phoenix, 293T, and Fugene 6 (Roche) for MEFs.
Immunoblotting, immunoprecipitation (IP), and IF
WB, IP, and IF experiments were performed as described previously (Colombo et al., 2002). All protein total lysates for WB analysis were performed in Laemmli buffer. The primary antibodies used for WB were: monoclonal anti-NPM (NPMa that recognizes both WT and mutant NPM, and NPMc that recognizes only the WT NPM1 isoform; Cordell et al., 1999); monoclonal anti-NPMmut specific (homemade according to the protocol described in Quentmeier et al., 2005), anti-Myc (provided by S. Hann, Vanderbilt University School of Medicine, Nashville, TN), anti-flag (Sigma-Aldrich), monoclonal anti-HA (Covance), anti-actin (Sigma-Aldrich), anti-tubulin (Santa Cruz Biotechnology, Inc.). The primary antibodies used for IPs were: monoclonal anti-NPM (Invitrogen), monoclonal anti-NPMmut, monoclonal anti-flag M2 (Sigma-Aldrich), anti-fibrillarin (Santa Cruz Biotechnology, Inc.), and monoclonal anti-HA (Covance). Secondary antibodies conjugated to FITC or Cy3 fluorochromes were used for immunofluorescence.
Images were acquired at room temperature using a camera (C4742-95; Hamamatsu) on a microscope (BX61; Olympus) with 60× 1.40 NA oil objective lenses (Olympus). The acquisition software was Cell^F (Olympus).
Plasmids
Fbw7α, Fbw7β, and Fbw7γ flag-tagged plasmids were provided by B. Clurmann (Fred Hutchinson Cancer Research Center, Seattle, WA). Fbw7α and Fbw7γ cDNAs were subcloned in the pCDNA3.1 mammalian expression vector (Invitrogen). Fbw7γ was also subcloned in the pBabe-flag-HA retroviral vector. The mutated form of NPM (NPMmut) was cloned in the pRRLsin.hPGK.IRES.EGFP.Wpre vector (Follenzi et al., 2000) using BamH1 restriction sites. The plasmid expressing HA-ubiquitin was kindly provided by S. Polo (Istituto FIRC di Oncologia Molecolare [IFOM], Milan, Italy).
Apoptosis assay
Cells were harvested and double fixed in 1% formaldehyde and then cold ethanol. After permeabilization in 0.1% Triton X-100 for 10 min, cells were stained with anti-cleaved caspase3 antibody (Cell Signaling Technology), washed, and incubated with cy5-conjugated secondary antibody. The percentage of positive cells was evaluated by FACS analysis.
Nuclear and cytoplasmic fractionation
MEFs were washed twice in ice-cold hypotonic buffer (20 mM Hepes, pH 7, 5 mM potassium-acetate, 0.5 mM MgCl2, and 0.5 mM DTT) supplemented with protease inhibitors, and then swollen on ice for 10 min. After buffer removal, cells were collected by scraping and disrupted by vigorous pipetting. The integrity of nuclei was checked with the microscope after trypan blue staining. Nuclei were pelleted at 4,000 rpm for 5 min, and supernatants were stored as cytoplasmic fraction. The nuclear fraction was washed twice with hypotonic buffer and then lysed in Laemmli buffer. Equal volumes of nuclear and cytosolic fractions were loaded on an SDS-polyacrylamide gel for WB analysis.
Statistical analysis
Statistical significance was evaluated by nonparametric Mann-Whitney U test (Wilcoxon-Kruskal Wallis) to analyze variables that were not normally distributed. Significance was defined at P < 0.05 (two-tailed test).
Online supplemental material
Fig. S1 shows control experiments regarding the role of p53 in regulating c-Myc and Fbw7γ half-lives, the role of NPM in controlling Skp2 protein localization and stability, and the expression levels of different proteins upon NPMmut expression in MEFs. Fig. S2 shows the effect of NPMmut expression in dKO MEFs on Fbw7γ localization and stability. Table S1 shows all the primers used in the QPCR experiments. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200711040/DC1.
Supplementary Material
Acknowledgments
We thank Steve Hann for helpful discussions and providing anti c-Myc antibodies; components of the Amati group for technical contributions, helpful suggestions, and discussions; and Simona Ronzoni for technical support.
This work was supported by grants from Associazione Italiana Ricerca sul Cancro and the European Community (Intact Consortium No. 506803). The authors declare no competing financial interests.
P.G. Pelicci and E. Colombo contributed equally to this paper.
Abbreviations used in this paper: AML, acute myelogenous leukemia; ChIP, chromatin immunoprecipitation; CHX, cycloheximide; LMB, leptomycin B; MEF, mouse embryonic fibroblast; Myc-ER, Myc–estrogen receptor fusion protein; NPM, nucleophosmin; NPMmut, NPM mutant; OHT, hydroxytamoxifen; QPCR, quantitative PCR; WB, Western blot; WT, wild type.
References
- Adhikary, S., and M. Eilers. 2005. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 6:635–645. [DOI] [PubMed] [Google Scholar]
- Bartkova, J., Z. Horejsi, K. Koed, A. Kramer, F. Tort, K. Zieger, P. Guldberg, M. Sehested, J.M. Nesland, C. Lukas, et al. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 434:864–870. [DOI] [PubMed] [Google Scholar]
- Borer, R.A., C.F. Lehner, H.M. Eppenberger, and E.A. Nigg. 1989. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 56:379–390. [DOI] [PubMed] [Google Scholar]
- Colombo, E., J.C. Marine, D. Danovi, B. Falini, and P.G. Pelicci. 2002. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 4:529–533. [DOI] [PubMed] [Google Scholar]
- Colombo, E., P. Bonetti, E. Lazzerini Denchi, P. Martinelli, R. Zamponi, J.C. Marine, K. Helin, B. Falini, and P.G. Pelicci. 2005. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol. Cell. Biol. 25:8874–8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, E., P. Martinelli, R. Zamponi, D.C. Shing, P. Bonetti, L. Luzi, S. Volorio, L. Bernard, G. Pruneri, M. Alcalay, and P.G. Pelicci. 2006. Delocalization and destabilization of the Arf tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 66:3044–3050. [DOI] [PubMed] [Google Scholar]
- Cordell, J.L., K.A. Pulford, B. Bigerna, G. Roncador, A. Banham, E. Colombo, P.G. Pelicci, D.Y. Mason, and B. Falini. 1999. Detection of normal and chimeric nucleophosmin in human cells. Blood. 93:632–642. [PubMed] [Google Scholar]
- Di Micco, R., M. Fumagalli, A. Cicalese, S. Piccinin, P. Gasparini, C. Luise, C. Schurra, M. Garre, P.G. Nuciforo, A. Bensimon, R. Maestro, P.G. Pelicci, and F. d'Adda di Fagagna. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 444:638–642. [DOI] [PubMed] [Google Scholar]
- Dominguez-Sola, D., C.Y. Ying, C. Grandori, L. Ruggiero, B. Chen, M. Li, D.A. Galloway, W. Gu, J. Gautier, and R. Dalla-Favera. 2007. Non-transcriptional control of DNA replication by c-Myc. Nature. 448:445–451. [DOI] [PubMed] [Google Scholar]
- Falini, B., C. Mecucci, E. Tiacci, M. Alcalay, R. Rosati, L. Pasqualucci, R. La Starza, D. Diverio, E. Colombo, A. Santucci, et al. 2005. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352:254–266. [DOI] [PubMed] [Google Scholar]
- Follenzi, A., L.E. Ailles, S. Bakovic, M. Geuna, and L. Naldini. 2000. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 25:217–222. [DOI] [PubMed] [Google Scholar]
- Frank, S.R., M. Schroeder, P. Fernandez, S. Taubert, and B. Amati. 2001. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 15:2069–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, M.A., and S.R. Hann. 2000. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt's lymphoma cells. Mol. Cell. Biol. 20:2423–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisendi, S., R. Bernardi, M. Rossi, K. Cheng, L. Khandker, K. Manova, and P.P. Pandolfi. 2005. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 437:147–153. [DOI] [PubMed] [Google Scholar]
- Hann, S.R. 2006. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 16:288–302. [DOI] [PubMed] [Google Scholar]
- Ho, J.S., W. Ma, D.Y. Mao, and S. Benchimol. 2005. p53-dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol. Cell. Biol. 25:7423–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, B., A. Amanullah, M. Shafarenko, and D.A. Liebermann. 2002. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene. 21:3414–3421. [DOI] [PubMed] [Google Scholar]
- Itahana, K., K.P. Bhat, A. Jin, Y. Itahana, D. Hawke, R. Kobayashi, and Y. Zhang. 2003. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell. 12:1151–1164. [DOI] [PubMed] [Google Scholar]
- Kuo, M.L., W. den Besten, D. Bertwistle, M.F. Roussel, and C.J. Sherr. 2004. N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev. 18:1862–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurki, S., K. Peltonen, L. Latonen, T.M. Kiviharju, P.M. Ojala, D. Meek, and M. Laiho. 2004. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 5:465–475. [DOI] [PubMed] [Google Scholar]
- Luo, H., Q. Li, J. O'Neal, F. Kreisel, M.M. Le Beau, and M.H. Tomasson. 2005. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood. 106:2452–2461. [DOI] [PubMed] [Google Scholar]
- Mariano, A.R., E. Colombo, L. Luzi, P. Martinelli, S. Volorio, L. Bernard, N. Meani, R. Bergomas, M. Alcalay, and P.G. Pelicci. 2006. Cytoplasmic localization of NPM in myeloid leukemias is dictated by gain-of-function mutations that create a functional nuclear export signal. Oncogene. 25:4376–4380. [DOI] [PubMed] [Google Scholar]
- Nakayama, K.I., and K. Nakayama. 2005. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin. Cell Dev. Biol. 16:323–333. [DOI] [PubMed] [Google Scholar]
- Okuwaki, M., K. Matsumoto, M. Tsujimoto, and K. Nagata. 2001. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 506:272–276. [DOI] [PubMed] [Google Scholar]
- Popov, N., M. Wanzel, M. Madiredjo, D. Zhang, R. Beijersbergen, R. Bernards, R. Moll, S.J. Elledge, and M. Eilers. 2007. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 9:765–774. [DOI] [PubMed] [Google Scholar]
- Qi, Y., M.A. Gregory, Z. Li, J.P. Brousal, K. West, and S.R. Hann. 2004. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature. 431:712–717. [DOI] [PubMed] [Google Scholar]
- Quentmeier, H., M.P. Martelli, W.G. Dirks, N. Bolli, A. Liso, R.A. Macleod, I. Nicoletti, R. Mannucci, A. Pucciarini, B. Bigerna, et al. 2005. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 19:1760–1767. [DOI] [PubMed] [Google Scholar]
- Salghetti, S.E., S.Y. Kim, and W.P. Tansey. 1999. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 18:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebeni, A., and M.O. Olson. 1999. Nucleolar protein B23 has molecular chaperone activities. Protein Sci. 8:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy, F., C.M. Eischen, D.H. Randle, T. Kamijo, J.L. Cleveland, C.J. Sherr, and M.F. Roussel. 1998. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12:2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}