

Abstract
Bioassay-guided fractionation of the combined fruits, leaves, and twigs (fruiting branches) of Callicarpa americana, collected from a plot in a forested area in southern Florida, led to the isolation of six new clerodane diterpenes (1–6) and eight known compounds. The structures of 1–6 [12(S),16ξ-dihydroxycleroda-3,13-dien-15,16-olide (1), 12(S)-hydroxy-16ξ-methoxycleroda-3,13-dien-15,16-olide (2), 12(S)-hydroxycleroda-3,13-dien-15,16-olide (3), 16 ξ-hydroxycleroda-3,11(E),13-trien-15,16-olide (4), 3β,12(S)-dihydroxycleroda-4(18),13-dien-15,16-olide (5), and 12(S)-hydroxycleroda-3,13-dien-16,15-olide (6)] were elucidated by interpretation of spectroscopic data and chemical methods. The absolute configuration at C-12 in 1 and 3 was ascertained using the Mosher ester technique. The cytotoxicity of all isolates was tested against a panel of human cancer cell lines, and compounds 1, 4, and 6, and the known compounds genkwanin, 16ξ-hydroxycleroda-3,13-dien-15,16-olide, and2-formyl-16ξ-hydroxy-3-A-norcleroda-2,13-dien-15,16-olide were active (ED50 <5 μg/mL). However, 1 was found to be inactive against human cancer cells implanted in mice using a hollow-fiber tumor model.
Callicarpa americana L. is a shrub native to the southeastern United States.2 Although the genus has been considered traditionally as a member of the plant family Verbenaceae, molecular and micro-morphological observations have prompted contemporary taxonomic authorities to reclassify Callicarpa as a member of the family Lamiaceae.3–5 Preparations of the bark of C. americana have been used to treat fever,6 the leaves to treat dropsy,7 and the roots to alleviate colic,8 dysentery,9 and skin cancer.10 The roots and branches have been used in preparations intended to relieve malaria, rheumatism, and fever.8 The leaf essential oils of C. americana have antialgal and mosquito-deterrent properties, and numerous essential oil components have been identified from the leaves of C. americana.11–12
As part of an ongoing effort to discover novel anticancer agents from plants,13 a chloroform-soluble extract of the combined fruits, leaves, and twigs of C. americana was investigated, using cytotoxicity against hormone-dependent prostate cancer cells (LNCaP) to guide the isolation of active constituents. This plant material was obtained from a forest plot in southern Florida using a plot-based collection method (briefly reviewed in ref.13).
Results and Discussion
The dried fruits, leaves, and twigs of Callicarpa americana L. were extracted with methanol and partitioned following a previously described protocol.14 Based on the cytotoxic activity of the chloroform-soluble portion, bioassay-guided fractionation using a number of chromatographic techniques was carried out, guided by activity against LNCaP cells. This investigation resulted in the isolation of six new clerodane-type diterpenes (1–6), the structures of which were elucidated using a range of spectroscopic techniques, including ID and 2D NMR and accurate mass measurement. Additionally, eight known compounds were identified by comparison of their measured spectroscopic data with literature values. The known compounds comprised three substances that were previously reported to occur in the genus Callicarpa [calliterpenone,15,16 euscaphic acid,17,18 and salvigenin19,20], and five new to the taxon [genkwanin,21 3β,16ξ-dihydroxycleroda-4(18), 13-dien-15,16-olide,22 16ξ-hydroxycleroda-3,13-dien-15,16-olide,23 5-hydroxy-7,4′-dimethoxyflavone,24 and 2-formyl-16ξ-hydroxy-3-A-norcleroda-2,13-dien-15,16-olide25,26].
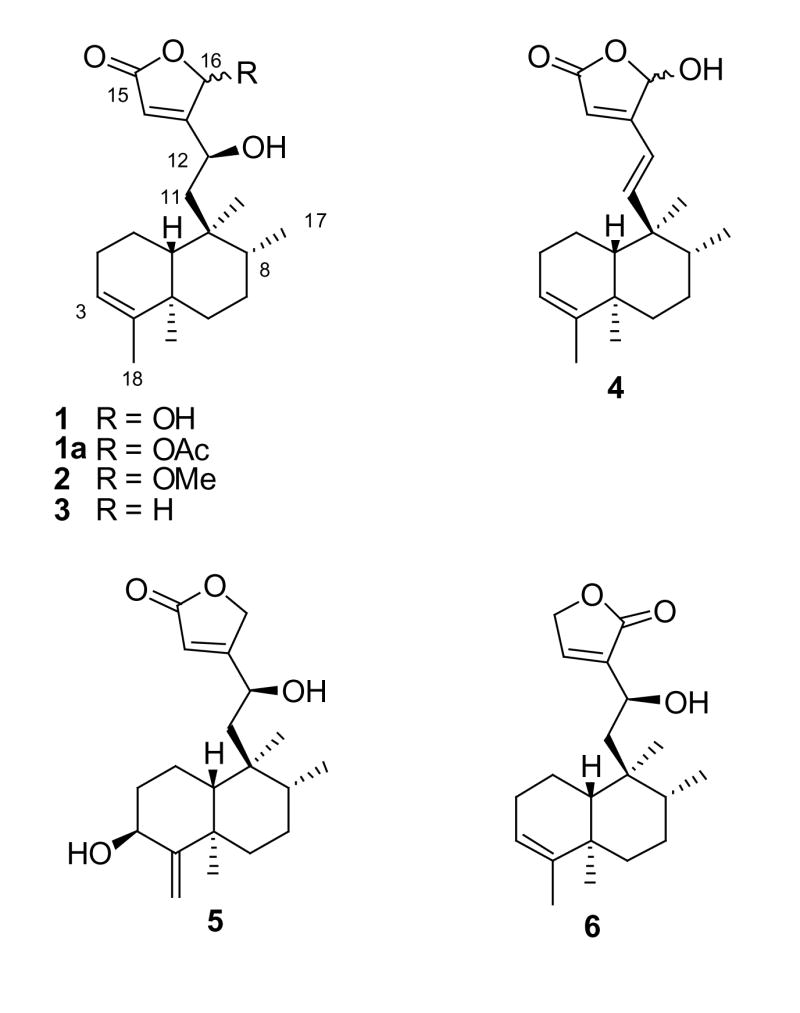
A molecular formula of C20H30O4 and six degrees of unsaturation were determined for 1 based on accurate mass measurement. Comparison of the 13C NMR and DEPT135 NMR spectra for 1 indicated the presence of four methyl groups, five methylenes, three aliphatic methines, two olefinic methines, one methine with a chemical shift typical of a hemiacetal (δc 98.4), and five quaternary carbons. The 1H NMR spectrum for 1 contained signals for four downfield methines and four methyl groups, as well as a number of signals attributable to methylene protons.
HMBC NMR correlations from the vinylic methyl (H-18) to C-3, C-4, and C-5, from H-19 to C-4, C-5, C-6, and C-10, from the doublet methyl (H-17) to C-7, C-8, and C-9, from the H-20 methyl singlet to C-8, C-9, and C-10, from the H-10 bridgehead methine to C-2 and C-19, and from the H-3 proton to C-1, C-2, and C-5 helped to establish the structure of the substituted Δ3(4)-decalin ring system, accounting for 14 carbon signals and three of the six degrees of unsaturation. Although the bridgehead proton signal overlapped the vinylic methyl (δH 1.58) signal, the correlations for these two proton resonances could be distinguished in the HMBC NMR spectrum because of the width of the H-10 multiplet signal. The C-1 to C-10 linkage was evident from a cross-peak in the 1H-1H COSY NMR spectrum between H-l and H-10. Additional 1H-1H couplings were observed between H-3 and H-18 (vinylic) and H-2, and between H-17 and a buried multiplet for H-8.
The six remaining carbon signals in the 13C NMR spectrum of 1 comprised a six-carbon side chain [a methylene (δC 43.0, C-11), a carbinol (δC 64.6, C-12), a hemiacetal methine (δC 98.4, C-16), an olefinic methine (δC 116.8, C-14), a carbonyl (δC 172.0, C-15), and a quaternary olefinic carbon (δC 174.3, C-13)], which could be connected to the decalin ring system at C-9. HMBC correlations from the H-14 proton (δH 6.01) to C-16 (δC 98.4) and C-15 (δC 172.0) and from the H-12 carbinol proton (δH 4.72) to C-9 (δC 39.8), C-11 (δC 43.0), C-13 (δC 174.3), C-14 (δC 116.8), and C-16 (δC 98.4), as well as the 1H-1H COSY cross-peaks observed between H-12 (δH 4.72) and the H-11 protons, suggested the side chain structure to consist of an α, β-unsaturated γ-lactone ring connected to the decalin ring by a two-carbon linker. This side-chain moiety, plus the mono-unsaturated decalin ring system, accounted for the six degrees of unsaturation indicated by the molecular formula.
The 1H NMR chemical shifts of the C-5 methyl (H-19, δH 1.01) and the C-9 methyl (H-20, δH 0.75) in 1 indicated the ring junction to have a 5α,10β-trans relative configuration.27 This was also supported by consideration of the 13C NMR chemical shift of the C-19 methyl group, which resonated upfield relative to the predicted value for a cis-clerodane.28 The 1H and 13C NMR chemical shifts for the C-8 methyl (CH3-17) and C-6 (13C NMR shift) are indicative of the relative configuration of CH3-17 in trans-clerodanes,29 and the corresponding chemical shifts in 1 indicate that the CH3-17 has an α (equatorial) relative configuration.
The absolute configuration at C-12 of 1a (the 16-O-acetyl derivative of 1) was determined using the method first described by Mosher and colleagues30,31 and further elaborated by other research groups.32–34 Comparison of relevant 1H NMR chemical shift differences between the R- and S-MTPA esters of 1a [H-3 (−0.150), H-17 (−0.039), H-18 (−0.073), H-19 (−0.042), H-20 (−0.047), H-14 (+0.312), H-16 (+0.169), OAc-16 (−0.007)] showed that the 1H NMR signals associated with the decalin ring system in the R-MTPA ester were shifted downfield relative to those of the S-MTPA ester, and the opposite was true for the resonances of the lactone ring moiety (with the exception that essentially no difference in relative shift was observed for the protons of the acetyl group). This allowed 1 to be assigned structurally as 12(S),16ξ-dihydroxycleroda-3,13-dien-15,16-olide.
A molecular formula of C21H32O4 and six degrees of unsaturation were determined for 2 based on accurate mass measurement. The 1H NMR and 13C NMR spectra were nearly superimposable with those of 1, with the exception of an additional carbon signal at δC 57.5 and a proton signal at δC 3.59 in the spectra for 2, and the C-16 methine signal, which resonated at δC 98.4 in 1, was shifted to δC 103.1 in 2. The inference that 2 contains a 16-O-methyl ether was confirmed by the observation of an HMBC correlation (three-bond) from the methoxy methyl protons (δH 3.59) to the carbon resonance at δC 103.1 (C-16). The assignment of the 1H NMR and 13C NMR data was carried out by analysis of its 1H-1H COSY, HMBC, and HMQC NMR spectra. The absolute configuration of OH-12 was assigned as S on biogenetic grounds by analogy with 1 and 3 (see below). Thus 2 was assigned structurally as 12(S)-hydroxy-16ξ-methoxycleroda-3,13-dien-15,16-olide.
A molecular formula of C20H30O3 and six degrees of unsaturation were determined for 3 based on accurate mass measurement. The IR spectrum indicated the presence of hydroxyl and α,β-unsaturated carbonyl resonances [3426 (br) and 1743 cm−1]. The 1H, 13C, and DEPT135 NMR spectra for 3 suggested that this isolate is based on a clerodane diterpene skeleton, similar to 1. Comparison of the 13C NMR and DEPT135 NMR spectra indicated signals for four methyl carbons, six methylene carbons [δC 20.3, 27.6, 28.7, 37.9, 45.1, and 72.9 (oxymethylene)], two aliphatic, an oxymethine (& 65.8), two olefinic methines (δC 114.1, and 122.0), and five quaternary carbons (δC 39.5, 40.9, 144.9, 176.5, and 179.0). The 1H NMR spectrum showed the presence of three quaternary methyl groups, one secondary methyl, three methine resonances in the downfield region of the spectrum, and one pair of oxymethylene protons (δH 4.96). The assignment of the 1H NMR signals was carried out by analysis of the 1H NMR, 1H-1H COSY, and HSQC NMR spectra, and selective proton decoupling experiments, with the latter experiments being instrumental in determining the splitting patterns for the 1H NMR signals. Key 1H-13C correlations in the HMBC NMR spectrum of 3 were observed from H-12 to C-11, C-14, and C-16, and from H-16 to C-13 and C-14, thus confirming the structure of the side chain as being composed of an α, β-unsaturated butyrolactone with a two-carbon linker attached at the β-position.
Analysis of the 1H-1H J-values for the protons on the decalin ring system and 1H-1H correlations observed in the NOESY NMR of 3 indicated the same relative configuration as determined for 1. The absolute configuration at C-12 in 3 was determined using a modified Mosher technique as described for 1. The protons associated with the lactone ring portion of 3 (H-14, H-16a, and H-16b) showed a positive relative difference in chemical shift [Δδ (δS − δR], and signals associated with the decalin ring system (H-3, H-17, H-18, H-19, and H-20) showed a negative relative difference in chemical shift, thus, the absolute configuration of C-12 was determined as S, and the structure of 3 was established as 12(S)-hydroxycleroda-3,13-dien-15,16-olide.
A molecular formula of C20H28O3 and seven degrees of unsaturation were determined for 4 based on accurate mass measurement. The IR spectrum for this isolate displayed a broad absorption at 3426 cm−1 and an additional absorption at 1743 cm−1, indicative of the presence of hydroxyl and carbonyl functionalities (with the wavelength of the latter being consistent with an α,β-unsaturated γ-lactone ring).35 The NMR spectra for 4 were obtained in deuterated methanol (MeOD). However, signal crowding in the downfield region of the 1H NMR spectrum of 4 obtained in MeOD made assignment of the associated signals difficult. Thus, NMR spectra for 4 were also measured in pyridine-d5, making use of the phenomenon of pyridine-induced chemical shift modification.36 Comparison of the 13C NMR and DEPT135 NMR spectra for 4 indicated the presence of four methyl groups, four methylenes, seven methines, and an additional five quaternary carbons.
The 1H NMR and HMQC NMR spectra obtained for 4 in pyridine-d5 exhibited a pair of trans-coupled methine doublets at δH 6.39 and 6.52 (H-12 and H-11, J = 16.4 Hz), corresponding to an asymmetrically substituted olefinic bond, and these 1H NMR signals correlated in the HMQC NMR spectrum with 13C NMR resonances at δC 156.1 and 119.9 (C-11 and C-12, respectively). Analysis of the HMBC and HMQC NMR spectra confirmed that the connectivity and relative configuration of 4 were consistent with those of 1–3. Thus, 4 was assigned as 16ξ-hydroxycleroda-3,11,13-trien-15,16-olide.
A molecular formula of C20H30O4 and six degrees of unsaturation were determined for 5 based on accurate mass measurement. Comparison of the 13C NMR and DEPT135 NMR spectra for 5 indicated the presence of 20 carbons, including three methyls, seven methylenes, five methines, and five quaternary carbons. Visual inspection of the 1H, DEPT135, and 13C NMR spectra suggested a close homology between 5 and 1–4. HMBC NMR correlations for 5 were observed from H-14 to C-15 and C-16, from H-16 to C-13 and C-14, and from H-11 to C-9, C-12, and C-13, establishing the side chain structure as being the same as that of 3. Additional HMBC correlations from H-18 to C-5, C-3, and C-4, from H-17 to C-7 and C-9, from H-19 to C-3, C-4, C-5, and C-10, and from H-20 to C-8, C-9, C-10, C-11, and C-12 (4JC-H) helped to determine the structure of the decalin ring moiety. The assignment of C-1, C-2, and C-6 was complicated by the nearly overlapping 13C NMR signals for C-1 and C-19 and for C-2, C-6, and C-8. The 1H-13C assignments for these signals are thus based on interpretation of the DEPT135, HMBC, and HMQC NMR spectra, and comparison with published values for 3β,16ξ-dihydroxycleroda-4(18), 13-dien-15,16-olide.22
A similar compound, pentandranoic acid C, differing from 5 in the composition of the side chain, was reported by Xu and colleagues from Callicarpa pentandra Roxb.26 The orientation of OH-3 in pentandranoic acid C was determined as α, based on NOESY NMR data and X-ray crystallographic analysis. The H-3 chemical shift of 5 (δH 4.33, CDCl3) was in close agreement with that of pentandranoic acid C (δH 4.30, CDCl3), but the C-3 chemical shift was significantly different from that of pentandranoic acid C (δC 69.7 in 5, rather than δC 74.6). Furthermore, in 5 the 1H-1H splitting pattern for H-3 was a doublet of doublets (J = 5.4, 11.6 Hz), whereas Xu et al. reported a triplet (J = 3.0 Hz).26 The relative configuration of OH-3 in 3β,16ξ-dihydroxycleroda-4(18),13-dien-15,16-olide was determined by Ma et al.22 as β, based on the observation of a dd splitting pattern (J = 5.8, 12 Hz) for H-3, implying an α-axial orientation for H-3. Based on these considerations, OH-3 was assigned as β for 5. Thus, the structure of 5 was determined as 3′,12(S)-dihydroxycleroda-4(18),13-dien-15,16-olide.
A molecular formula of C20H30O3, indicating six degrees of unsaturation, was determined for 6 based on accurate mass measurement. The 1H NMR and DEPT135 NMR spectra of 6 were nearly identical to those of 3, and HMQC and HMBC experiments confirmed that, with the exception of the atoms associated with the γ-lactone ring, 6 was identical with 3. Two olefinic methines were observed in the DEPT135 NMR spectrum. One of these was assigned as the C-3 methine (δC 120.7), and this correlated with H-3 in the HMQC spectrum. The 13C NMR signal at δC 143.7 exhibited an apparent correlation with the residual solvent peak at δH 7.26 in the HMQC spectrum, and cross-peaks were observed in the 1H-1H COSY and HMBC NMR spectra between H-14 and the chloroform peak, indicating that a proton signal was overlapped with the residual solvent peak. After optimizing the window functions relating to Gaussian multiplication, two signals were resolved, revealing a methine at δH 7.28 (dd, J= 3.1, 1.6 Hz). HMBC NMR correlations indicated two- and three-bond coupling between H-15 and C-13 and C-14, and showed weak coupling with the carbonyl signal at δC 173.1 (C-16). The H-14 proton displayed an HMBC correlation with C-15.
Chemical shift considerations and HMBC NMR data were used to establish that the α,β-unsaturated lactone ring of 6 is attached to C-12 at the α-carbon, rather than at the β-carbon, thus differing in this respect from 1–5. In 6, the 1H NMR and 13C NMR chemical shifts associated with C-14 (δC 143.7) were significantly downfield (average Δδ = −34.3) and the 13C NMR resonance for C-13 (δC 138.3) was considerably upfield (average Δδ = +28.0) relative to the analogous signals in 1–5. These differences of NMR chemical shifts for 6 can be explained as resulting from the electron-withdrawing (deshielding) effect of the conjugated carbonyl on the β-carbon (and proton in 6). NMR data for several compounds having this structural feature exhibit similar 13C and 1H NMR patterns.37,38 Thus, the structure of 6 was elucidated as 12(S)-hydroxycleroda-3,13-dien-16,15-olide, assuming the same absolute configuration for OH-12 as determined for 1 and 3 on biogenetic grounds.
The isolates obtained in this investigation of C. americana were tested for cytotoxicity in a panel of human cancer cell lines. Cytotoxic activity (ED50 < 5 μg/mL) was observed in at least one cell line for 1, 4, 6, genkwanin, 16ξ-hydroxycleroda-3,13-dien-15,16-olide, and 2-formyl-16ξ-hydroxy-3-A-norcleroda-2,13-dien-15,16-olide (Table 1). A structure-activity relationship trend was observed for these clerodane diterpene isolates, in which compounds lacking a free hydroxy group at the 16-position were less cytotoxic than compounds with a γ-hydroxy group. The activity of the 3β-hydroxy-4(18)-exomethylene analogs was weaker compared with the Δ3(4)-clerodane diterpenes. Indeed, 5, which lacks an OH-16 group, was not active in the cell line panel. These trends suggest that the γ-OH in the α,β-unsaturated γ-lactone ring structure is necessary for activity, but that the structure of the decalin ring system also contributes to the cytotoxic potency.
Table 1.
Cytotoxicity Data for Isolates from C. americana in Several Human Cell Linesa
| Compound Code | Cell Lineb,c |
|||||
|---|---|---|---|---|---|---|
| MCF-7 | Lu1 | Col2 | LNCaP | hTERT-RPE1 | HUVEC | |
| 1 | - | 2.4 | 2.3 | 4.1 | 1.9 | 1.8 |
| 4 | 2.7 | 2.6 | - | 2.5 | - | 1.2 |
| 6 | 2.8 | 2.9 | - | 3.3 | - | - |
| Genkwanin | - | >20 | >20 | >20 | 3.9 | >20 |
| 16ξ-Hydroxycleroda-3,13-dien-15,16-olide | 3.5 | 3.7 | - | 3.3 | - | 4.1 |
| 2-Formyl-16ξ-hydroxy-3-A-norcleroda-2,13-dien-15,16-olide | 3.9 | 8.7 | 9.0 | 4.5 | 1.9 | 9.8 |
Compounds 2, 3, and 5, and the known compounds calliterpenone, 3(3,16ξ-dihydroxycleroda-4(18),13-dien-15,16-olide, euscaphic acid, 5-hydroxy-7,4′-dimethoxyflavone, and salvigenin were inactive in the cell lines tested.
Cell lines: MCF-7 = breast cancer; Lu1 = lung cancer; Col2 = colon cancer; LNCaP = hormone-dependent prostate cancer; hTERT-RPE1 = human telomerase reverse-transcriptase retinal pigment epithelium; HUVEC = human umbilical vein epithelial cells.
ED50 values are given in μg/mL, and values <5 μg/mL are considered to be active.
Owing to its initial cytotoxicity and the relatively large amount isolated, compound 1 was tested in vivo against LNCaP, Lul, and MCF-7 cells in a hollow fiber antitumor model39,40 at 6.25, 12.5, 25, and 50 mg/kg in mice. No activity was observed at either the i.p. or s.c. sites in any of the three cell lines, even at the highest dose tested (50 mg/kg), and two of the three mice died at each the two highest doses (25 mg/kg and 50 mg/kg) (data not shown).
The co-occurrence of clerodane diterpenes and methoxylated flavones was recently observed in an investigation of the cytotoxic constituents from Premna tomentosa,41 and the co-occurrence of these two structural classes in Callicarpa americana lends support to the previously noted close relationship with the genus Premna4 Furthermore, the occurrence of salvigenin, a 6-methoxylated flavonoid suggests a close alliance with Lamiaceae, as flavonoid substitution at C-6 is usually associated with members of this family.19,42 This is in agreement with the current taxonomic thinking that Callicarpa and several other closely related genera should be included in the Lamiaceae, rather than in the Verbenaceae (see disscussion above).43
Experimental Section
General Experimental Procedures
Melting points of the isolates were determined on a Thomas Hoover capillary melting point apparatus (Unimelt, Philadelphia, PA) and are uncorrected. Optical rotations of the isolates were measured using a Perkin-Elmer Model 241 polarimeter (Germany). Ultraviolet (UV) absorption spectra were recorded using a Beckman DU 640 spectrophotometer (Beckman Instruments, Fullerton, CA). Optical rotatory dispersion (ORD) measurements (for calliterpenone) were performed using a JASCO J-810 spectropolarimeter (Tokyo, Japan). Infrared absorption spectra (IR) of the isolates were recorded on a Nicolet Protegé 460 FTIR spectrophotometer (Thermo, Waltham, MA). The NMR spectra were obtained on Bruker Avance DPX-300 (300 MHz), DPX-360 (360 MHz), DRX-400 (400 MHz), and DRX-600 (600 MHz) NMR spectrometers. Spectroscopic-grade deuterated solvents (Sigma-Aldrich, St. Louis, MO) were used for all NMR experiments, and tetramethylsilane (TMS) was used as internal standard for all samples in CDCl3, and the residual solvent peak was used to calibrate samples in pyridine-d5 and MeOD (δH 8.71 and 3.31, respectively, for the corresponding solvent peaks). Spectrum (1H NMR) optimization for 6 was carried out using Bruker-provided Gaussian multiplication window function used LB = −0.98 and GB = 0.145. Low- and high-resolution (accurate mass) electrospray-ionization time-of-flight (ESITOF) mass spectra were recorded on a Micromass LCT (Milford, MA). Chemical ionization (CI), electron impact (EI), and fast-atom bombardment (FAB) mass spectra were obtained on a Finnigan MAT 90 mass spectrometer (Finnigan A.G., Bremen, Germany). Silica gel (Merck, Darmstadt, Germany) and polystyrene polymer (styrene-divinylbenzene; MCI Gel®, Sigma-Aldrich) were used for low-pressure chromatography. HPLC was carried out on a Waters HPLC system with two Waters 515 pumps and a Waters 2487 dual wavelength detector (Waters, Milford, MA), using an octadecylsilane (ODS) column (Waters, Sunfire 19 × 150 mm, 5 μm) and HPLC-grade solvents at a flow rate of 8 mL/min. Thin later chromatography (TLC) was performed on silica gel 60 F254 on glass plates (Merck) using various solvent systems.
Plant Material
The fruiting branches of Callicarpa americana L. (Lamiaceae) were collected from a forested area of Matheson Hammock, near Miami, Florida (permit #0014 from Natural Areas Management, Miami-Dade County Park and Recreation Department), and the plant material was air dried prior to milling. Voucher specimens (TL-71 and TL-93) were deposited at the Fairchild Tropical Garden herbarium in Florida and the herbarium of The Field Museum of Natural History in Chicago, IL. Assistance in identifying the plant material was provided by Mr. Roger Hammer, Director of Castellow Hammock Preserve and Nature Center, Miami, FL.
Extraction and Isolation
The milled plant material (634 g) was extracted with MeOH (overnight, 3 × 2.5 L, at room temp.), and the concentrated MeOH extract was suspended in 90% MeOH (500 mL). A series of previously described liquid/liquid partitioning steps13,14 (petroleum ether-90% MeOH; CHCl3-20% MeOH; CHCl3-aqueous 1% NaCl) were used to prepare a CHCl3 extract (15.5 g), defatted and essentially free of tannins. This CHCl3 extract was chromatographed over Si gel, eluting with a stepped gradient of MeOH in CHCl3, resulting in nine major fractions F01-F09. Of these, fractions F03-F06 were active against LNCaP cells.
Fraction F03 was chromatographed over MCI gel (eluting with 100% MeOH) resulting in six pooled fractions (F20-F25). F20 was chromatographed over Si gel (eluting with hexane/acetone), resulting in six combined fractions (F26-31). F26 (90% hexane) was separated by reversed-phase HPLC (80% MeOH) yielding 2, 3, 4, and 6 (tR 28.9, 25.3, 26.6, and 23.8 min, respectively), as well as 16ξ-hydroxycleroda-3,13-dien-15,16-olide (tR 31.1 min). 5-Hydroxy-7,4′-dimethoxyflavone was purified from F28 (hexane-acetone, 4:1) by preparative TLC (hexane-EtOAc-acetone, 7:3:1). F31 was chromatographed over silica gel (eruting with hexane-EtOAc, 1:1), and 5 and 30,16ξ-dihydroxycleroda-4(18),13-dien-15,16-olide were isolated by further purification of the subfractions obtained. 2-Formyl-16ξ-hydroxy-A-norcleroda-2,13-dien-15,16-olide was obtained by semi-preparative HPLC of F22 (77% MeOH, tR 10.7 min).
Fractions F04 and F05 were combined, and the residue (7.8 g) was chromatographed over coarse Si gel, eluting with a stepped gradient of increasing polarity from 100% CHCl3 to 40% MeOH in CHCl3, affording 64 fractions of 200 mL each, which were combined according to TLC profiles into ten pooled fractions (F10-F19). Of these, F14 yielded genkwanin, and calliterpenone crystallized from F15 on standing. Repeated Si gel chromatography of F15 resulted in the isolation of a fraction containing impure 1, which was further purified by re versed-phase HPLC (70% CHCN, tR 27 min). Calliterpenone, euscaphic acid, genkwanin, and salvigenin were also obtained from the combined fractions F04 and F05.
Fraction F06 (1.0 g) was chromatographed over MCI gel, but the resulting pooled fractions were essentially inactive, and further purification of the active principles was not performed.
12(S),16ξ-Dihydroxycleroda-3,13-dien-15,16-olide (1)
[α]20D -67 (c 0.18, MeOH); UV (MeOH) λmax; (log ε) 209 (3.17) nm; IR (dried film) υmax 3394 (broad), 2926, 1748, 1133, 949 cm−1; 1H NMR (CDCl3, 360 MHz) δ 6.13 (1H, br, H-16), 6.01 (1H, s, H-14), 5.19 (1H, s, H-3), 4.72 (1H, d, J = 8.0 Hz, H-12), 2.10 (2H, br, H-2), 1.87 (1H, m, H-11a), 1.77 (1H, m, H-1a), 1.74 (1H, m, H-6a), 1.60 (1H, m, H-10), 1.58 (3H, s, H-18), 1.50 (1H, m, H-8), 1.49 (1H, m, H-11b), 1.48 (1H, m, H-1b), 1.39–1.51 (2H, m, H-7), 1.22 (1H, m, H-6b), 1.01 (3H, s, H-19), 0.80 (3H, d, J = 6.1 Hz, H-17), 0.75 (3H, s, H-20); 13C NMR (CDCl3, 90 MHz) δ 174.3 (C, C-13), 172.0 (C, C-15), 144.2 (C, C-4), 120.8 (CH, C-3), 116.8 (CH, C-14), 98.4 (CH, C-16), 64.6 (CH, C-12), 47.1 (CH, C-10), 43.0 (CH2, C-11), 39.8 (C, C-9), 38.3 (C, C-5), 36.9 (CH, C-8), 36.5 (CH2, C-6), 27.5 (CH2, C-7), 26.6 (CH2, C-2), 20.1 (CH3, C-19), 19.2 (CH2, C-1), 18.1 (CH3, C-18), 17.6 (CH3, C-20), 16.1 (CH3, C-17); ESIMS m/z 333 [M-H]−; HRCIMS m/z 352.2475 [M+NH4]+ (calcd 352.2488 for C20H34O4). 1a-12-(R)-MTPA ester 1H NMR (pyridine-d5, 400 MHz) δ 7.357 (1H, s, H-16), 6.407 (1H, s, H-14), 6.219 (1H, dd, J = 7.2, 4.0 Hz, H-12), 5.173 (1H, s, H-3), 2.249 (1H, dd, J = 16.5, 7.2 Hz, H-11a), 2.180 (3H, s, OAc-16), 1.948 (1H, dd, J = 16.5, 4.0 Hz, H-11b), 1.538 (3H, s, H-18), 0.938 (3H, s, H-19), 0.807 (3H, d, J = 6.5 Hz H-17), 0.704 (3H, s, H-20). 1a-12-(S)-MTPA ester 1H NMR (pyridine-d5, 400 MHz) S 7.533 (1H, s, H-16), 6.719 (1H, s, H-14), 6.192 (1H, dd, J = 7.5, 3.9 Hz, H-12), 5.024 (1H, s, H-3), 2.265 (1H, dd, J = 16.4, 7.5 Hz, H-11a), 2.174 (3H, s, OAc-16), 1.909 (1H, dd, J = 16.4, 3.9 Hz, H-11b), 1.465 (3H, s, H-18), 0.895 (3H, s, H-19), 0.768 (3H, d, J = 6.4 Hz H-17), 0.657 (3H, s, H-20).
12(S)-Hydroxy-16ξ-methoxycleroda-3,13-dien-15,16-olide (2)
[α]20D -94 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 264 (3.41), 204 (4.23) nm; IR (dried film) υmax 3442 (broad), 2955, 2928, 2353 1756 cm−1; 1H NMR (CDCl3, 300 MHz) δH 6.07 (1H, d, J = 0.94 Hz, H-14), 5.71 (1H, d, J = 0.94 Hz, H-16), 5.20 (1H, br s, H-3), 4.71 (1H, br d, J = 8.0 Hz, H-12), 3.59 (3H, s, MeO-16), 1.59 (3H, br s, H-18), 1.53 (1H, m, H-8), 1.02 (3H, s, H-19), 0.81 (3H, d, J = 6.1 Hz, H-17), 0.76 (3H, s, H-20); 13C NMR (CDCl3, 75 MHz) δC 171.2 (C, C-13), 170.0 (C, C-15), 144.1 (C, C-4), 120.7 (CH, C-3), 117.5 (CH, C-14), 103.1 (CH, C-16), 65.0. (CH, C-12), 57.5 (CH3, MeO-16), 47.3 (CH, C-10), 43.4 (CH2, C-11), 39.8 (C, C-9), 38.4 (C, C-5), 37.0 (CH, C-8), 36.5 (CH2, C-6), 27.5 (CH2, C-7), 26.7 (CH2, C-2), 20.1 (CH3, C-19), 19.2 (CH2, C-1), 18.0 (CH3, C-18), 17.6 (CH3, C-17), 16.0 (CH3, C-20); ESITOFMS m/z 371.2204 [M + Na]+ (calcd 371.2198 for C21H32O4Na).
12(S)-Hydroxycleroda-3,13-dien-15,16-olide (3)
[α]20D -38 (c 0.20, MeOH); UV (MeOH) λmax (&log ε) 206 (4.22) nm; IR (dried film) υmax 3426 (broad), 2958, 2922, 1743 cm−1; 1HNMR (MeOD, 600 MHz) 85.93 (1H, dt, J = 1.8, 1.2 Hz, H-14), 5.17 (1H, br s, H-3), 4.96 (2H, d, J = 1.8 Hz, H-16), 4.71 (1H, dd, J = 8.4, 1.8 Hz, H-12), 2.08 (1H, ddddq, J = 17.6, 9.3, 6.8, 2.9, 1.5 Hz, H-2β), 1.95 (1H, dddq, J = 17.6, 4.6, 4.6, 1.4 Hz, H-2α), 1.89(1H, dd, 7=16.4, 8.4 Hz, H-11a), 1.83 (1H, dd, J = 12.5, 6.8 Hz, H-1β), 1.74 (1H, ddd, J = 12.8, 3.2, 3.2 Hz, H-6α), 1.72 (1H, dd, 11.8, 1.5 Hz, H-10), 1.60 (1H, dqd, 11.2, 6.7, 4.6 Hz, H-8), 1.58 (3H, m, H-18), 1.53 (1H, dd, J = 15.9, 1.8 Hz, H-11b), 1.47 (2H, m, H-7), 1.44 (1H, m, H-1α), 1.24 (1H, ddd, J = 12.8, 12.8, 4.5 Hz, H-6p), 1.04 (3H, s, H-19), 0.83 (3H, d, J = 6.7 Hz, H-17), 0.78 (3H, s, H-20); 13C NMR (MeOD, 75 MHz) δ 179.0 (C, C-13), 176.5 (C, C-15), 144.9 (C, C-4), 122.0 (CH, C-3), 114.1 (CH, C-14), 72.9 (CH2, C-16), 65.8 (C, C-12), 48.3 (CH, C-10), 45.1 (CH2, C-11), 40.9 (C, C-9), 39.5 (C, C-5), 38.2 (CH, C-8), 37.9 (CH2, C-6), 28.7 (CH2, C-7), 27.6 (CH2, C-2), 20.7 (CH3, C-19), 20.3 (CH2, C-1), 18.3 (CH3, C-18), 18.2 (CH3, C-20), 16.5 (CH3, C-17); ESITOFMS m/z 341.2085 [M + Na]+ (calcd 341.2093 for C20H30O3Na).
16ξ-Hydroxycleroda-3,11(E),13-trien-15,16-olide (4)
[α]20D -56 (c 0.085, MeOH); UV (MeOH) λmax (&log ε) 267 (4.47) nm; IR (dried film) υmax 3426 (broad), 2958, 2922, 1743 cm−1; 1H NMR (pyridine-rf5, 300 MHz) δ 6.74 (1H, s, H-16), 6.52 (1H, d, J = 16.4 Hz, H-11), 6.39 (1H, d, J = 16.4 Hz, H-12), 6.22 (1H, s, H-14), 5.22 (1H, s, H-3), 1.89 (2H, m, H-2), 1.58 (3H, brs, H-18), 1.42 (2H, m, H-1), 1.37 (1H, H-8), 1.37 (2H, m, H-7), 1.30–1.43 (1H, m, H-10), 0.96 (3H, s, H-19), 0.86 (3H, s, H-20), 0.72 (3H, d, J = 4.5 Hz, H-17); 13C NMR (pyridine-rf5, 75 MHz) δ 171.8 (C, C-15), 163.3 (C, C-13), 156.1 (CH, C-11), 143.6 (C, C-4), 121.0 (CH, C-3), 119.9 (CH, C-12), 115.4 (CH, C-14), 99.3 (CH, C-16), 50.3 (CH, C-10), 45.2 (C, C-9), 41.2 (CH, C-8), 37.6 (CH2, C-6), 36.6 (C, C-5), 27.0 (CH2, C-2), 26.9 (CH2, C-7), 20.4 (CH2, C-1), 19.9 (CH3, C-19), 18.1 (CH3, C-18), 17.0 (CH3, C-17), 12.1 (CH3, C-20); 1H NMR (MeOD, 300 MHz) 8 6.29 (1H, s, H-11), 6.29 (1H, s, H-12), 6.27 (1H, s, H-16), 5.93 (1H, s, H-14), 5.18 (1H, s, H-3), 1.81 (1H, m, H-2a), 1.60 (3H, brs, H-18), 1.44 (2H, m, H-1), 1.48 (1H, H-8), 1.52 (2H, m, H-7), 1.38 (1H, m, H-10), 1.06 (3H, s, H-19), 0.98 (3H, s, H-20), 0.76 (3H, d, J = 4.5 Hz, H-17); 13C NMR (MeOD, 75 MHz) 8 173.9 (C, C-15), 164.4 (C, C-13), 157.6 (CH, C-11), 144.8 (C, C-4), 121.8 (CH, C-3), 120.5 (CH, C-12), 115.7 (CH, C-14), 100.3 (CH, C-16), 51.8 (CH, C-10), 46.2 (C, C-9), 42.4 (CH, C-8), 38.6 (C, C-5), 36.6 (CH2, C-6), 27.9 (CH2, C-2), 27.8 (CH2, C-7), 21.3 (CH2, C-1), 20.3 (CH3, C-19), 18.2 (CH3, C-18), 17.2 (CH3, C-17), 12.5 (CH3, C-20); ESITOFMS 339.1941 [M + Na]+ (calcd 339.1936 for C20H28O3Na).
3beta;,12(S)-Dihydroxycleroda-4(18),13-dien-15,16-olide (5)
[α]20D +18 (c 0.13, MeOH); UV (MeOH) λmax (&log ε) 207 (4.10) nm; IR (dried film) υmax 3400 (broad), 2920, 2850, 1746, 1028 cm−1; 1H NMR (CDCl3, 300 MHz) 8 5.90 (1H, dd, J = 3.0, 1.7 Hz, H-14), 4.91 (1H, d, J = 1.2 Hz, H-18a), 4.86 (2H, dd, J = 1.9, 1.9 Hz, H-16), 4.73 (1H, br d, J = 8.0 Hz, H-12), 4.72 (1H, s, H-18b), 4.33 (1H, dd, J = 11.6, 5.4 Hz, H-3), 2.15–2.25 (2H, m, H-2), 1.92 (1H, m, H-1a), 1.88 (1H, dd, J = 15.6, 8.0 Hz, H-11a), 1.58 (1H, m, H-6a), 1.54 (1H, m, H-8), 1.54 (2H, m, H-7), 1.54 (1H, H-lb), 1.47 (1H, dd, 15.6, 1.1 Hz, H-11b), 1.38 (1H, dd, J = 12.1, 2.1 Hz, H-10), 1.18 (1H, dd, 12.8, 4.3 Hz, H6b), 1.05 (3H, s, H-19), 0.80 (3H, d, J = 5.6 Hz, H-17), 0.76 (3H, s, H-20); 13C NMR (CDCl3, 75 MHz) δ 174.3 (C, C-13), 173.6 (C, C-15), 162.0 (C, C-4), 114.2 (CH, C-14), 99.6 (CH2, C-18), 70.9 (CH2, C-16), 69.7 (CH, C-3), 65.2 (CH, C-12), 49.2 (CH, C-10), 44.1 (CH2, C-11), 40.4 (C, C-9), 40.2 (C, C-5), 37.4 (CH, C-8), 37.31* (CH2, C-2), 37.30* (CH2, C-6), 27.2 (CH2, C-7), 21.24 (CH3, C-19), 21.16 (CH2, C-1), 17.6 (CH3, C-20), 16.0 (CH3, C-17) (* these resonances are interchangeable); ESITOFMS m/z 357.2051 [M + Na]+ (calcd 357.2042 for C20H30O4Na).
12(S)-Hydroxycleroda-3,13-dien-16,15-olide (6)
[α]20D -67 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 209 (3.87) nm; IR (dried film) υmax 3436 (broad), 2955, 2928, 1744 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.28 (1H, dd, J = 3.1, 1.6 Hz, H-14), 5.19 (1H, br s, H-3), 4.82 (2H, dd, J = 1.7, 1.7 Hz, H-15), 4.69 (1H, br d, J = 8.4 Hz, C-12), 2.05 (2H, m, H-2), 1.87 (1H, dd, J = 15.6, 8.6 Hz, H-11a), 1.80 (1H, m, H-1a), 1.73 (1H, m, H-6a), 1.68 (1H, m, H-10), 1.67 (1H, dd, J = 15.6, 2.3 Hz, H-11b), 1.62 (1H, m, H-8), 1.59 (3H, dd, J = 3.4, 1.9 Hz, H-18), 1.47 (1H, d, J = 17.0 Hz, H-1b), 1.45 (2H, m, H-7), 1.22 (1H, m, H-6b), 1.01 (3H, s, H-19), 0.83 (3H, d, J = 6.6 Hz, H-17), 0.75 (3H, s, H-20); 13C NMR (CDC13, 75 MHz) δ 173.1 (C, C-16), 144.2 (C, C-4), 143.7 (CH, C-14), 138.3 (C, C-13), 120.7 (CH, C-3), 70.3 (CH2, C-15), 63.8 (CH, C-12), 47.4 (CH, C-10), 43.3 (CH2, C-11), 39.7 (C, C-9), 38.4 (C, C-5), 37.1 (CH, C-8), 36.7 (CH2, C-6), 27.6 (CH2, C-7), 26.7 (CH2, C-2), 20.2 (CH3, C-19), 19.2 (CH2, C-1), 18.0 (CH3, C-18), 17.8 (CH3, C-20), 16.1 (CH3, C-17); ESITOFMS m/z 341.2099 [M + Na]+ (calcd 341.2093 for C20H30O3Na).
Determination of Absolute Configuration of OH-12 in 1 and 3
Prior to the esterification of OH-12 with MTPA chloride, OH-16 of 1 was acetylated using pyridine and acetic anhydride, with reaction conditions (0 °C, 5 min) that favored the acetylation of the less hindered hydroxyl group (OH-16) rather than the more sterically crowded OH-12. After HPLC separation of the mixture of epimers at C-16 (present in approximately a 2:3 ratio), the more abundant of the epimers 1a was selected for acylation using the S- and R-MTPA chloride (separately). The reactions were carried out in deuterated pyridine under nitrogen, using NMR tubes as the reaction vessel, as previously described.44 Compound 3 was reacted in the same manner with MTPA chloride, but with no need for prior acetylation. 39,40
Bioassay Evaluation
The isolates obtained were evaluated for cytotoxicity against a panel of human cancer cell lines according to established protocols.45,46 The hollow fiber assay was performed following an established protocol,39 with minor modification.40 Briefly, the hollow fiber assay was performed using NCr nu/nu athymic mice (five to six-weeks-old), obtained from the National Cancer Institute-Frederick Cancer Research Facility (Frederick, MD). LNCaP, Lu1, and MCF-7 cells were implanted at intraperitoneal and subcutaneous sites (i.p. and s.c.) enclosed in sealed, semi-porous hollow fibers. The test compound 1 was administered i.p. at 6.25, 12.5, 25, and 50 mg/kg (three mice per dose group). Each mouse received each of the three cell lines implanted at both physiological sites. The mice were handled and cared for humanely, following a protocol (ACC No. 01-124) approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago.
Acknowledgments
The research described herein was supported by grant U19-CA52956, funded by the National Cancer Institute, NIH, Bethesda, MD. We thank the staff at the Miami-Dade County Parks and Recreation Department for granting permission to collect the plant material used in this investigation, and we also thank Mr. Roger Hammer, Director of the Castellow Hammock Preserve and Nature Center, Miami, FL for assistance in plant identification. Mass spectra were obtained and processed by Dr. Keith Fagerquist, Department of Chemistry, University of Minnesota, Dr. John L. Anderson, Research Resources Center, UIC, and Dr. Kari Green-Church, Campus Chemical Instrument Center, OSU. Assistance in acquiring spectra on the Bruker Avance DRX-600 NMR was provided by Dr. Charles E. Cottrell, Campus Chemical Instrument Center, OSU. Dr. Raymond W. Doskotch is gratefully acknowledged for helpful discussions and his insight regarding the 600 MHz NMR data for compound 3.
Footnotes
Dedicated to the late Dr. Kenneth L. Rinehart of the University of Illinois at Urbana-Champaign for his pioneering work on bioactive natural products.
References and Notes
- 1.The results presented here have appeared in part in: Jones WPA. Pharmacognostic Investigation of Callicarpa Americana for Potential Anticancer Agents. University of Illinois at Chicago; Chicago, IL: 2006. Ph.D. Dissertation.
- 2.Mabberly DJ. The Plant-Book: A Portable Dictionary of the Vascular Plants. 2. Cambridge University Press; Cambridge, UK: 1997. p. 69. [Google Scholar]
- 3.Thorne RF. Bot Rev. 1992;58:225–350. [Google Scholar]
- 4.Zomlefer WB. Guide to Flowering Plant Families. University of North Carolina Press; Chapel Hill: 1994. p. 430. [Google Scholar]
- 5.Cantino PD. Ann Missouri Bot Card. 1992;79:361–379. [Google Scholar]
- 6.Crellin JK, Philpott JA. Reference Guide to Medicinal Plants: Herbal Medicine Past and Present. Duke University Press; Durham, NC: 1997. p. 551. [Google Scholar]
- 7.Rafinesque CS. Medical Flora; or Manual of the Medical Botany of the United States of North America. Atkinson and Alexander; Philadelphia: 1830. p. 276. [Google Scholar]
- 8.Moerman DE. American Medical Ethnobotany. A Reference Dictionary. Garland Publishing; New York: 1977. p. 527. [Google Scholar]
- 9.Taylor LA. Plants Used as Curatives by Certain Southeastern Tribes. Botanical Museum, Harvard University; Cambridge, MA: 1940. p. 88. [Google Scholar]
- 10.Hartwell JL. Plants Used Against Cancer. A Survey. Quarterman; Lawrence, MA: 1982. p. 710. [Google Scholar]
- 11.Tellez MR, Dayan FE, Schrader KK, Wedge DE, Duke SOJ. Agric Food Chem. 2000;48:3008–3012. doi: 10.1021/jf991026g. [DOI] [PubMed] [Google Scholar]
- 12.Cantrell CL, Klun JA, Bryson CT, Kobaisy M, Duke SOJ. Agric Food Chem. 2005;53:5948–5953. doi: 10.1021/jf0509308. [DOI] [PubMed] [Google Scholar]
- 13.Kinghorn AD, Farnsworth NR, Soejarto DD, Cordell GA, Swanson SM, Pezzuto JM, Wani MC, Wall ME, Oberlies NH, Kroll DJ, Kramer RA, Rose WC, Vite GD, Fairchild CR, Peterson RW, Wild R. Pharm Biol. 2003;41(Suppl):53–67. [Google Scholar]
- 14.Wall ME, Wani MC, Brown DM, Fullas F, Oswald JB, Josephson FF, Thornton NM, Pezzuto JM, Beecher CWW, Farnsworth NR, Cordell GA, Kinghorn AD. Phytomedicine. 1996;3:281–285. doi: 10.1016/S0944-7113(96)80067-5. [DOI] [PubMed] [Google Scholar]
- 15.Chatterjee A, Desmukh SK, Chandrasekharan S. Tetrahedron. 1972;28:4319–4323. [Google Scholar]
- 16.Agrawal PK, Singh AK, Bhakuni RS. Indian J Chem B. 1996;35B:803–805. [Google Scholar]
- 17.Kuang HX, Kasai R, Ohtani K, Liu ZS, Yuan CS, Tanaka O. Chem Pharm Bull. 1989;37:2232–2233. doi: 10.1248/cpb.37.2232. [DOI] [PubMed] [Google Scholar]
- 18.Seto T, Tanaka T, Tanaka O, Naruhashi N. Phytochemistry. 1984;23:2829–2834. [Google Scholar]
- 19.Ulubelen A, Özttirk S, Isíldatici S. J Pharm Sci. 1968:1037–1038. doi: 10.1002/jps.2600570630. [DOI] [PubMed] [Google Scholar]
- 20.Wollenweber E, Wassum M. Tetrahedron Lett. 1972:797–800. [Google Scholar]
- 21.Brieskorn CH, Biechele W. Tetrahedron Lett. 1969:2603–2605. doi: 10.1016/s0040-4039(01)88222-8. [DOI] [PubMed] [Google Scholar]
- 22.Ma X, Lee IS, Chai HB, Zaw K, Farnsworth NR, Soejarto DD, Cordell GA, Pezzuto JM, Kinghorn AD. Phytochemistry. 1994;37:1659–1662. doi: 10.1016/s0031-9422(00)89587-4. [DOI] [PubMed] [Google Scholar]
- 23.Phadnis AP, Patwardhan SA, Dhaneshwar NN, Tavale SS, Guru Row TN. Phytochemistry. 1988;27:2899–2901. [Google Scholar]
- 24.Achari B, Chaudhuri C, Saha CR, Dutta PK, Padrashi SC. Phytochemistry. 1990;29:3671–3673. [Google Scholar]
- 25.Kijjoa A, Pinto MMM, Pinho PMM, Tantisewie B, Herz W. Phytochemistry. 1990;29:653–655. [Google Scholar]
- 26.Xu J, Harrison LJ, Vittal JJ, Xu YJ, Goh SHJ. Nat Prod. 2000;63:1062–1065. doi: 10.1021/np990584m. [DOI] [PubMed] [Google Scholar]
- 27.Seaman R, Bohlmann R, Zdero C, Mabry TJ. Diterpenes of Flowering Plants: Compositae (Asteraceae) Springer-Verlag; New York: 1990. p. 638. [Google Scholar]
- 28.Manabe S, Nishino C. Tetrahedron. 1986;42:3461–3470. [Google Scholar]
- 29.Nogueira RT, Shepherd GJ, Laverde A, Marsaioli AJ, Imamura PM. Phytochemistry. 2001;58:1153–1157. doi: 10.1016/s0031-9422(01)00303-x. [DOI] [PubMed] [Google Scholar]
- 30.Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519. [Google Scholar]
- 31.Sullivan GR, Dale JA, Mosher HS. J Org Chem. 1973;38:2143–2147. [Google Scholar]
- 32.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 33.Ohtani I, Kusumi T, Kashman Y, Kakisawa HJ. Org Chem. 1991;56:1296–1298. [Google Scholar]
- 34.Yamaguchi S, Yasuhara F, Kabuto K. Tetrahedron. 1976;32:1363–1367. [Google Scholar]
- 35.Silverstein RM, Webster FX. Spectrometric Identification of Organic Compounds. 6. John Wiley & Sons; New York: 1998. p. 482. [Google Scholar]
- 36.Demarco PV, Farkas E, Doddrell D, Mylari BL, Wenkert E. J Am Chem Soc. 1968;90:5480–5486. [Google Scholar]
- 37.Ahmad VU, Farooq U, Abbaskhan A, Hussain J, Abbasi HA, Nawaz SA, Choudhary MI. Helv Chim Acta. 2004;87:682–689. [Google Scholar]
- 38.Martin TS, Ohtani K, Kasai R, Yamasaki K. Phytochemistry. 1995;40:1729–1736. [Google Scholar]
- 39.Hollingshead MG, Alley MC, Camalier RF, Abbott BJ, Mayo JG, Malspeis L, Grever MR. Life Sci. 1995;57:131–141. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
- 40.Mi Q, Lantvit D, Reyes-Lim E, Chai HB, Zhao WM, Lee IS, Peraza-Sanchez S, Ngassapa O, Kardono LBS, Riswan S, Hollingshead MG, Mayo JG, Farnsworth NR, Cordell GA, Kinghorn AD, Pezzuto JMJ. Nat Prod. 2002;65:842–850. doi: 10.1021/np010322w. [DOI] [PubMed] [Google Scholar]
- 41.Chin YW, Jones WP, Mi Q, Rachman L, Riswan S, Kardono LBS, Chai HB, Farnsworth NR, Cordell GA, Swanson SM, Cassady JM, Kinghorn AD. Phytochemistry. 2006;67:1243–1248. doi: 10.1016/j.phytochem.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 42.Brieskorn CH, Biechele W. Tetrahedron Lett. 1969:2603–2605. doi: 10.1016/s0040-4039(01)88222-8. [DOI] [PubMed] [Google Scholar]
- 43.Cantino PD. In: Advances in Labiate Science. Harley RM, Reynolds T, editors. Royal Botanic Gardens; Kew: 1992. pp. 27–37. [Google Scholar]
- 44.Su BN, Park EJ, Mbwambo ZH, Santarsiero BD, Mesecar AD, Fong HHS, Pezzuto JM, Kinghorn AD. J Nat Prod. 2002;65:1278–1282. doi: 10.1021/np0202475. [DOI] [PubMed] [Google Scholar]
- 45.Likhitwitayawuid K, Angerhofer CK, Cordell GA, Pezzuto JM, Ruangrungsi N. J Nat Prod. 1993;56:30–38. doi: 10.1021/np50091a005. [DOI] [PubMed] [Google Scholar]
- 46.Seo EK, Kim NC, Mi Q, Chai H, Wall ME, Wani MC, Navarro HA, Burgess JP, Graham JG, Cabieses F, Tan GT, Farnsworth NR, Pezzuto JM, Kinghorn AD. J Nat Prod. 2001;64:1483–1485. doi: 10.1021/np0103158. [DOI] [PubMed] [Google Scholar]