

Abstract
MicroRNAs (miRNAs) are noncoding RNAs with important roles in regulating gene expression. In studying the earliest nuclear steps of miRNA biogenesis, we observe that primary miRNA (pri-miRNA) transcripts retained at transcription sites due to the deletion of 3′-end processing signals are converted more efficiently into precursor miRNAs (pre-miRNAs) than pri-miRNAs that are cleaved, polyadenylated, and released. Flanking exons, which also increase retention at transcription sites, likewise contribute to increased levels of intronic pri-miRNAs. Consistently, efficiently processed endogenous pri-miRNAs are enriched in chromatin-associated nuclear fractions. In contrast, pri-miRNAs that accumulate to high nuclear levels after cleavage and polyadenylation because of the presence of a viral RNA element (the ENE of the Kaposi's sarcoma–associated herpes virus polyadenylated nuclear RNA) are not efficiently processed to precursor or mature miRNAs. Exogenous pri-miRNAs unexpectedly localize to nuclear foci containing splicing factor SC35; yet these foci are unlikely to represent sites of miRNA transcription or processing. Together, our results suggest that pri-miRNA processing is enhanced by coupling to transcription.
Introduction
MicroRNAs (miRNAs) are a class of noncoding RNAs that play critical roles in regulating gene expression at the posttranscriptional level. Mature miRNAs of ∼22 nt act by base pairing with the 3′ untranslated region of target mRNAs, resulting in degradation or translational control of the mRNA targeted (Pillai et al., 2007; Vasudevan et al., 2007). Many cellular functions are regulated by miRNAs, and aberrant miRNA expression has been linked to disease (for reviews see Calin and Croce, 2006; Kloosterman and Plasterk, 2006).
MiRNAs are initially embedded in long primary miRNAs (pri-miRNAs), usually transcribed by RNA polymerase II (Cai et al., 2004; Lee et al., 2004). The pri-miRNA is recognized by a protein complex termed the Microprocessor (Denli et al., 2004; Gregory et al., 2004), which minimally contains the RNase III–like enzyme Drosha and the double-stranded RNA–binding protein DGCR8. Drosha cleavage releases an intermediate hairpin precursor miRNA (pre-miRNA), which is transported to the cytoplasm by exportin-5 and further processed by another RNase III–like enzyme, Dicer, to the mature miRNA (for review see Kim, 2005).
Pri-miRNA processing is a regulated event. Early in development, as well as in several tumor types, multiple pri-miRNA transcripts are expressed, but many are not processed to precursor or mature miRNAs (Thomson et al., 2006; Kluiver et al., 2007); the molecular mechanisms underlying this regulation remain unclear. Here, we have examined the processing and localization of pri-miRNAs in human cells. Our results suggest that pri-miRNAs undergo initial processing at the site of transcription. Exogenous pri-miRNAs that escape processing at transcription sites appear immune to cleavage by Drosha and accumulate in SC35-containing foci, as do some endogenous pri-miRNAs. Our results provide insights into the regulation of miRNA expression at the level of cleavage by Drosha.
Results
Pri-miRNAs containing a stabilizing viral RNA element do not undergo efficient processing
Because most endogenous pri-miRNAs are expressed at low levels, we began by studying the processing of ectopically expressed pri-miRNA constructs in human HeLa cells. Approximately 150 nt of endogenous flanking sequence on either side of the pre-miRNA hairpin for human pri-let-7a-1 (here referred to as pri-let-7) and Caenorhabditis elegans pri-lin-4 (Fig. 1 A) were inserted into a vector containing the cytomegalovirus (CMV) RNA polymerase II promoter and the bovine growth hormone (BGH) cleavage and polyadenylation (CPA) signal. After transient transfection, pri-let-7 was barely detectable by Northern blotting (Fig. 1 B, lane 2), even though its level was increased by ∼10-fold over endogenous pri-let-7 (Fig. 1 C, lanes 4 and 5). Similarly, transfected pri-lin-4 was detected only at low levels (Fig. 1 B, lane 6); no endogenous lin-4 is expressed in HeLa cells (Fig. 1 B, lane 5).
Figure 1.

Pri-miRNAs containing a stabilizing viral RNA element, the ENE, accumulate to high levels in the nucleus but are not efficiently processed. (A) Schematic of pri-miRNA constructs. CMV and BGH pA denote the CMV promoter and BGH CPA signal. The pri-miRNA is shown in black, with the mature miRNA indicated in red. Red boxes show five copies of the 79-nt ENE in either the forward or reverse complement (r) orientation. Blue lines below the pri-miRNA sequence specify DIG-tailed oligonucleotide probes used for ISH. (B) Northern blot analysis of pri-miRNA processing. Constructs diagramed in A for pri-let-7 (lanes 1–4) or pri-lin-4 (lanes 5–8) were transfected into HeLa cells, and total RNA was analyzed by Northern blotting using either a denaturing formaldehyde-agarose gel to detect pri-miRNAs (top) or a 15% denaturing polyacrylamide gel to visualize the precursor and mature miRNAs (bottom). Pri-miRNA Northern blots were probed for 7SK as a loading control and for the neomycin resistance gene RNA (NeoR) as a transfection efficiency control. Mature miRNA Northern blots were probed for U6 snRNA as a loading control. 18S denotes cross-hybridization with 18S ribosomal RNA. (C) RT-PCR of pri-let-7. RNA from cells transfected as in B was reverse transcribed followed by PCR with primers designed to amplify pri-let-7. β-actin mRNA was amplified for normalization. Lanes 1–3 show samples treated identically to those in lanes 4–6 but without addition of reverse transcriptase (RT). (D) In vitro processing reactions were performed according to Lee et al. (2002) on pri-let-7 (lanes 1–3) and pri-lin-4 (lanes 4–6) with or without the ENE. Lanes 7–12 show pri-miRNA substrates not incubated in cell extract. Bands marked with asterisks represent processing intermediates or nonspecific degradation products. DNA size markers are in lane 13. (E) Localization of pri-miRNAs in transfected HeLa cells was assessed by ISH using DIG-labeled probes (A, blue lines). Bar, 10 μm.
To further enhance the levels of pri-miRNAs, we took advantage of a viral RNA element termed the ENE (Conrad and Steitz, 2005). The ENE is a 79-nt region from a polyadenylated nuclear RNA that is expressed at high levels during lytic growth of Kaposi's sarcoma–associated herpes virus (Sun et al., 1996; Song et al., 2001). The ENE increases the nuclear abundance of intronless, polyadenylated transcripts by inhibiting their entry into a rapid deadenylation-dependent decay pathway (Conrad et al., 2006).
Fig. 1 B shows that the five copies of the 79-nt ENE inserted near the 3′ end of the pri-miRNA construct (diagramed in Fig. 1 A) indeed dramatically increase the levels of pri-let-7 in transfected HeLa cells (∼10-fold; Fig. 1 B, lane 3) relative to a construct lacking the ENE or containing five copies of the ENE in the reverse orientation (Fig. 1 B, lanes 2 and 4). Similarly, the ENE increased levels of pri-lin-4 >10-fold (Fig. 1 B, compare lanes 6, 7, and 8).
Surprisingly, despite striking increases in pri-miRNA levels, the amounts of precursor and mature miRNAs were not significantly increased by the ENE. Mature let-7 remained relatively unchanged (Fig. 1 B, lane 3), as did mature lin-4 (Fig. 1 B, lane 7), when compared with levels in cells expressing pri-miRNAs lacking the ENE or with the ENE in the reverse orientation (Fig. 1 B, lanes 2, 4, 6, and 8).
A possible explanation for the inefficient processing of pri-miRNAs containing the ENE is that Microprocessor recognition is blocked. If true, the ENE would be predicted to interfere with pri-miRNA processing in vitro as well. Therefore, in vitro reactions (Lee et al., 2002) were performed. Fig. 1 D reveals that both pri-let-7 and pri-lin-4 containing the ENE are processed as efficiently in vitro as pri-miRNAs lacking the ENE (Fig. 1 D, pri-let-7, lanes 1–3; and Fig. 1 D, pri-lin-4, lanes 4–6). Thus, it is unlikely that the ENE blocks pri-miRNA recognition or processing by the Microprocessor.
Another explanation for the failure of ENE-containing pri-miRNAs to be processed is that the ENE might localize pri-miRNAs to nuclear regions inaccessible to Drosha and DGCR8. We therefore investigated the localization of pri-miRNAs by performing in situ hybridization (ISH) using probes diagramed in Fig. 1 A. Unexpectedly, pri-let-7 or pri-lin-4 transcripts expressed from transfected plasmids concentrated in three to eight distinct foci per nucleus, with lower signal throughout the nucleoplasm (Fig. 1 E, a and d). In contrast, pri-let-7 and pri-lin-4 containing five copies of the ENE in the forward orientation exhibited diffuse nucleoplasmic distribution (Fig. 1 E, b and e). Pri-miRNAs containing the ENE in the reverse orientation also localized to nuclear foci (Fig. 1 E, c and f). Identification of these dots as SC35-containing foci will be described (see Fig. 6).
Figure 6.

Overexpressed pri-miRNAs accumulate in nuclear foci containing splicing factor SC35 and Microprocessor components. (A) Pri-miRNA–containing nuclear foci colocalize with SC35. ISH was performed on transfected cells, followed by IF for SC35. (B and C) IF for Drosha (B) or DGCR8 (C) was performed on transfected cells, followed by ISH for the pri-miRNA. Arrowheads indicate untransfected cells. (D) IF for Drosha followed by ISH was performed on cells transfected with a β-globin cDNA construct. Bars, 10 μm.
Pri-miRNAs lacking a CPA signal are processed with increased efficiency and do not localize to nuclear foci
To explore the significance of localization of transfected pri-miRNAs to nuclear foci, we asked whether these foci might represent novel sites of transcription created upon transfection. We therefore checked the localization of DNA plasmids carrying pri-let-7 and pri-lin-4, but they appeared much more diffuse in the nucleoplasm than the focal pattern of the RNA transcripts (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.20080311/DC1).
We then reasoned that if pri-miRNA–containing foci represented sites of pri-miRNA synthesis, their signal should be enhanced by retaining transfected pri-miRNAs at the site of transcription. We deleted the AAUAAA CPA signal from the BGH sequence that resides downstream of the pri-miRNA insert (Fig. 2 A) because this should prevent recruitment of the CPA machinery and impair release of the nascent transcript from the transcription site (Custodio et al., 1999; for reviews see Zhao et al., 1999; Edmonds, 2002). Pri-let-7ΔpA and pri-lin-4ΔpA (Fig. 2 A) were transfected into HeLa cells, and ISH was performed.
Figure 2.

Pri-miRNAs lacking a CPA signal do not localize to nuclear foci and are processed with increased efficiency. (A) Pri-miRNA constructs are diagramed as in Fig. 1 A; arrowheads indicate the location of five copies of the ENE in the forward or reverse complement (r) orientation. Deleted vector-encoded CPA signals are indicated by ΔAAUAAA. (B) ISH performed on cells transfected with constructs diagramed in A. Bar, 10 μm. (C and D) Northern blot analysis of RNA from cells transfected as in B. Note that transfections in C (lanes 6–8) are repeated in D (lanes 2, 5, and 6) to allow comparison of miRNA levels. Cross-hybridization with 18S and 28S ribosomal RNA is indicated. RT, readthrough transcripts. (E) Comparison of mature miRNA levels produced from the constructs in A, quantified from Northern blots similar to those shown in C and D. miRNA levels were normalized to U6 snRNA; levels of endogenous let-7 (top) or levels of lin-4 from pri-lin-4 transfections (bottom) were set to 1. Error bars indicate standard deviations (pri-let-7, n = 3; pri-lin-4, n = 5).
Consistent with the localization of plasmid DNA (Fig. S1), Fig. 2 B shows that pri-let-7ΔpA and pri-lin-4ΔpA transcripts did not exhibit the punctate pattern of cleaved and polyadenylated pri-let-7 and pri-lin-4 (Fig. 2 B, a and d) but showed more diffuse nuclear localization (Fig. 2 B, b and e). Northern blot analysis confirmed that cleavage was defective for the ΔpA primary transcripts; pri-let-7ΔpA and pri-lin-4ΔpA migrated more slowly than transcripts expressed from plasmids containing the CPA signal (Fig. 2 C, compare lanes 2 and 3 and lanes 6 and 7). Together, these data suggest that pri-miRNA–containing nuclear foci are not sites of transcription but instead sites where pri-miRNAs that have been cleaved, polyadenylated, and released accumulate.
Most interestingly, Northern blot analysis of miRNA species in cells expressing retained pri-let-7ΔpA and pri-lin-4ΔpA transcripts revealed approximately twofold higher levels of both the precursor and mature miRNA compared with levels in cells expressing the corresponding cleaved and polyadenylated pri-miRNAs (Fig. 2 C, lanes 2 and 3 and lanes 6 and 7; and quantitation in Fig. 2 E). Deletion of the simian virus 40 (SV40) CPA signal downstream of the vector-encoded neomycin resistance gene (to create pri-let-7ΔpAx2 and pri-lin-4ΔpAx2; Fig. 2 A) generated even longer transcripts (Fig. 2 C, lanes 4 and 8). Importantly, the processing efficiency of pri-let-7ΔpAx2 and pri-lin-4ΔpAx2 transcripts increased further (three- to fourfold; Fig. 2 C, lanes 4 and 8; and quantitation in Fig. 2 E). Pri-miRNAs with both CPA signals deleted again displayed diffuse nuclear localization (Fig. 2 B, c and f), which supports the conclusion that pri-miRNA–containing nuclear foci are not sites of transcription. Moreover, because pri-miRNAs with deleted CPA signals are processed with increased efficiency but do not localize to nuclear foci, these foci are unlikely to be major sites of pri-miRNA processing.
The significant increase in processing efficiency of pri-miRNAs predicted to be retained at the site of transcription provides a possible explanation for why pri-miRNAs containing the ENE are not efficiently processed (Fig. 1 B). Because the ENE counteracts decay dependent on the presence of a polyadenylate (polyA) tail (Conrad et al., 2006), only those pri-miRNA transcripts that have already been cleaved, polyadenylated, and released from the site of transcription are stabilized. Therefore, if pri-miRNA processing normally occurs on nascent pri-miRNAs before CPA, we would expect released pri-miRNAs containing the ENE to be processed inefficiently, as is observed (Fig. 1 B).
To confirm this interpretation, we examined the processing of pri-miRNAs that contain the ENE but are also deleted for CPA signals (Fig. 2 A, arrowheads indicate the position of five copies of the ENE as diagramed in Fig. 1 A). Fig. 2 D shows that pri-lin-4ENEΔpA is processed more efficiently than pri-lin-4ENE. Pri-lin-4ENEΔpAx2 exhibits a further increase in processing efficiency. Similar results were observed for pri-let-7ENEΔpA and pri-let-7ENEΔpAx2 (quantitation in Fig. 2 E). Therefore, pri-miRNAs containing the ENE can be efficiently processed if they lack a CPA signal.
Pri-miRNAs lacking a CPA signal are retained at the site of transcription, whereas ENE-containing pri-miRNAs are released
Pri-miRNA transcripts lacking a functional CPA signal should remain tethered to the DNA template, whereas ENE-containing pri-miRNAs are predicted to be stabilized after release. To confirm these predictions, we fractionated nuclear RNAs using a procedure that separates nascent, chromatin-associated transcripts from transcripts that have been released into the nucleoplasm (Wuarin and Schibler, 1994; Dye et al., 2006). Nuclear extracts from HeLa cells transfected with pri-miRNA constructs diagramed in Fig. 2 A were centrifuged to obtain released (supernatant) and chromatin-associated (pellet) fractions; RNA was isolated and then reverse transcribed using random primers.
Controls for efficient fractionation were provided by examining endogenous β-actin precursor mRNA (pre-mRNA) and fully spliced mRNA by PCR (primers indicated in Fig. 3 A).Fig. 3 B (middle) demonstrates that nascent, unspliced β-actin pre-mRNA is found almost exclusively in the chromatin-containing pellet, as is expected, because splicing occurs predominantly cotranscriptionally. In contrast, β-actin–spliced mRNA is found primarily in the released, nucleoplasmic supernatant (Fig. 3 B, bottom). These results complement previous findings using similar fractionation procedures (Mili et al., 2001; Dye et al., 2006).
Figure 3.

Pri-miRNAs lacking a CPA signal are retained in chromatin, whereas ENE-containing pri-miRNAs are released. (A) Schematic of primers used for PCR in B and C. (B and C) Nuclear location of pri-miRNAs. Cells transfected with constructs diagramed in Fig. 2 A for pri-let-7 (B) or pri-lin-4 (C) were fractionated into released, nucleoplasmic RNAs (supernatant [S]) and chromatin-associated RNAs (pellet [P]). Fractionated RNA was reverse transcribed and amplified by PCR. β-actin pre-mRNA and spliced mRNA were amplified as controls for fractionation efficiency.
The distributions of pri-miRNA transcripts generated from each construct were then analyzed by PCR using primers specific for the pri-miRNA sequence (Fig. 3 A). Fig. 3 B (top) shows that the majority of pri-let-7 transcripts containing the CPA signal are in the nucleoplasmic supernatant, which indicates efficient release (Fig. 3 B, lanes 7 and 8). An even larger fraction of the highly abundant pri-let-7ENE transcripts is in the nucleoplasmic supernatant, which confirms that the ENE causes accumulation after release from the DNA template (Fig. 3 B, lanes 9 and 10). In contrast, pri-let-7ΔpAx2 transcripts are more abundant in the chromatin-associated pellet, which demonstrates that pri-miRNAs lacking a CPA signal are indeed retained at sites of transcription (Fig. 3 B, lanes 11 and 12).
Fractionation of nuclei from cells expressing pri-lin-4 constructs likewise demonstrated that accumulated pri-lin-4ENE transcripts are most abundant in the nucleoplasmic supernatant (Fig. 3 C, lanes 9 and 10), whereas pri-lin-4ΔpAx2 transcripts remain in the chromatin-associated pellet (Fig. 3 C, lanes 11 and 12). Pri-lin-4 transcripts were more abundant in the chromatin-associated fractions than the released fractions (Fig. 3 C, lanes 7 and 8), which may be caused by sequence or other variables and is consistent with the greater processing efficiency of pri-lin-4 relative to pri-let-7 (Fig. 1 B).
The data in Figs. 2 and 3 indicate that retention of ectopically expressed pri-miRNAs at the site of transcription correlates with increased processing efficiency. Furthermore, pri-miRNAs containing the ENE are inefficiently processed not because the ENE intrinsically inhibits processing, but more likely because they are released from the DNA template. Moreover, it is unlikely that the Microprocessor is saturated for processing by the high levels of ENE-containing pri-miRNAs because significantly higher amounts of mature miRNA can be generated from pri-miRNA transcripts when they are retained at sites of transcription.
Intronic miRNA levels are increased by retention at the site of transcription via flanking exons or deletion of the CPA signal
The majority of miRNA sequences (∼80%) reside within intronic regions of mammalian transcription units (Rodriguez et al., 2004; Kim and Kim, 2007). We therefore investigated processing of the intronic miR-26b, which is expressed at low levels in HeLa cells. We initially compared processing of two constructs: pri-miR-26bFE, which contains the miR-26b–harboring intron as well as the entirety of the two flanking exons; and pri-miR-26b, which contains only the intronic sequence (Fig. 4 A). Northern blot analysis demonstrated that pri-miR-26b is poorly expressed (Fig. 4 B, lane 3), whereas the presence of flanking exons increased the levels of pri-miRNA (Fig. 4 B, lane 2). Deletion of the BGH CPA signal to create transcription site–retained pri-miR-26bΔpA (diagramed in Fig. 4 A) also increased pri-miRNA expression levels (Fig. 4 B, lane 4). Importantly, pri-miRNAs containing flanking exons or with a deleted CPA signal yielded increased levels of precursor and mature miRNA (three- to fourfold; Fig. 4 B, lanes 2 and 4; and quantitation shown on Fig. 4 B, bottom). The higher levels of pri-miR-26bFE and pri-miR-26bΔpA transcripts detected in Fig. 4 B (top) cannot alone explain increased precursor and mature miRNA levels (middle) because addition of the ENE yields abundant pri-miR-26bENE but not more precursor or mature miR-26b (Fig. 4 B, lane 5).
Figure 4.

Flanking exons or deletion of the CPA signal results in increased levels of an intronic pri-miRNA. (A) Schematic of pri-miR-26b constructs. Flanking exons (E) are represented as blue rectangles. Probes used for ISH are shown as blue lines under pri-miR-26bFE. (B, top and middle) Northern blotting was performed on cells transfected with constructs in A as described in Fig 1. RT, readthrough transcripts. (B, bottom) Mature miRNA levels were quantitated and normalized to the U6 snRNA, and the amount of miR-26b produced from pri-miR-26b was set to 1. Error bars represent standard deviations (n = 4). (C) Nuclear fractionation of released and chromatin-associated transcripts. Cells transfected as in B were fractionated as described in Fig 3. P, pellet; S, supernatant. (D) ISH of pri-miR-26b transcripts in cells transfected as in B using DIG-labeled probes (Fig. 4 A, blue lines). Nuclei were stained with DAPI and merged images are shown in panels e–h. Bar, 10 μm.
Analysis of intronic miRNA biogenesis has demonstrated that pre-mRNA splicing is not required for pri-miRNA processing (Kim and Kim, 2007). However, splicing of flanking exons may be rate-limiting for the release of nascent transcripts (Custodio et al., 1999) and may thereby increase the time pri-miRNAs spend at transcription sites. To test this hypothesis, we transfected cells with constructs diagramed in Fig. 4 A and fractionated nuclei into nucleoplasmic and chromatin-associated RNAs. The majority of pri-miR-26bΔpA transcripts were localized in the chromatin-associated pellet (Fig. 4 C, lanes 12 and 13), whereas most pri-miR-26bENE transcripts were released into the nucleoplasmic supernatant (Fig. 4 C, lanes 14 and 15). Importantly, pri-miR-26bFE transcripts were enriched in the chromatin-associated pellet (Fig. 4 C, lanes 8 and 9). These data support the idea that flanking exons may facilitate processing by increasing the time pri-miRNAs spend tethered to the DNA template relative to ENE-containing pri-miRNAs (see Discussion).
ISH of pri-miR-26b transcripts revealed that they localize to nuclear foci (Fig. 4 D, b; and Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.20080311/DC1), whereas pri-miR-26bENE (Fig. 4 D, d) and pri-miR-26bΔpA (Fig. 4 D, c) show nucleoplasmic localization. Pri-miR-26bFE transcripts were found in nuclear foci and in the cytoplasm (Fig. 4 D, a; and Fig. S2); however, the nucleoplasmic signal for these transcripts was enhanced over that of pri-miR-26b, which is consistent with the idea that pri-miR-26bFE is retained longer at transcription sites.
Efficiently processed endogenous pri-miRNAs are enriched in chromatin-associated nuclear fractions
Our observation that exogenous pri-miRNAs are processed with enhanced efficiency when retained at transcription sites suggests that pri-miRNA processing may normally occur cotranscriptionally. We therefore asked whether endogenous pri-miRNAs are also tethered to chromatin. HeLa cells or U2OS cells were fractionated into chromatin-associated and nucleoplasmic RNAs (Wuarin and Schibler, 1994; Dye et al., 2006), and several well-expressed endogenous pri-miRNAs (diagramed in Fig. 5 A) were amplified by RT-PCR.Fig. 5 B shows that the endogenous pri-miRNAs investigated are indeed enriched in the chromatin-associated pellet (Fig. 5 B; lanes 2 and 4) as compared with the nucleoplasmic supernatant (Fig. 5 B, lanes 1 and 3) in both cell types, which suggests that they undergo processing before release from the DNA template.
Figure 5.

Endogenous pri-miRNAs that are efficiently processed are enriched in chromatin. (A) Schematic of endogenous pri-miRNAs. Exons are represented as gray boxes; hairpins indicate miRNA locations. Arrowheads indicate the location of primers used for PCR in B, D, and F. Promoters for EBNA (Cp, Wp, and Qp) and BHRF1 (BHRF1p) transcripts are represented by arrows. Binding sites for p53 (p53 BS) are indicated. Schematics are not shown to scale. (B) Nuclear fractionation of endogenous pri-miRNAs. HeLa or U2OS cells were fractionated, RNA was reverse transcribed, and endogenous pri-miRNAs were amplified by PCR using primers diagramed in A from the released, nucleoplasmic supernatant (S; lanes 1 and 3) and the chromatin-associated pellet (P; lanes 2 and 4). β-globin pre-mRNA, β-globin fully spliced mRNA, and TAFII30 fully spliced mRNA served as controls for fractionation efficiency. (C) Northern blot analysis of primary and mature miR-34a expression in HeLa cells after UV irradiation. (D) Nuclear fractionation of HeLa cells 8 h after UV irradiation or mock treatment (–). (E) Northern blot analysis of pri-miR-BHRF1-2∼3 after treatment of Raji or B95-8 cells with DMSO (–) or PMA and Ionomycin (P/I). (F) Nuclear fractionation of Raji cells after 24 h of DMSO (–) or P/I treatment. For all PCRs, the size of the amplified fragment (in base pairs) is indicated at the right of the gel.
Further evidence for enhanced processing of endogenous pri-miRNAs at the site of transcription was provided by analysis of a pri-miRNA that can be induced to significantly higher expression levels. Pri-miR-34a is a transcriptional target of p53 (for review see He et al., 2007); UV irradiation of HeLa cells induces expression of both primary and mature miR-34a (Fig. 5 C). Nuclear fractionation revealed that in UV-irradiated cells, the majority of induced pri-miR-34a transcripts are in the chromatin-associated pellet (Fig. 5 D, lane 6), which mirrors the β-actin pre-mRNA control. Therefore, increased production of miR-34a after p53 induction correlates with increased levels of pri-miR-34a associated with chromatin, whereas very little is released into the nucleoplasm (Fig. 5 D; lane 5). Similar results were observed for pri-miR-34b∼34c, which is also a transcriptional target of p53 (Fig. 5 D; for review see He et al., 2007).
Finally, we investigated the association with chromatin of a viral pri-miRNA encoded by latent Epstein Barr virus (EBV) upon induction to high levels. EBV miRNAs miR-BHRF1-1, -2, and -3, are encoded within introns of the EBV nuclear antigen (EBNA) transcripts produced from the Cp or Wp promoters of EBV (diagramed in Fig. 5 A; Cai et al., 2006; Xing and Kieff, 2007). Two of these viral miRNAs are also present in the 3′ untranslated region of the overlapping BHRF1-1 transcript, which is expressed only upon induction of viral replication. When EBV replication was induced in Raji B cells and B95-8 cells, increased expression of two BHRF1 mRNA/pri-miRNA transcripts (∼1.3 and 1.4 kb; Fig. 5 E, top) was observed; however, levels of precursor and mature BHRF1 miRNAs did not increase, which is consistent with previous results (Fig. 5 E, bottom; Xing and Kieff, 2007). Nuclear fractionation of Raji cells before induction of viral replication showed that the majority of pri-miR-BHRF1-2∼3 was in the chromatin-associated pellet (Fig. 5 F, lane 4), which is consistent with its efficient processing (Fig. 5 E, lane 1). After induction of viral replication, although the total level of pri-miR-BHRF1-2∼3 increased (Fig. 5 F, compare lanes 1 and 2), the amount of pri-miR-BHRF1-2∼3 in the chromatin-associated pellet decreased (Fig. 5 F, lane 6), which is consistent with the observed decrease in precursor and mature BHRF1 miRNAs. The accumulated BHRF1 mRNA/pri-miRNA transcripts were not present in the nucleoplasmic supernatant but rather were detected abundantly in the cytoplasmic fraction (unpublished data), which is consistent with their translation into the BHRF1 protein under these conditions.
The data in Fig. 5 demonstrate that efficiently processed endogenous pri-miRNAs are found most abundantly in chromatin-associated nuclear fractions, whereas poorly processed pri-miRNAs are more abundant in released fractions. Thus, as for transfected pri-miRNAs, association with the DNA template is correlated with efficient processing of endogenous pri-miRNAs.
Cleaved and polyadenylated pri-miRNAs localize to nuclear foci containing the splicing factor SC35 and Microprocessor components
The data presented in Figs. 2 and 4 suggest that pri-miRNA–containing nuclear foci in transfected cells are not sites of transcription or major sites of pri-miRNA processing. To explore the functional significance of pri-miRNA localization to these foci, we asked what proteins colocalize. Because unspliced pre-mRNAs have been shown to accumulate in nuclear foci that contain splicing factor SC35 (Huang and Spector, 1996), HeLa cells were transfected with pri-miRNA constructs and subjected to ISH followed by indirect immunofluorescence (IF) with anti-SC35 antibodies. Indeed, SC35 colocalized in nuclear foci with pri-let-7 (Fig. 6 A, a–c), pri-lin-4 (unpublished data), and pri-miR-26b transcripts with or without flanking exons (Fig. S2). In contrast, neither the Cajal body marker protein coilin, nor the Cajal and Gem marker protein SMN colocalized with pri-miRNAs (unpublished data). Pri-miRNAs containing the ENE did not colocalize with SC35 (Fig. 6 A, d–f) even after transfection of 10-fold less pri-miRNA-ENE plasmid resulting in levels of pri-miRNA comparable to those of pri-miRNAs without the ENE (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.20080311/DC1).
We investigated the localization of the Microprocessor components Drosha and DGCR8 in cells overexpressing pri-miRNA transcripts as well. In untransfected HeLa cells, both Drosha and DGCR8 exhibit diffuse nuclear staining (Fig. 6 B, b and e; and Fig. 6 C, b; untransfected nuclei are indicated by arrowheads). However, in cells overexpressing pri-let-7, Drosha was recruited to pri-miRNA/SC35–containing foci (Fig. 6 B, a–c; and Fig. S2), as was DGCR8 (Fig. 6 C). Quantitative measurements showed that ∼7.5 and ∼5.4% of Drosha and DGCR8, respectively, were recruited to foci in pri-miRNA–transfected cells. Expression of pri-miRNAs containing the ENE, however, did not alter the diffuse nucleoplasmic localization of Drosha (Fig. 6 B, d–f) or of DGCR8 (unpublished data). The relocalization of Drosha and DGCR8 to SC35-containing foci is specific for pri-miRNA transcripts because overexpression of GFP-Pin1 cDNA (unpublished data) or β-globin cDNA, does not result in relocalization of Drosha (Fig. 6 D) or of DGCR8 (unpublished data).
These localization data suggest that pri-miRNAs that are not processed at transcription sites accumulate in SC35-containing foci. One interpretation is that Drosha and DGCR8 bind pri-miRNAs and are carried to SC35-containing foci; however, after release from the DNA template, their processing efficiency may be reduced (see Discussion).
Overexpressed pri-miRNAs transcribed by RNA polymerase III do not localize in SC35 foci
Further confirmation of our conclusion that SC35-containing foci are not sites of transcription or major sites of processing of the transfected pri-miRNAs came from comparison of the localization of a pri-miRNA transcribed by RNA polymerase III with that of pri-miRNAs transcribed by RNA polymerase II. If the foci contained aggregates of plasmid DNA or were sites of processing, pri-miRNAs would be predicted to accumulate there independent of the promoter used.
We therefore removed the RNA polymerase II promoter and the CPA signal from the pri-lin-4 plasmid and replaced them with the RNA polymerase III U6 small nuclear RNA (snRNA) promoter and an RNA polymerase III termination signal (Fig. 7 A). ISH revealed that transcripts generated from U6-pri-lin-4 show a nucleoplasmic localization pattern with far fewer areas of intense staining (Fig. 7 B, b) than the RNA polymerase II–transcribed CMV-pri-lin-4 (Fig. 7 B, a).
Figure 7.

Pri-miRNAs transcribed by RNA polymerase III do not localize to nuclear foci. (A) miRNA constructs used for transfection. U6 denotes the RNA polymerase III promoter; the Pol III termination signal consisted of five thymidylate residues (T). (B) ISH was performed as detailed in Fig. 1 E for pri-lin-4 constructs (a and b) or pri-miR-30 constructs (c and d). (C) Northern blot analysis of mature miRNA species produced from transfections with decreasing amounts of CMV-pri-miR-30 (lanes 2–4) or U6-pri-miR-30 (lanes 6–8) constructs compared with empty vector (−; lanes 1 and 5). (D) Cells transfected as in B were fixed, and IF was performed for Drosha followed by ISH as described in Fig. 6 B. Nuclei were stained with DAPI. Bars, 10 μm.
We also investigated the localization of an shRNA construct (shRNA-mir; Silva et al., 2005) commonly used for RNAi that contains the flanking sequence of miR-30 driven by the U6 RNA polymerase III promoter, here termed U6-pri-miR-30. For comparison, we cloned the pri-miR-30 insert of this construct into an RNA polymerase II–driven vector to generate CMV-pri-miR-30. CMV-pri-miR-30 localized in distinct nuclear foci (Fig. 7 B, c), whereas U6-pri-miR-30 showed a diffuse nucleoplasmic localization (Fig. 7 B, d). Despite their different localization patterns, CMV-pri-miR-30 and U6-pri-miR-30 were processed to mature miRNAs with similar efficiency (Fig. 7 C). Moreover, in cells expressing U6-pri-miR-30, Drosha showed a localization pattern that is diffuse throughout the nucleoplasm (Fig. 7 D, f) rather than relocalizing to nuclear foci as in cells expressing CMV-pri-miR-30 (Fig. 7 D, b). The observation that RNA polymerase III–transcribed pri-miRNAs do not localize to SC35-containing foci further confirms the interpretation that these foci are not sites of plasmid aggregation and transcription or obligatory sites of pri-miRNA processing.
Endogenous pri-miRNAs exhibit various nuclear localization patterns correlated with abundance
Finally, to investigate whether the unexpected localization of transfected pri-miRNAs to SC35-containing foci is relevant to nontransfected cells, we investigated the localization of several endogenous pri-miRNAs (Fig. 8). Endogenous pri-miRNAs expressed at low levels (not detectable by Northern blotting), including pri-let-7a-1, pri-let-7g, and the pri-miR-17∼92 cluster, could only be visualized by amplifying the ISH signal, and were found to localize in one or two foci per cell (Fig. 8 A), which likely represent sites of transcription.
Figure 8.

Localization of endogenous pri-miRNAs. (A) Pri-miRNAs expressed at low levels were detected using amplification and localize to one or two foci per cell. (B) Pri-miR-34a is not detected in untreated HeLa cells (a–d) but shows diffuse nucleoplasmic localization in HeLa cells after UV irradiation (e–h). Costaining for SC35 is shown in panels b and f. (C) BIC/pri-miR-155 localizes throughout the nucleoplasm in Raji (a–d) and B95-8 cells treated with PMA and ionomycin (P/I) for 24 h, as well as in nuclear foci in B95-8 cells (e–h), many of which overlap with SC35 (g, arrows). (D) Pri-miR-BHRF1-2∼3 shows diffuse nucleoplasmic and cytoplasmic distribution in Raji cells (a–d) but is concentrated in nuclear foci in B95-8 cells (e–h), some of which overlap with SC35 (g, arrows). (E) EBV BHRF1 DNA localizes diffusely in the nucleus and cytoplasm (due to viral release). Bars, 10 μm.
We also determined the localization of two inducible pri-miRNAs that are present at high cellular levels under specific conditions: (1) pri-miR-34a and (2) B cell integration cluster (BIC)/pri-miR-155. ISH of HeLa cells that were UV-irradiated to induce pri-miR-34a expression revealed diffuse nucleoplasmic localization (Fig. 8 B, a–h). Similarly, the noncoding RNA BIC, which encodes miR-155, revealed diffuse nucleoplasmic localization in the majority of Raji cells (Fig. 8 C, a–d). In B95-8 cells, BIC also localized diffusely throughout the nucleoplasm, and in ∼5% of cells, BIC was concentrated in several speckle-shaped foci, overlapping SC35 in some cases (Fig. 8 C, e–h; and Fig. 8 C, g, arrow).
Finally, we localized EBV-encoded pri-miR-BHRF1-2∼3 (diagramed in Fig. 5 A). ISH of pri-miR-BHRF1-2∼3 in Raji cells after induction of viral replication revealed a diffuse nucleoplasmic and cytoplasmic signal (Fig. 8 D, a–d). However, in B95-8 cells, pri-miR-BHRF1-2∼3 was observed diffusely in the nucleus as well as concentrated in several nuclear foci in ∼60% of cells; most of these pri-miRNA–containing foci showed partial overlap with SC35 (Fig. 8 D, e–h). Consistently, EBNA transcripts have previously been found associated with SC35 domains in Raji and Namalwa cells (Melcak et al., 2000). The observed foci did not colocalize with episomal viral DNA (Fig. 8 E), which suggests that transcripts concentrated in SC35-containing foci are cleaved, polyadenylated, and released. Furthermore, because no increase in mature miR-BHRF1-2 or -3 was observed after induction of viral replication (Fig. 5 E, bottom panels), pri-miR-BHRF1-2∼3 transcripts that accumulate in or near SC35 foci are likely to represent unprocessed transcripts.
Pri-miR-155 and pri-miR-BHRF1-2∼3 therefore represent physiological examples of cellular pri-miRNAs localizing in or near SC35 domains. However, unlike cleaved and polyadenylated transfected pri-miRNAs, endogenous pri-miRNAs expressed at low or moderate levels (Fig. 8, A–C) are predominantly localized at transcription sites or localized diffusely in the nucleoplasm. Furthermore, the colocalization of highly expressed endogenous pri-miR-155 and pri-miR-BHRF1-2∼3 with SC35 was not as complete as that of transfected pri-miRNAs. Therefore, high expression levels appear to result in association with SC35 domains but transfection may cause this colocalization to be more dramatic. For both transfected and endogenous pri-miRNAs, the association with SC35-containing foci is not likely to be important for processing.
Discussion
We have presented evidence suggesting that pri-miRNA processing is coordinated with transcription. Overexpressed, nascent pri-miRNA transcripts that remain tethered to the DNA template by deletion of the CPA signal are processed more efficiently than pri-miRNAs that are cleaved, polyadenylated, and released (Figs. 2–4). This explains why overexpressed pri-miRNAs containing the ENE accumulate to high levels in the nucleoplasm but do not generate higher levels of precursor and mature miRNAs (Figs. 1 and 4). Flanking exons, like deletion of the CPA signal, increase miRNA levels; increased residence time of such pri-miRNAs at their sites of transcription as splicing occurs may underlie this observation (Fig. 4). Endogenous pri-miRNAs that are efficiently processed are enriched in chromatin-associated nuclear fractions, which suggests that their processing occurs while they are physically associated with the DNA template (Fig. 5). Overexpressed polyadenylated pri-miRNAs that escape processing at transcription sites unexpectedly accumulate in nuclear foci containing splicing factor SC35 along with Microprocessor components (Fig. 6); some highly expressed endogenous pri-miRNAs also accumulate in foci that partially overlap with SC35 (Fig. 8). However, because pri-miRNAs retained at transcription sites do not localize to SC35-containing foci but are processed with increased efficiency, these foci are unlikely to represent either sites of transcription or obligate sites of processing (Figs. 2 and 4).
Enhanced pri-miRNA processing at sites of transcription
Much evidence indicates that mRNA processing steps, including capping, splicing, and 3′-end formation occur largely at the site of transcription (Zhang, et al., 1994; McCracken et al., 1997; Roberts et al., 1998). Our observations that transfected pri-miRNAs retained at transcription sites are processed more efficiently than released pri-miRNAs (Figs. 2–4) extend a previous finding of increased levels of synthetic miRNA from the pri-miR-155 backbone upon deletion of the CPA signal (Chung et al., 2006). Furthermore, we observe that endogenous pri-miRNAs are most abundant in chromatin-associated nuclear fractions (Fig. 5), which suggests that the majority of pri-miRNAs are processed before release from the DNA template. Our finding that highly abundant ENE-containing transcripts do not undergo efficient processing even though they exhibit the same localization pattern as Microprocessor components (Fig. 6) suggests that the mere existence of a pre-miRNA hairpin is not sufficient for processing. Instead, recruitment of Drosha to nascent pri-miRNAs at the transcription site may facilitate entry into the pri-miRNA processing pathway. Similarly, in cells expressing an RNA polymerase II large subunit lacking the carboxy terminal domain, unspliced pre-mRNAs accumulate in the nucleoplasm, despite the fact that most splicing factors also have a diffuse nucleoplasmic distribution (Misteli and Spector, 1999). Our data argue that Drosha action should be added to the list of RNA processing steps that occur cotranscriptionally.
Kim and Kim (2007) recently demonstrated that the Microprocessor can excise intronic pre-miRNAs before the completion of splicing; however, the splicing of flanking exons is not required for miRNA processing. Our finding that the presence of flanking exons increases pri-miRNA processing efficiency as compared with that of ENE-containing pri-miRNAs (Fig. 4) is therefore best explained if increased retention time at transcription sites required for spliceosome assembly and catalysis favors pri-miRNA processing. Further investigation will be necessary to elucidate the molecular mechanisms of cotranscriptional miRNA processing.
Unprocessed pri-miRNAs and Microprocessor components accumulate in SC35-containing foci
Our findings raise the question of why high-level pri-miRNAs accumulate in SC35-containing foci. Interestingly, transiently expressed β-globin cDNA transcripts, which undergo rapid decay (Conrad et al., 2006), can also be detected in nuclear foci (Fig. 6 D). Similarly, intron-containing β-globin transcripts that contain splice-site mutations accumulate in SC35-containing nuclear foci (unpublished data), as do endogenous collagen 1A1 transcripts with splice-site mutations (Johnson et al., 2000). Together, these observations indicate that localization to SC35-containing foci is a feature common to aberrant RNAs that have not been properly processed. In fact, SC35 domains contain a population of polyadenylated RNAs proposed to represent defective transcripts that should not be exported (Lamond and Spector, 2003; Hall et al., 2006). Thus, inefficient processing of overexpressed pri-miRNAs at transcription sites may lead to their accumulation in SC35 domains as a result of quality control mechanisms.
Relocalization of Drosha and DGCR8 to SC35-containing foci may consequently reflect their binding to overexpressed pri-miRNAs. In vitro, DGCR8 specifically binds pri-miRNAs, whereas Drosha interacts only transiently (Han et al., 2006). Perhaps, if a pri-miRNA is released from the site of transcription before processing, Drosha activity, but not binding, is inhibited. Indeed, Drosha and DGCR8 have been previously reported to bind processing-defective pri-miRNAs with mutations in the hairpin (Duan et al., 2007; Kim and Kim, 2007). In Arabidopsis thaliana, exogenously expressed proteins involved in pri-miRNA processing, as well as an introduced pri-miRNA, localize in discrete nuclear bodies (Fang and Spector, 2007; Fujioka et al., 2007; Song et al., 2007), but it remains unclear whether these are sites of processing or sites of storage of the miRNA-processing machinery.
Pri-miRNAs containing the ENE may fail to localize to SC35-containing nuclear foci (Figs. 1 and S3) because of inaccessibility of the polyA tail. The ENE increases the levels of nuclear RNAs by engaging the polyA tail (Conrad et al., 2006, 2007), which might prevent recognition by factors that normally recruit polyadenylated RNAs to SC35 domains. In support of this hypothesis, pri-miRNAs generated by RNA polymerase III, which lack a polyA tail, fail to localize in SC35-containing foci (Fig. 7).
Regulation of pri-miRNA processing
Our conclusion that Drosha prefers to cleave pri-miRNAs at the site of transcription has implications for the regulation of pri-miRNA processing, which is critical to development and disease (Thomson et al., 2006). Pre-mRNA processing events such as alternative splicing and 3′-end formation are regulated through cotranscriptional recruitment of processing factors or varying rates of RNA polymerase II elongation (Listerman et al., 2006; for reviews see Neugebauer, 2002; Kornblihtt et al., 2004). Thus, analogous to splicing, alterations in chromatin structure or transcription factors during development or in cancerous cells may influence Drosha recruitment to pri-miRNAs at transcription sites.
Consistently, enhanced recruitment of Drosha to specific pri-miRNA chromosomal loci through interaction with leukemogenic All1/Af4 and All1/Af9 fusion genes is associated with increased expression of these miRNAs in several acute lymphoblastic leukemia cell lines (Nakamura et al., 2007). Because Drosha and DGCR8 copurify (Gregory et al., 2004; Fukuda et al., 2007) with proteins that regulate transcription and mRNA biogenesis, these proteins may serve as adaptors to coordinate transcription and pri-miRNA processing. Further investigation of how pri-miRNA processing is spatially and temporally coordinated with transcription should reveal the molecular mechanisms underlying the regulation of miRNA expression that are critical to development and disease.
Materials and methods
Plasmid construction
The plasmid pri-let-7 was generated by PCR amplification from HeLa cell genomic DNA of a 395-nt region surrounding let-7a-1 using primers Pri-let-7FH and Pri-let-7RA, which add a HindIII and an ApaI site, respectively (see Table S1). Similarly, the pri-lin-4 plasmid was made by PCR amplification of a region surrounding the lin-4 miRNA from C. elegans genomic DNA using primers Pri-lin4FH and Pri-lin4RA. Both pri-miRNA amplicons were digested with HindIII and ApaI and cloned into pcDNA3 (Invitrogen) digested with HindIII and ApaI.
To generate pri-miRNAs containing the ENE in the forward or reverse complement orientation, five copies of the 79-nt ENE were removed from βΔ1,2-79Fx5 or βΔ1,2-79Rx5 (Conrad and Steitz, 2005) by digestion with BglII, blunt ending with Klenow polymerase, and digestion with HindIII. The ENE digestion products were inserted into pri-let-7 or pri-lin-4 after digestion with ApaI, blunting using T4 DNA polymerase, and digestion with HindIII to create the plasmids pri-let-7ENE (prepared by N.K. Conrad; University of Texas Southwestern, Dallas, TX), pri-let-7rENE, pri-lin-4ENE, and pri-lin-4rENE.
The plasmids pri-let-7ΔpA, pri-lin-4ΔpA, pri-let-7ENEΔpA, and pri-lin-4ENEΔpA were made by subcloning a region from the plasmid βΔ1,2delAAUAAA (a gift from N.K. Conrad) into the pri-miRNA constructs. The βΔ1,2delAAUAAA plasmid contains a deletion of the AATAAA sequence from the pcDNA3-encoded BGH CPA signal from the vector βΔ1,2 (Conrad et al., 2006). The AAUAAA deletion was generated using the QuikChange Site-Directed Mutagenesis kit (Stratagene) with oligonucleotides NC275 and NC276. A 1,077–base pair region encompassing the AATAAA deletion was removed from βΔ1,2delAAUAAA using StuI and ApaI. Pri-let-7 and pri-lin-4 were likewise digested with StuI and ApaI to remove the corresponding region and treated with alkaline phosphatase, then the insert containing the AATAAA deletion from βΔ1,2delAAUAAA was ligated into these vectors to create pri-let-7ΔpA and pri-lin-4ΔpA. Deletion of the BGH CPA signal was performed in an identical manner for pri-let-7ENE and pri-lin-4ENE except that both the plasmids and the inserts were digested with StuI and XhoI and blunted with T4 DNA polymerase before ligation (due to the presence of ApaI sites in the ENE constructs).
To generate the constructs pri-let-7ΔpAx2, pri-lin-4ΔpAx2, pri-let-7ENEΔpAx2, and pri-lin-4ENEΔpAx2, the SV40 CPA signal was deleted from the corresponding pri-miRNAΔpA plasmids by digesting each plasmid with BstBI and BsmI followed by blunt ending using T4 DNA polymerase and ligation using T4 DNA ligase. The digestion removes a 239–base pair region that includes both SV40 AAUAAA hexamers but leaves the GU-rich region downstream of the final hexamer intact.
Pri-miR-26bFE was PCR amplified from HeLa genomic DNA using primers Pri26bFEF and Pri26bFER. Pri-miR-26b was PCR amplified from this plasmid using primers Pri26bIF and Pri26bIR. Five copies of the ENE were inserted into pri-miR-26b in the forward or reverse orientation by digestion of βΔ1,2-79Fx5 or βΔ1,2-79Rx5 (Conrad and Steitz, 2005) with BglII, blunt ending with Klenow polymerase, and digestion with HindIII. The ENE digestion products were inserted into pri-miR-26b after digestion with XhoI, blunting using T4 DNA polymerase, and digestion with HindIII. Deletion of the BGH CPA signal was performed by digesting βΔ1,2delAAUAAA with StuI and XhoI, blunting with T4 DNA polymerase, ligation into pri-miR-26b that had been digested with StuI and XhoI and blunted as well.
CMV-pri-miR-30 was derived from the construct pSM2-αGSTpi (here referred to as U6-pri-miR-30; a gift from S. Vasudevan, Yale University, New Haven, CT; Open Biosystems), which contains a U6 snRNA promoter and a pri-miR-30 insert that contains genomic flanking sequences except in regions of the pre-miRNA hairpin, which were replaced with sequences designed to target the mRNA encoding GST-Pi. The pri-miR-30 region of PSM2-αGSTpi was released from the parent vector with SalI and MluI, blunt ended with T4 DNA polymerase, and ligated into the pcDNA3 vector digested with EcoRV to create the final product CMV-pri-miR-30. Note that the mature miRNA sequence probed in Fig. 7 C corresponds to an artificial miRNA that is complementary to a region of the GST-Pi mRNA (see probe sequence in Table S1).
U6-pri-Lin-4 was generated by first removing the CMV promoter from pri-Lin-4 by digestion with NruI and HindIII followed by blunt ending with T4 DNA polymerase and treatment with alkaline phosphatase. The U6 promoter including the 27-nt leader sequence was digested out of the pSM2 vector using BamHI and SalI, blunt ended with T4 DNA polymerase, and inserted into the pri-Lin-4 vector in place of the CMV promoter. The AATAAA hexamer from the BGH CPA signal of U6-pri-Lin-4 was then mutated to TTTTTT using the QuikChange Site-Directed Mutagenesis kit.
Cell lines, transfection, and cell treatments
HeLa cells and U2OS cells were cultured in Dulbecco's Modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (BD Biosciences), 2 mM l-glutamine, and 1× penicillin streptomycin solution (Sigma-Aldrich) at 37°C in 5% carbon dioxide. Raji and B95-8 cells were cultured in RPMI supplemented as above. Transfections were performed with HeLa MONSTER TransIT Reagent (Mirus Bio Corporation) according to the manufacturer's instructions. Transfection efficiencies were ∼50–60%, and only experiments in this range were analyzed.
To induce expression of pri-miR-34a, HeLa cells plated at ∼5 × 105 cells/ml in 10-cm plates or in 8-well chamber slides were washed once in PBS, then placed in PBS and cross-linked in a UV cross-linker (Stratagene) at 20 J/m2. PBS was replaced with fresh media and cells were incubated for 0–12 h. All experiments included mock-treated cells as a control, which were treated as described above, but without exposure to UV. To induce expression of BIC/pri-miR-155 or pri-miR-BHRF1-2∼3 in Raji and B95-8 cells, cells seeded at 4 × 105 cells/ml were treated with 1 nM PMA and 0.5 μM Ionomycin both dissolved in DMSO, or DMSO alone as a control, for 24 or 48 h. After treatments, cells were either subjected to ISH, nuclear fractionation was performed, or RNA was isolated and analyzed by Northern blotting.
RNA isolation and Northern blot analysis
Total RNA was isolated using Trizol reagent (Invitrogen). RNA from each sample was run on two gels to detect (1) pri-miRNAs or (2) precursor and mature miRNAs by Northern blotting. For pri-miRNAs, 3–5 μg of total RNA was resolved on a 1.2% agarose/6.5% formaldehyde gel and transferred overnight to positively charged Zeta probe membranes (Bio-Rad Laboratories) by capillary transfer using 20× SSC. After UV cross linking, membranes were hybridized to in vitro transcribed riboprobes overnight at 55–60°C in hybridization buffer (50% formamide, 6× SSC buffer, 5× Denhardt's solution, 25 mM sodium phosphate, pH 6.5, 0.5% sodium dodecylsulfate, and 150 μg/ml yeast total RNA). For precursor and mature miRNAs, 3–5 μg of total RNA was separated on a 15% polyacrylamide/8 M urea/1× TBE gel followed by semidry electroblotting onto Hybond N+ nylon membranes (GE Healthcare). Membranes were cross linked and prehybridized for at least 1 h in ExpressHyb Solution (Clontech Laboratories, Inc.). Oligonucleotide probes complementary to the mature miRNA of interest were end labeled with T4 polynucleotide kinase in the presence of [γ-32P]ATP. Results were quantitated using a Storm PhosphorImager (Molecular Dynamics). Sequences of probes are detailed in Table S1.
In vitro pri-miRNA processing assays
In vitro pri-miRNA processing assays were performed based on Lee et al. (2002). In brief, whole cell extract was made by sonicating HEK293 cells in buffer E (20 mM Hepes-KCl, 100 mM KCl, 0.2 mM EDTA, 10% glycerol, and 1 mM DTT), followed by centrifugation according to Kataoka and Dreyfuss (2004). Pri-miRNA templates were generated by PCR of the pri-miRNA–encoding plasmids using primers that added a T7 RNA polymerase promoter and either no polyA tail or a stretch of 60 adenylates to create an artificial polyA tail. Pri-miRNAs were in vitro transcribed from these templates in the presence of [α-32P]UTP using T7 RNA polymerase followed by gel purification and ethanol precipitation. Approximately 6.5 × 104 cpm (∼1 fmol) of pri-miRNA substrate was used per processing reaction. The resulting RNA was run on 10% polyacrylamide/8 M urea/1× TBE gels and viewed using a Storm PhosphorImager (Molecular Dynamics). Processing assays performed on in vitro transcribed pri-miRNAs either lacking or containing a polyA tail of 60 nt gave identical results, as did processing assays performed on pri-miRNA transcripts that had been heated to 95°C followed by slow cooling to ensure proper folding of pri-miRNA substrates.
Fractionation of nucleoplasmic and chromatin-associated transcripts
Fraction of HeLa or U2OS nuclei was performed according to Wuarin and Schibler (1994) and Dye et al. (2006). All fractionation steps were performed on ice in chilled buffers. In brief, 24 h after transfection, cells were washed twice in PBS and collected in PBS with a cell scraper. To isolate nuclei, scraped cells were spun at 3,000 g and resuspended in RLB buffer (10 mM Tris, pH 7.5, 140 mM NaCl, 1.5 mM MgCl2, and 0.5% Nonidet P-40). Cells were incubated on ice for 5 min, layered onto a 24% (wt/vol) sucrose cushion in RLB, and spun at 13,000 g for 10 min. To fractionate nuclei, the nuclear pellet was resuspended in NUN1 buffer (20 mM Tris, pH 7.9, 75 mM NaCl, 0.5 mM EDTA, 0.125 mM PMSF, 50% glycerol, and 0.1 mg/ml tRNA). Nuclear lysis was achieved by adding 10 volumes of NUN2 buffer (20 mM Hepes, pH 7.6, 7.5 mM MgCl2, 0.2 mM EDTA, 0.1 mg/ml tRNA, 0.3 M NaCl, 1 M urea, 1% Nonidet P-40, and 1 mM DTT) followed by incubation on ice for 15 min with occasional vortexing. Fractionated nuclei were spun at 13,000 rpm for 15 min. The nucleoplasmic supernatant was removed, phenol chloroform was extracted, and ethanol was precipitated. The chromatin-associated pellet fraction was resuspended in a high-salt buffer (10 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM MgCl2, and 10 mM CaCl2) plus 10 U RQ1 DNase (Promega) and incubated for 10 min at 37°C. Proteinase K was then added to a final concentration of 200 μg/ml, and the chromatin-associated fraction was further incubated at 37°C for 30 min followed by phenol chloroform extraction and ethanol precipitation. The precipitates from both fractions were resuspended twice in RQ1 DNase Buffer plus 20 U of RQ1 DNase and incubated at 37°C for 30 min, then phenol chloroform was extracted and ethanol was precipitated. Fractionated RNA was then resuspended in distilled water.
RT-PCR
After nuclear fractionation, cDNA was generated from 2.5 μg RNA using SuperScript II reverse transcriptase (Invitrogen) in a 20-μl reaction volume according to the manufacturer's instructions. 1 μl of the reaction mix was amplified by PCR for 24–33 cycles using the primers indicated in Table S1. TAFII30 mRNA, β-actin mRNA, and β-actin pre-mRNA primers are the same as those used by Mili et al. (2001). Because of differences in cellular abundance, β-actin pre-mRNA was amplified for 30 cycles, whereas mature mRNA was amplified for 28 cycles. No PCR products were obtained from control RT reactions that omitted Superscript II reverse transcriptase.
ISH in HeLa cells
ISH in HeLa cells was performed based the protocol of Kendirgi et al. (2003). Cells were grown on sterile 8-well chamber slides, and, 24–28 h after transfection, fixed in 4% formaldehyde/1× PBS for 30 min. Fixed cells were then washed three times for 5 min each in cold PBS followed by permeabilization for 10 min in 0.5% Triton X-100/PBS on ice. Permeabilized cells were washed once with PBS followed by a final wash with 2× SSC, both at room temperature. Cells were prehybridized in Phil's prehybridization buffer (50% formamide, 10% dextran sulfate, 2× SSC, 0.1% RNase-free BSA, 500 μg/ml salmon sperm DNA, 125 μg/ml E. coli tRNA, and 1 mM vanadyl ribonucleoside complexes; Forrester et al., 1992) for at least 1 h at 37°C, and then hybridized overnight with 25 μM each of four digoxygenin (DIG)-labeled oligonucleotide probes that recognize sequences flanking the pre-miRNA hairpin within the pri-miRNA of interest (see Table S1). Probes were labeled with DIG-dUTP using the 3′ DIG Oligonucleotide Tailing kit (Roche) according to the manufacturer's instructions. After hybridization, slides were washed three times for 10 min each in 2× SSC at 37°C followed by three 10-min washes in 1× SSC at room temperature. Cells were fixed again in 4% formaldehyde/PBS and washed twice in PBS, and hybridized probes were detected using a 1:200 dilution of anti-DIG antibody conjugated to fluorescein (green; Roche) in 10 mg/ml BSA/0.2% Triton X-100/PBS at room temperature for 1 h. Cells were then washed twice for 10 min each in PBS and once in 0.2 μg/ml DAPI/PBS for nuclear visualization followed by a final wash in PBS and mounting for fluorescence microscopy using Vectashield mounting media (Vector Laboratories). Note that in Fig. S2, an alternate ISH procedure was used (see DNA fluorescence ISH).
ISH using catalyzed reporter deposition
For detection of low-level pri-miRNAs (in Fig. 6 B), an amplification procedure was used. HeLa cells were grown on glass slides and fixed, washed, and permeabilized as described in the previous section, with the exception that endogenous peroxidase activity was quenched by incubating cells in 0.1% hydrogen peroxide/PBS for 10 min at room temperature before incubation in 2× SSC. Slides were hybridized to 4 μM of each of four DIG-labeled oligonucleotide probes as described in the previous section (see Table S1). 20–24 h later, amplification of the hybridization signal was performed using the Tyramide Signal Amplification kit No. 22 (Invitrogen) according to the manufacturer's instructions, with the exception that anti-DIG-hydrogen peroxidase (Roche) was used in place of the streptavidin-hydrogen peroxidase included in the kit.
ISH in B cell lines and marmoset leukocyte B95-8 cells
For ISH in B cell lines an alternate ISH procedure was used according to Lawrence and Singer (1985, 1986) with the exception that slides were hybridized to probes in Phil's prehybridization buffer (see ISH in HeLa cells) at 37°C for 15–18 h. Post-hybridzation washes and IF were performed as described for HeLa cells. Probe sequences are found in Table S1. For all ISH procedures, no signal was detected in control in situs that were performed in parallel, which either omitted probes or used nonspecific DIG-labeled probes. For detection of EBV DNA, slides were treated identically, except that they were incubated in RNase A for 2 h at 37°C and heat denatured at 85°C for 10 min in 2× SSC immediately before addition of probes in hybridization buffer. For ISH using Raji cells, slides were coated with poly-l-lysine (Sigma-Aldrich) according to the manufacturer's instructions before use.
IF and quantitation of Drosha and DGCR8 in foci
Immunostaining of SC35 was performed immediately after the ISH procedure. After detection of DIG-labeled probes and washes with PBS, cells were fixed again in 4% formaldehyde/PBS, washed twice in PBS for 5 min each, and permeabilized in 0.2% Triton X-100/PBS for 10 min on ice. After two 5-min washes with 1× PBS, blocking was performed for 30 min at room temperature in blocking solution (10 mg/ml BSA/PBS). Cells were then incubated with mouse monoclonal anti-SC35 antibody (Sigma-Aldrich) at a dilution of 1:200 in blocking solution for 1 h at room temperature. Slides were washed with PBS three times for 10 min each, followed by incubation with Alexa Fluor 595–conjugated goat anti–mouse (red; Invitrogen) in blocking solution for 45 min at room temperature. Slides were then washed twice with PBS for 10 min, once with 0.2 μg/ml DAPI/PBS solution, and a final time with PBS for 10 min each, followed by mounting for fluorescence microscopy.
Immunostaining of Drosha and DGCR8 was performed immediately before the ISH procedure (note that the order of analysis was reversed because the epitope recognized by the anti-Drosha and anti-DGCR8 antibodies is denatured by the ISH procedure). 24–28 h after transfection, cells were fixed, washed, and permeabilized as described for the ISH procedure. After permeabilization, cells were washed twice in PBS for 5 min each and incubated in blocking solution for 30 min at room temperature. Either rabbit polyclonal anti-Drosha (Millipore) or rabbit polyclonal anti-DGCR8 (Abcam) were added to the slides at a 1:200 dilution in blocking solution and incubated at room temperature for 1 h. Slides were washed with PBS three times for 10 min each followed by incubation with Alexa Fluor 595–conjugated goat anti–rabbit (red; Invitrogen) in blocking solution for 45 min at room temperature. Slides were washed three times in PBS for 10 min each and fixed in 4% formaldehyde/PBS. ISH to the pri-miRNA was then performed as described previously (see ISH in HeLa cells).
To obtain rough quantitations of the amounts of Drosha and DGCR8 in foci, the program ImageJ was used. Background-corrected intensities of the entire cell and each focus were determined, and the sum of intensities of the foci was divided by the total intensity of the cell to obtain the percent of Drosha or DGCR8 in foci.
Image acquisition
Fluorescent images were captured with a microscope (Axioplan II; Carl Zeiss, Inc.) using a 40× or 63× 1.3 NA oil immersion objective (Plan-Neofluar; Carl Zeiss, Inc.) at room temperature. Image acquisition was performed with a digital charge-coupled device camera (C4742-95-12; Hamamatsu) using Openlab software (PerkinElmer). Raw data images were adjusted for image size, brightness, and contrast using Openlab (PerkinElmer) or Photoshop CS software (Adobe).
Nick translation of probes for DNA FISH (Fig. S1)
Templates for nick translation were either the pri-let-7 plasmid or the pri-lin-4 plasmid that had been digested with SmaI and BsmI to remove the neomycin resistance gene insert (to prevent hybridization of the nick-translated probes with the neomycin resistance gene transcript in the RNA ISH performed in parallel; see DNA fluorescence ISH). Approximately 1 μg of template DNA was incubated in a reaction consisting of nick translation buffer (50 mM Tris-HCl, pH 8.0, 5 mM MgCl2, and 50 μg/ml BSA), 10 mM β-mercaptoethanol, 10 mM d(A,C,G)TP, 6.5 mM dUTP, 3.5 mM DIG-dUTP (Roche), 10 U/ml E. coli DNA polymerase I (New England Biolabs, Inc.), and 10 ng/ml RQ1 DNase. Reactions were incubated at 16°C for ∼4.5 h and visualized on an agarose gel stained with ethidium bromide to ensure that probe fragments were in the size range of 250–750 nt. Probes were then ethanol precipitated and resuspended in formamide. Immediately before being added to the slides, probes were denatured by heating to 80°C for 5 min, and 140 ng of probe per slide was added in Phil's prehybridization buffer.
DNA fluorescence ISH (Fig. S1)
Transfected cells grown on glass slides were fixed and permeabilized as previously described in “ISH in HeLa cells.” After permeabilization, slides were washed twice in PBS, dehydrated in sequential washes with 80%, 95%, and 100% ethanol for 3 min each, and allowed to air dry for ∼2 min. Slides were incubated in 0.2 M sodium hydroxide for 15 min to degrade RNA and denature DNA, followed by two washes in 2× SSC for 10 min each. Dehydration was then repeated as described, and further denaturation of the DNA using heat was performed by addition of 50% formamide/2× SSC, pH 7.2, and incubation at 85°C for 10 min. DIG-labeled nick-translated probes in Phil's prehybridization solution were then added directly to the slides and hybridized overnight at 37°C in a humid chamber. 14–18 h later, slides were washed three times in 50% formamide/2× SSC for 10 min each at 37°C followed by three 5-min washes with 2× SSC at room temperature. Slides were then blocked in 4× SSC/0.1% Tween/1% BSA for 30 min followed by incubation with fluorescein-conjugated anti-DIG antibody in 4× SSC/0.1% Tween/1% BSA for 1 h. Slides were washed three times for 5 min each with 2× SSC, incubated with 0.2 μg/ml DAPI in 2× SSC for 10 min, and finally washed twice in 2× SSC for 5 min each. Coverslips were then mounted. Treatment of samples with DNase before probe hybridization abrogated the signal observed. RNA ISHs shown in Fig. S1 were performed using the same protocol as DNA fluorescence ISH using nick-translated probes except that the sodium hydroxide and heat denaturation steps were omitted.
Online supplemental material
Fig. S1 shows that transfected pri-miRNA–encoding plasmid DNA does not localize to nuclear foci. Fig. S2 shows the localization of transcripts produced from pri-miR-26b constructs in relation to SC35 and directly demonstrates that Drosha is recruited to SC35-containing foci in cells expressing a pri-miRNA. Fig. S3 demonstrates that pri-Lin-4ENE expressed at low levels by transfection of 10–20-fold less plasmid DNA does not localize to nuclear foci. Table S1 provides sequences of PCR primers and oligonucleotide probes. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.20080311/DC1.
Supplementary Material
Acknowledgments
We gratefully acknowledge N.K. Conrad for plasmids and helpful suggestions. We thank all members of the Steitz laboratory for stimulating discussions throughout this work and constructive criticisms of this manuscript.
This work was supported by grant R01GM026154 from the National Institute of General Medical Sciences (NIGMS) and a National Science Foundation Graduate Research Fellowship to J.M. Pawlicki. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the National Institutes of Health. J.A. Steitz is an investigator of the Howard Hughes Medical Institute.
Abbreviations used in this paper: BGH, bovine growth hormone; BIC, B cell integration cluster; CMV, cytomegalovirus; CPA, cleavage and polyadenylation; DIG, digoxygenin; EBNA, EBV nuclear antigen; EBV, Epstein Barr virus; IF, immunofluorescence; ISH; in situ hybridization; miRNA, microRNA; PolyA, polyadenylate; pre-miRNA, precursor miRNA; pre-mRNA, precursor mRNA; pri-miRNA, primary miRNA; snRNA, small nuclear RNA; SV40, simian virus 40.
References
- Cai, X., C.H. Hagedorn, and B.R. Cullen. 2004. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 10:1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, X., A. Schafer, S. Lu, J.P. Bilello, R.C. Desrosiers, R. Edwards, N. Raab-Traub, and B.R. Cullen. 2006. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin, G.A., and C.M. Croce. 2006. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 66:7390–7394. [DOI] [PubMed] [Google Scholar]
- Chung, K.H., C.C. Hart, S. Al-Bassam, A. Avery, J. Taylor, P.D. Patel, A.B. Vojtek, and D.L. Turner. 2006. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 34:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad, N.K., and J.A. Steitz. 2005. A Kaposi's sarcoma virus RNA element that increases the nuclear abundance of intronless transcripts. EMBO J. 24:1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad, N.K., S. Mili, E.L. Marshall, M.D. Shu, and J.A. Steitz. 2006. Identification of a rapid mammalian deadenylation-dependent decay pathway and its inhibition by a viral RNA element. Mol. Cell. 24:943–953. [DOI] [PubMed] [Google Scholar]
- Conrad, N.K., M.D. Shu, K.E. Uyhazi, and J.A. Steitz. 2007. Mutational analysis of a viral RNA element that counteracts rapid RNA decay by interaction with the polyadenylate tail. Proc. Natl. Acad. Sci. USA. 104:10412–10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custodio, N., M. Carmo-Fonseca, F. Geraghty, H.S. Pereira, F. Grosveld, and M. Antoniou. 1999. Inefficient processing impairs release of RNA from the site of transcription. EMBO J. 18:2855–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli, A.M., B.B. Tops, R.H. Plasterk, R.F. Ketting, and G.J. Hannon. 2004. Processing of primary microRNAs by the Microprocessor complex. Nature. 432:231–235. [DOI] [PubMed] [Google Scholar]
- Duan, R., C. Pak, and P. Jin. 2007. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum. Mol. Genet. 16:1124–1131. [DOI] [PubMed] [Google Scholar]
- Dye, M.J., N. Gromak, and N.J. Proudfoot. 2006. Exon tethering in transcription by RNA polymerase II. Mol. Cell. 21:849–859. [DOI] [PubMed] [Google Scholar]
- Edmonds, M. 2002. A history of poly A sequences: from formation to factors to function. Prog. Nucleic Acid Res. Mol. Biol. 71:285–389. [DOI] [PubMed] [Google Scholar]
- Fang, Y., and D.L. Spector. 2007. Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr. Biol. 17:818–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester, W., F. Stutz, M. Rosbash, and M. Wickens. 1992. Defects in mRNA 3′-end formation, transcription initiation, and mRNA transport associated with the yeast mutation prp20: possible coupling of mRNA processing and chromatin structure. Genes Dev. 6:1914–1926. [DOI] [PubMed] [Google Scholar]
- Fujioka, Y., M. Utsumi, Y. Ohba, and Y. Watanabe. 2007. Location of possible miRNA processing site in SmD3/SmB nuclear bodies in Arabidopsis. Plant Cell Physiol. 48:1243–1253. [DOI] [PubMed] [Google Scholar]
- Fukuda, T., K. Yamagata, S. Fujiyama, T. Matsumoto, I. Koshida, K. Yoshimura, M. Mihara, M. Naitou, H. Endoh, T. Nakamura, et al. 2007. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat. Cell Biol. 48:604–611. [DOI] [PubMed] [Google Scholar]
- Gregory, R.I., K.P. Yan, G. Amuthan, T. Chendrimada, B. Doratotaj, N. Cooch, and R. Shiekhattar. 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature. 432:235–240. [DOI] [PubMed] [Google Scholar]
- Hall, L.L., K.P. Smith, M. Byron, and J.B. Lawrence. 2006. Molecular anatomy of a speckle. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288:664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J., Y. Lee, K.H. Yeom, J.W. Nam, I. Heo, J.K. Rhee, S.Y. Sohn, Y. Cho, B.T. Zhang, and V.N. Kim. 2006. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 125:887–901. [DOI] [PubMed] [Google Scholar]
- He, L., X. He, S.W. Lowe, and G.J. Hannon. 2007. microRNAs join the p53 network—another piece in the tumour-suppression puzzle. Nat. Rev. Cancer. 7:819–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S., and D.L. Spector. 1996. Intron-dependent recruitment of pre-mRNA splicing factors to sites of transcription. J. Cell Biol. 133:719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, C., D. Primorac, M. McKinstry, J. McNeil, D. Rowe, and J.B. Lawrence. 2000. Tracking COL1A1 RNA in osteogenesis imperfecta. splice-defective transcripts initiate transport from the gene but are retained within the SC35 domain. J. Cell Biol. 150:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka, N., and G. Dreyfuss. 2004. A simple whole cell lysate system for in vitro splicing reveals a stepwise assembly of the exon-exon junction complex. J. Biol. Chem. 279:7009–7013. [DOI] [PubMed] [Google Scholar]
- Kendirgi, F., D.M. Barry, E.R. Griffis, M.A. Powers, and S.R. Wente. 2003. An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J. Cell Biol. 160:1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, V.N. 2005. MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 6:376–385. [DOI] [PubMed] [Google Scholar]
- Kim, Y.K., and V.N. Kim. 2007. Processing of intronic microRNAs. EMBO J. 26:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman, W.P., and R.H. Plasterk. 2006. The diverse functions of microRNAs in animal development and disease. Dev. Cell. 11:441–450. [DOI] [PubMed] [Google Scholar]
- Kluiver, J., A. van den Berg, D. de Jong, T. Blokzijl, G. Harms, E. Bouwman, S. Jacobs, S. Poppema, and B.J. Kroesen. 2007. Regulation of pri-microRNA BIC transcription and processing in Burkitt lymphoma. Oncogene. 26:3769–3776. [DOI] [PubMed] [Google Scholar]
- Kornblihtt, A.R., M. de la Mata, J.P. Fededa, M.J. Munoz, and G. Nogues. 2004. Multiple links between transcription and splicing. RNA. 10:1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond, A.I., and D.L. Spector. 2003. Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4:605–612. [DOI] [PubMed] [Google Scholar]
- Lawrence, J.B., and R.H. Singer. 1985. Quantitative analysis of ISH methods for the detection of actin gene expression. Nucleic Acids Res. 13:1777–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, J.B., and R.H. Singer. 1986. Intracellular localization of messenger RNAs for cytoskeletal proteins. Cell. 45:407–415. [DOI] [PubMed] [Google Scholar]
- Lee, Y., K. Jeon, J.T. Lee, S. Kim, and V.N. Kim. 2002. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 21:4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y., M. Kim, J. Han, K.H. Yeom, S. Lee, S.H. Baek, and V.N. Kim. 2004. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23:4051–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listerman, I., A.K. Sapra, and K.M. Neugebauer. 2006. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat. Struct. Mol. Biol. 13:815–822. [DOI] [PubMed] [Google Scholar]
- McCracken, S., N. Fong, E. Rosonina, K. Yankulov, G. Brothers, D. Siderovski, A. Hessel, S. Foster, S. Shuman, and D.L. Bentley. 1997. 5′-capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 11:3306–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcak, I., S. Cermanova, K. Jirsova, K. Koberna, J. Malinsky, and I. Raska. 2000. Nuclear pre-mRNA compartmentalization: trafficking of released transcripts to splicing factor reservoirs. Mol. Biol. Cell. 11:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili, S., H.J. Shu, Y. Zhao, and S. Pinol-Roma. 2001. Distinct RNP complexes of shuttling hnRNP proteins with pre-mRNA and mRNA: candidate intermediates in formation and export of mRNA. Mol. Cell. Biol. 21:7307–7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., and D.L. Spector. 1999. RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell. 3:697–705. [DOI] [PubMed] [Google Scholar]
- Nakamura, T., E. Canaani, and C.M. Croce. 2007. Oncogenic All1 fusion proteins target Drosha-mediated microRNA processing. Proc. Natl. Acad. Sci. USA. 104:10980–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer, K.M. 2002. On the importance of being co-transcriptional. J. Cell Sci. 115:3865–3871. [DOI] [PubMed] [Google Scholar]
- Pillai, R.S., S.N. Bhattacharyya, and W. Filipowicz. 2007. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 17:118–126. [DOI] [PubMed] [Google Scholar]
- Roberts, G.C., C. Gooding, H.Y. Mak, N.J. Proudfoot, and C.W. Smith. 1998. Co-transcriptional commitment to alternative splice site selection. Nucleic Acids Res. 26:5568–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, A., S. Griffiths-Jones, J.L. Ashurst, and A. Bradley. 2004. Identification of mammalian microRNA host genes and transcription units. Genome Res. 14:1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, J.M., M.Z. Li, K. Chang, W. Ge, M.C. Golding, R.J. Rickles, D. Siolas, G. Hu, P.J. Paddison, M.R. Schlabach, et al. 2005. Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 37:1281–1288. [DOI] [PubMed] [Google Scholar]
- Song, L., M.H. Han, J. Lesicka, and N. Fedoroff. 2007. Arabidopsis primary microRNA processing proteins HYL1 and DCL1 define a nuclear body distinct from the Cajal body. Proc. Natl. Acad. Sci. USA. 104:5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, M.J., H.J. Brown, T.T. Wu, and R. Sun. 2001. Transcription activation of polyadenylated nuclear RNA by RTA in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:3129–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, R., S.F. Lin, L. Gradoville, and G. Miller. 1996. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA. 93:11883–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, J.M., M. Newman, J.S. Parker, E.M. Morin-Kensicki, T. Wright, and S.M. Hammond. 2006. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 20:2202–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan, S., Y. Tong, and J.A. Steitz. 2007. Switching from repression to activation: microRNAs can up-regulate translation. Science. 318:1931–1934. [DOI] [PubMed] [Google Scholar]
- Wuarin, J., and U. Schibler. 1994. Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol. Cell. Biol. 14:7219–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, L., and E. Kieff. 2007. Epstein-Barr virus BHRF1 micro- and stable RNAs during latency III and after induction of replication. J. Virol. 81:9967–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G., K.L. Taneja, R.H. Singer, and M.R. Green. 1994. Localization of pre-mRNA splicing in mammalian nuclei. Nature. 372:809–812. [DOI] [PubMed] [Google Scholar]
- Zhao, J., L. Hyman, and C. Moore. 1999. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev. 63:405–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
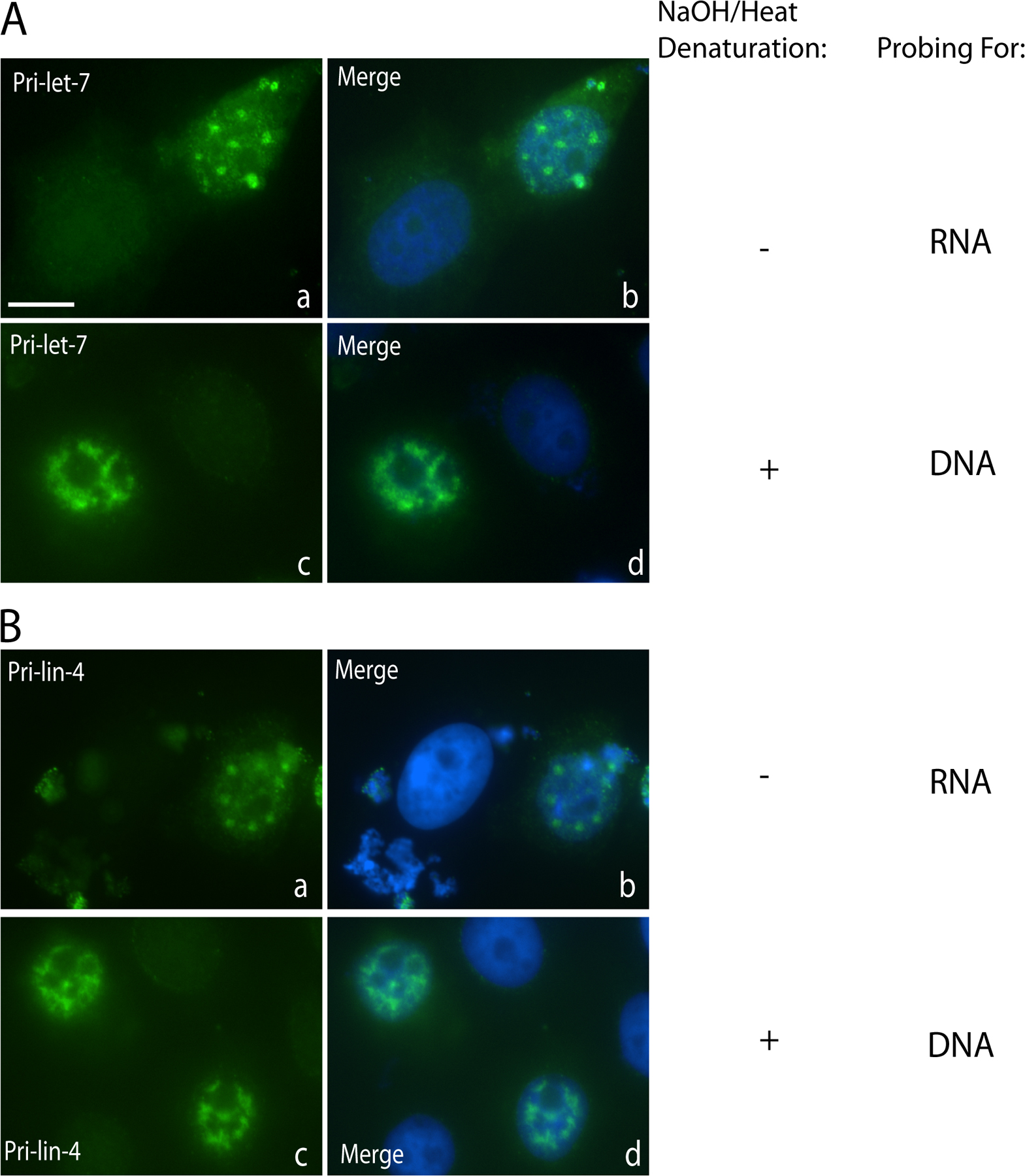{kind=link}
{kind=link}
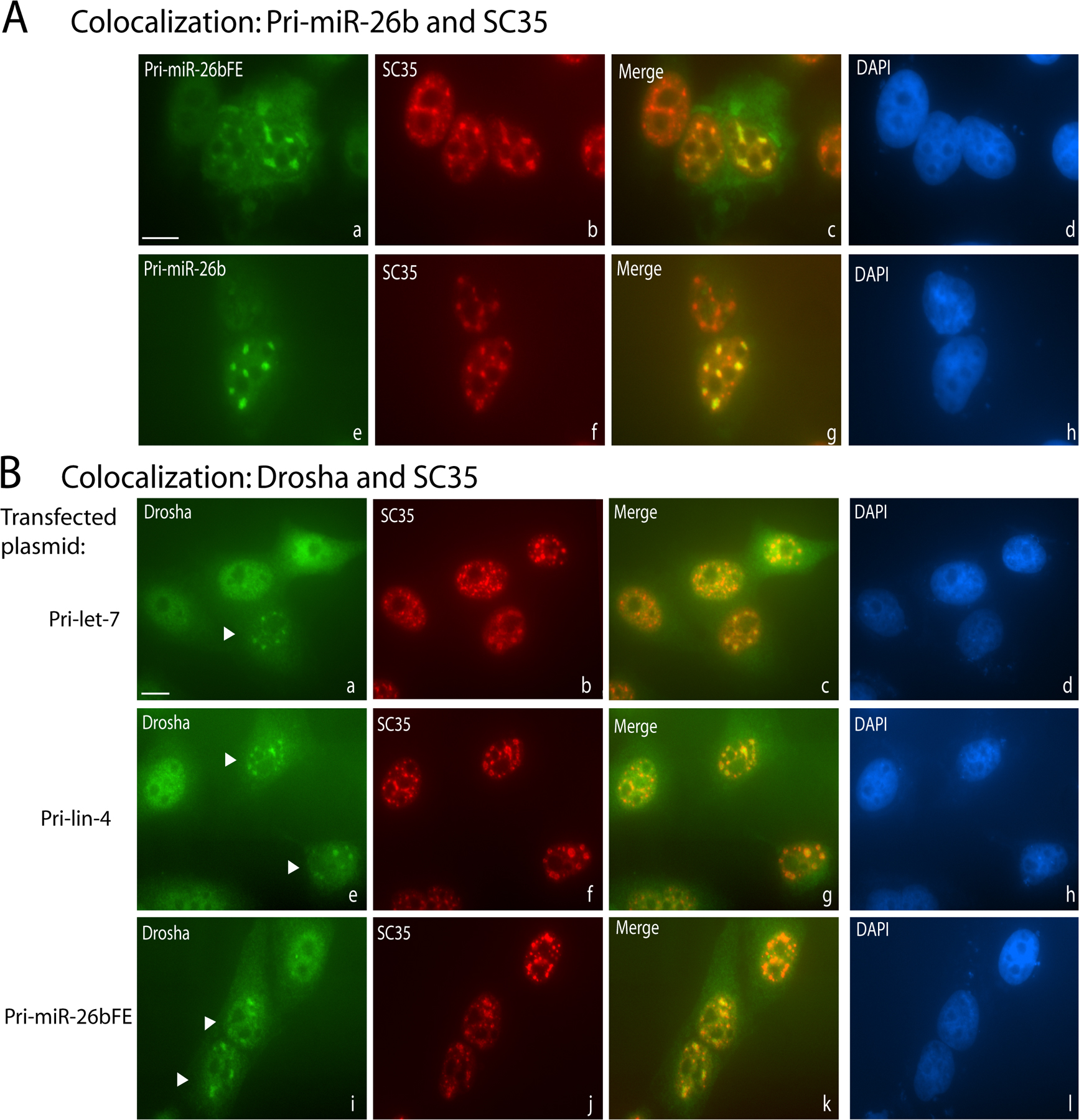{kind=link}
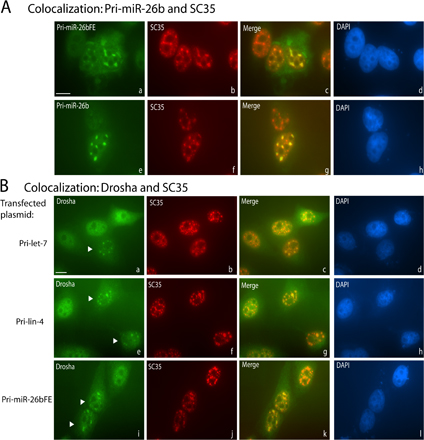{kind=link}
{kind=link}
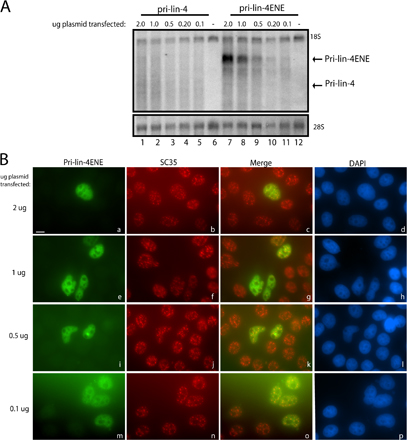{kind=link}