

Abstract
Typical 2-Cys peroxiredoxins (Prxs) are ubiquitous peroxidases that are involved in peroxide scavenging and/or the regulation of peroxide signaling in eukaryotes. Despite their prevalence, very few Prxs have been reliably characterized in terms of their substrate specificity profile and redox potential even though these values are important for gaining insight into physiological function. Here, we present such studies focusing on Salmonella typhimurium alkyl hydroperoxide reductase C component (StAhpC), an enzyme that has proven to be an excellent prototype of this largest and most widespread class of Prxs that includes mammalian Prx I–Prx IV. The catalytic efficiencies of StAhpC (kcat/Km) are >107 M−1·s−1 for inorganic and primary hydroperoxide substrates and ≈100-fold less for tertiary hydroperoxides, with the difference being exclusively caused by changes in Km. The oxidative inactivation of AhpC through reaction with a second molecule of peroxide shows parallel substrate specificity. The midpoint reduction potential of StAhpC is determined to be −178 ± 0.4 mV, a value much higher than most other thiol-based redox proteins. The relevance of these results for our understanding of Prx and the physiological role of StAhpC is discussed.
Keywords: antioxidants, peroxidases, redox regulation, redox-active disulfide
Peroxiredoxins (Prxs) are a ubiquitous group of cysteine-based peroxidases that appear to have evolved from a thioredoxin (Trx)-like precursor (1–3). For the evolution of the peroxidase function, one of the catalytic Trx Cys residues was lost, and the other came to be located at the base of an active site pocket that includes an Arg, a Thr, and a Pro residue conserved in all Prxs (4, 5). This so-called peroxidatic Cys residue (CP) directly reduces the peroxide substrate and becomes oxidized to a sulfenic acid (Fig. 1). The fate of the CP-sulfenic acid differs in various types of Prx enzymes, either forming an intramolecular disulfide with another Cys residue (the resolving Cys; CR) or forming an intermolecular disulfide with a Cys in a redox donor such as Trx, glutaredoxin (Grx), or reduced glutathione (GSH) (5, 6).
Fig. 1.

Reaction cycle of 2-Cys Prxs. The peroxidatic Cys of the Prx is depicted as a thiol (SPH) or sulfenic acid (SPOH), or in a disulfide with the resolving Cys (SRH). Colors distinguish the Cys residues from different subunits of the dimer in the typical 2-Cys Prxs, and the striped bar represents the intersubunit disulfide bond (intrasubunit in the case of the atypical 2-Cys Prxs). The disulfide reductase system that regenerates the active Prx varies with the organism and specific Prx but is Trx reductase and Trx in many eukaryotic systems and AhpF in the bacterial AhpC system. The pathway in the pink box represents regulation by oxidative inactivation (toward the right) and reactivation of hyperoxidized 2-Cys Prxs via sulfiredoxins (Srx) (toward the left).
The most widely distributed and abundant types of Prxs are the typical 2-Cys Prxs. In mammalian cells they are on the order of 1% of the soluble protein (5, 7, 8). They occur as active octameric, decameric, or dodecameric rings (3) that in some cases dissociate into less active dimers during the catalytic cycle (9–11). Prokaryotic typical 2-Cys Prxs are thought to have a primary role as peroxide scavengers (12), but since the discovery that many eukaryotic Prxs have an evolutionarily selected sensitivity to inactivation by peroxide (13) increasing evidence has accumulated supporting the hypothesis that the primary role of these enzymes is to regulate peroxide signaling (for recent reviews see refs. 8, 14, and 15).
The abilities of Prxs to detoxify various peroxides and regulate peroxide signaling will be impacted by their substrate specificity and redox potential, yet these properties have been only poorly studied. To date, no crystal structures have been published of substrate or product complexes, although some insight into substrate binding has been recently derived from the fortuitous binding of benzoate in one Prx crystal structure (3, 16). Whereas Prxs have gained a reputation as broad specificity peroxidases, reducing hydrogen peroxide, small alkyl hydroperoxides, and also peroxynitrite and phospholipid-associated or free fatty acid hydroperoxides (4, 17), for the vast majority of Prxs, substrate specificity has been tested only with single concentrations of peroxide substrates and standard steady-state linked assays. In most of these studies the catalytic rates toward the smallest possible hydroperoxide substrate, H2O2, vary <10-fold from the values for the larger t-butyl hydroperoxide and cumene hydroperoxide substrates (18–25). Exceptions include Escherichia coli thiol peroxidase (Tpx; also known as p20) and a Toxoplasma gondii 2-Cys Prx, with 30- to 200-fold greater activity toward bulky substrates than toward H2O2 (26, 27). A problem with many of these studies, however, is that even large differences in kcat or Km values for given substrates may be missed, particularly if, as is commonly the case, the assay conditions are such that Prx rather than peroxide reduction is rate-limiting (see Fig. 1) (10, 28). Recently, we have developed an assay for the bacterial Prx Salmonella typhimurium alkyl hydroperoxide reductase C component (StAhpC) that overcomes this limitation (10) and determined a Km for H2O2 of 1.4 μM and a kcat/Km of 4 × 107 that is ≈100-fold higher than previous estimates for Prxs (4). With this assay, it is now possible to properly evaluate the specificity of AhpC for various hydroperoxide substrates.
To our knowledge, redox potentials have only been studied for plant Prxs, where 2-Cys Prxs and PrxQ from chloroplasts have been found to exhibit redox potentials between −288 and −325 mV (7, 29). These values are quite low compared with other thiol-based redox proteins that typically possess reduction potentials near −250 to −270 mV, appropriately higher than those of their upstream pyridine nucleotide donors (NADH or NADPH, at −320 mV), but lower than GSH (at −240 mV) (30, 31). As the plant Prxs may possess very specific functions associated with photosynthesis, they may not be particularly representative of the wide range of Prxs in various organisms with important functions in antioxidant defense and/or regulation of redox signaling.
Here, we present a characterization of the substrate specificity and redox potential of StAhpC, a well studied model bacterial 2-Cys Prx (10, 32). Our studies reveal much higher catalytic efficiencies for inorganic and primary hydroperoxide substrates compared with bulky, tertiary hydroperoxides. The better substrates also show a parallel enhanced potency for oxidative inactivation of the enzyme. In terms of midpoint reduction potential, StAhpC shows a value much higher than the common thiol-based redox proteins, proving that the low redox potentials of the chloroplast 2-Cys Prxs and PrxQ are not representative of all Prxs.
Results
Specificity of AhpC For Hydroperoxide Substrates.
To accurately measure enzymatic activity of StAhpC with various hydroperoxide substrates, we used a sensitive, fluorescence-based assay previously developed by us to eliminate the issue typically plaguing these assays, rate-limiting reduction during catalytic turnover that obscures the rapid step of peroxide reduction (33). With this assay, we can directly measure oxidation of the electron donor to AhpC during turnover assays with the peroxide. Thus, full assays, which would normally be comprised of NADH, AhpF, AhpC, and the peroxide substrate, are replaced by a more simplified set of components. Furthermore, full-length AhpF (521 aa), which shuttles electrons from NADH to AhpC through three redox centers (FAD and two redox disulfide centers) within tethered Trx reductase-like and Trx-like modules, can be truncated to express only the Trx-like N-terminal domain (NTD; 202 aa), and this domain, when prereduced, can provide electrons directly to AhpC (34). To provide for fluorescence-based monitoring of redox state of the NTD, a mutation to introduce a tryptophan residue just in front of the catalytic CXXC motif, S128W, was also introduced into the expression vector for the NTD, making this protein more like its Trx homologue (10). Fluorescence-based assays could then be carried out with this prereduced domain provided at several fixed low-μM concentrations, the AhpC enzyme at concentrations of 100 nM or less, and the hydroperoxide substrate added at various μM concentrations, allowing for even very low Km values for hydroperoxide substrates to be accurately measured (10).
In the present studies, this same assay was carried out with ethyl hydroperoxide, t-butyl hydroperoxide, and cumene hydroperoxide in addition to hydrogen peroxide to assess the effects on specific catalytic parameters of the larger size and various structural differences surrounding the hydroperoxide moiety that undergoes reduction during catalysis (Fig. 2). Cumene hydroperoxide and t-butyl hydroperoxide are often used as mimics of lipid hydroperoxides, although in reality the latter are generally long-chain, secondary hydroperoxides. Still, the relatively high hydrophobicity and multiple linkages to the carbon bearing the hydroperoxide seem to be reasonably representative in at least some cases of the true lipid substrates that are more difficult to study given solubility and critical micellar concentration issues that affect the physical state of the lipid hydroperoxide substrate (26, 35, 36).
Fig. 2.

Structures of the four hydroperoxide substrates for AhpC studied herein (Top and Middle) and an oxidized lipid substrate, a linoleic acid hydroperoxide (Bottom).
For each hydroperoxide substrate, all of the data from multiple determinations of the bisubstrate kinetic profiles were simultaneously fit to Eq. 1 to give the true kcat for turnover and the Km values:
for each of the substrates, S128W NTD and the hydroperoxide (two single sets shown in Fig. 3 as examples). A summary of the final, globally fitted parameters from all of these analyses is provided in Table 1. Most striking is the observation that, although kcat/Km for the peroxides varies by as much as 160-fold, neither the Km for the reductant nor the overall kcat change very much. Instead, it is specifically the Km for the hydroperoxide substrate, and thus perhaps the binding within the peroxidatic active site, that is most affected by structural differences among the hydroperoxides (Table 1).
Fig. 3.

Differential activities of AhpC with two hydroperoxide substrates. Peroxidase activity was measured by mixing S128W NTD (prereduced by DTT) and AhpC (50–200 nM) in 50 mM potassium phosphate pH 7.0, 0.5 mM EDTA, and 100 mM ammonium sulfate with various concentrations of peroxide substrate in a stopped-flow spectrophotometer at 25°C. The reaction was followed by monitoring the decrease in fluorescence of S128W NTD (the modified NTD of AhpF, the direct electron donor to AhpC) with excitation at 280 nm and emission >320 nm. Fixed concentrations of S128W NTD used over a range of ethyl hydroperoxide (A) and t-butyl hydroperoxide (B) concentrations were 2.5 μM (■), 5 μM (○), 10 μM (▴), 15 μM (□), 20 μM (◆), and 30 μM (◇).
Table 1.
Kinetic parameters for reaction of AhpC with various peroxide substrates
| Peroxide substrate | kcat, s−1 | KmROOH, μM | KmS128W, μM | kcat/Km, M−1·s−1 |
|---|---|---|---|---|
| HOOH | 52.4 ± 1.7 | 1.4 ± 0.1 | 5.9 ± 0.6 | 3.7 × 107 |
| Ethyl-OOH | 52.7 ± 1.3 | 4.5 ± 0.3 | 4.7 ± 0.3 | 1.2 × 107 |
| Cumene-OOH | 52.0 ± 1.8 | 107 ± 8 | 4.0 ± 0.3 | 4.9 × 105 |
| t-Butyl-OOH | 54.7 ± 1.0 | 238 ± 8 | 4.1 ± 0.2 | 2.3 × 105 |
Varying Sensitivity of AhpC Toward Turnover-Induced Inactivation by Different Hydroperoxide Substrates.
As we found that AhpC demonstrates higher activity with small hydroperoxides like ethyl hydroperoxide and H2O2 but significantly decreased catalytic efficiency with the bulky, tertiary hydroperoxides, we investigated the relative potency of each of these substrates in inactivating the enzyme during turnover. This characteristic of Prxs tends to be less pronounced for prokaryotic enzymes than for eukaryotic Prxs, but inactivation during turnover is still observed for AhpC at high H2O2 concentrations (10, 13).
Similar to the results from the turnover assays, ethyl hydroperoxide demonstrates about the same tendency to inactivate AhpC at high concentrations as does H2O2 (Fig. 4A). However, neither t-butyl hydroperoxide (Fig. 4B) nor cumene hydroperoxide (data not shown) demonstrate any observable inactivation of the enzyme, even when present at concentrations as high as 60 mM and when compared over the same number of turnovers with the two better substrates. For cumene hydroperoxide, the analysis was somewhat more troublesome because of the necessity to predilute this substrate into DMSO, an inhibitor of the enzymatic turnover. However, control assays demonstrated that all decreased activity observed at high cumene hydroperoxide concentrations was attributable to the DMSO present.
Fig. 4.

AhpC shows a susceptibility to inactivation by hydrogen peroxide and ethyl hydroperoxide, but not to t-butyl hydroperoxide and cumene hydroperoxide. NADPH oxidation was measured (by monitoring loss of 340-nm absorbance) at 25°C in the presence of 50 mM Hepes-NaOH, pH 7.0, with 1 mM EDTA and 0.1 M ammonium sulfate, and with 80 nM Trx reductase, 2.5 μM Trx, 6 μM AhpC and ethyl hydroperoxide (A) at 0 mM (●), 1 mM (■), 5 mM (□), 10 mM (▴), and 30 mM (◇). The same conditions were used to examine overoxidation with t-butyl hydroperoxide (B), except that concentrations of this substrate were at 0 mM (●), 2 mM (▾), 10 mM (▵), 20 mM (◆), and 60 mM (○). Results with hydrogen peroxide were very similar to those with ethyl hydroperoxide (A), whereas results with cumene hydroperoxide were quite similar to those with t-butyl hydroperoxide (B), except for issues with lower solubility and DMSO effects in the latter case.
Determination of the Redox Potential of AhpC.
The redox potential of AhpC was determined by direct protein–protein interactions with a protein of known redox potential, given the convenience and considerable advantages offered by this method over equilibration with GSH/GSH disulfide redox buffers (31, 37). Initial experiments to determine the redox potential of AhpC using direct protein–protein equilibration with E. coli Trx showed that AhpC was completely reduced by Trx, indicating that the redox potential of AhpC was at least ≈50 mV higher than that of Trx1 (−270 mV). After reoptimization of the HPLC method to separate oxidized and reduced forms of both AhpC and E. coli Grx1, we mixed reduced Grx1 and oxidized AhpC at pH 7 and room temperature, allowed the mixture to equilibrate for 6 h (an equilibration time that was verified to be sufficient in preliminary experiments), then separated and quantified the respective redox forms of the two proteins (Fig. 5). The reverse experiment, reduced AhpC mixed with oxidized Grx1, was also conducted. The redox potential was calculated by using a derivation of the Nernst equation (Eq. 2)
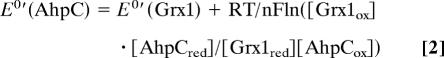 |
and a value of −233 mV for the Grx1 redox potential (30). From multiple independent experiments, the redox potential of wild-type AhpC was determined to be −178 ± 0.4 mV.
Fig. 5.

HPLC profile of the separation of reduced and oxidized forms of StAhpC and E. coli Grx1. Reduced and oxidized AhpC and Grx1 (each at 50 μM) were allowed to equilibrate for 6 h at room temperature in 100 mM potassium phosphate, pH 7.0, with 1 mM EDTA. The protein mixtures were quenched with phosphoric acid and immediately separated by HPLC as described in Experimental Procedures. The red trace illustrates the mixture after oxidized Grx1 was equilibrated with reduced AhpC and is labeled to show where the redox forms of each of the proteins elute. The blue, dotted trace is displaced by 5 min and 0.05 absorbance units and shows the mixture after reduced Grx1 was equilibrated with oxidized AhpC.
Discussion
This study explores the catalytically important attributes of substrate specificity and redox potential for the potent bacterial Prx AhpC, the primary scavenger of endogenously generated hydrogen peroxide in S. typhimurium and E. coli (12). Both attributes have been relatively overlooked or only superficially investigated in the burgeoning number of studies of the widely distributed Prx family of enzymes. As mentioned, only plant Prx redox potentials have been measured in vitro to date, and these included mainly 2-Cys Prxs and PrxQ from chloroplasts with very low redox potentials, in a range of −288 to −325 mV, which are, for the most part, even more negative than their putative reductants (e.g., Trx f at −290 mV and Trx m at −300 mV) (7, 38). As pointed out by Dietz and colleagues (7, 38), these plant Prxs are not likely to be “typical,” but rather are specialized to be regulated within the oxygenic environment of this photosynthetic organelle, with redox potentials lying between the range of redox potentials within which enzymes of the Calvin cycle are regulated (−280 to −305 mV) and the switching of the malate valve for export of excess reducing equivalents to the cytosol (−330 mV).
Our data pinning the redox potential of AhpC at −178 mV (Fig. 5) is a unique finding in the Prx field. Its significant difference compared with the values measured for chloroplast Prxs supports the notion that the redox potentials of those Prxs are specialized for regulation in the environment of the chloroplast. This value for AhpC, which is high in comparison with many redox disulfide-containing proteins, differs from an earlier in vivo estimate of around −250 mV that was based on the relative amounts of the thiol and disulfide forms of AhpC as measured in cell extracts of acid-quenched E. coli cultures and the assumption that AhpC is in perfect equilibrium with the GSH/oxidized GSH (GSSG) redox pair. Because AhpC is reduced in vivo by AhpF and/or Trx-linked pathways, we suggest that the discrepancy may exist because AhpC is not generally in a tightly linked equilibrium with the GSH/GSSG pair and the various intracellular redox buffers are not necessarily in equilibrium in vivo (39). Furthermore, the ratio of reduced to oxidized AhpC subunits may also be influenced by the presence of oxidants in this complex cellular system at the time of lysis and labeling.
In contrast to the very low redox potential for chloroplast Prxs, the relatively high redox potential of AhpC appears to make it well suited to act as an efficient scavenger of hydrogen peroxide under most conditions of the cell, even if subjected to oxidative stress. Indeed, the peroxide-sensing transcriptional regulator in E. coli, OxyR, has a redox potential slightly more negative than that of AhpC, at −185 mV, and has been shown to be activated not just by hydrogen peroxide sensing, but also through alterations in the thiol-disulfide redox status in mutant bacteria defective in the cellular disulfide-reducing systems (40). Thus, normally both of these highly peroxide-sensitive proteins, AhpC and OxyR, would be maintained in a reduced state given a typical cellular redox potential (based on Trx reduction status) of −260 to −298 mV for E. coli (40–42), but as a cell becomes increasingly oxidatively stressed, OxyR would be oxidized to form a disulfide bond by equilibration with the cellular redox potential and thus activated to mobilize a cellular response at a potential where AhpC would remain reduced and active (between −185 and −178 mV). It will be of great interest to discover the ranges of redox potentials of the mammalian typical 2-Cys Prxs, Prx I through Prx IV, and other Prxs that may serve roles as redox sensors, and see what insight that gives into their physiological roles. We note that, with a standard midpoint reduction potential (E°) of +1.77 V for the two-electron reduction of hydrogen peroxide to water (43), the thermodynamic driving force for the reduction of hydrogen peroxide by thiol-containing proteins will always be highly favorable.
With regard to substrate specificity, the results with our assay show that AhpC displays standard Michaelis–Menten kinetics with kcat ≈53 s−1 virtually independent of the type of hydroperoxide substrate and Km values ≈100-fold larger for t-butyl and cumene hydroperoxide compared with hydrogen peroxide and ethyl hydroperoxide. These results contrast with our earlier report (44) that AhpC has little discrimination between hydrogen peroxide and cumene hydroperoxide and underscore the concern raised in the introduction that many published substrate specificity studies of Prxs may be unreliable, especially those that rely only on single concentrations of both reductant and hydroperoxide to assess a catalytic rate across a panel of various peroxide substrates. In the case of AhpC, even though the reduction of AhpC by AhpF is rather efficient, with a kcat/Km value (varying AhpC to obtain the Km for that protein as a substrate of AhpF) of ≈1 × 107 M−1·s−1 (33), the typical assay conditions we used were incapable of discriminating between good and poor substrates because the reduction of AhpC by AhpF was still rate limiting. In addition, the early assays used high concentrations (1 mM) of the peroxide substrates and were not useful in assessing rates at the extremely low concentrations that would be needed to measure the very low μM Km values for the small peroxides (44). Only with the advent of the stopped-flow assay (10) used here were we able to achieve low enough concentrations of all of the components and acquire rapid enough assay data to determine the StAhpC Km of 1.4 μM for H2O2. This effect can also be seen from the data gathered for our overoxidation sensitivity studies conducted herein (Fig. 4); the initial rate data obtained at 100–200 μM hydroperoxide substrate gave turnover rates of ≈0.25 μM NADH oxidized s−1 (μM AhpC)−1 for both ethyl hydroperoxide and t-butyl hydroperoxide, even though we know those to be very distinctly different substrates from the specificity studies. It is clear, then, that large differences in the specificity constant, kcat/Km, for different substrates (as seen here) are often missed when substrate specificity is evaluated at a single concentration of substrate and/or with an assay limited by reduction rates.
It might be thought that the use of high concentrations of a nonphysiological reductant would be able to overcome this problem, but this can also give misleading results. For instance, with 1,4-dithiothreitol (DTT) as reductant, rates of Tpx with μM concentrations of H2O2 and t-butyl hydroperoxide substrates were about comparable to one another (and, surprisingly, much faster than those observed for AhpC in the same assays) (45), even though earlier studies using 10 μM Trx indicated apparent Km values of 648 and 66.6 μM for H2O2 and t-butyl hydroperoxide, respectively (26). As we have observed that the various substrates tested with AhpC vary in their Km values but not significantly in their kcat values, it is also clear that a single concentration of hydroperoxide used in an assay would likely not be a very good indicator of catalytic efficiency differences between substrates in such cases.
Surveying the literature, we now find that the large majority of studies of Prx substrate specificity are subject to one or more of the above problems and that very few studies have carefully defined the true substrate specificity in enzymological terms (28). Although a few studies using single physiological reductant and/or single hydroperoxide concentrations in the assays have suggested some degree of specificity toward or against hydrogen peroxide relative to cumene hydroperoxide or lipid hydroperoxides (27, 36, 46–52), most have reasonably fit the theme of broad specificity for Prxs. One study using detailed bisubstrate kinetic analyses of the typical 2-Cys Prx proteins from Schistosoma mansoni has specifically demonstrated broad specificity of these enzymes for hydroperoxide substrates; interestingly, these enzymes also accept both Trx and GSH as reductants (22). In another case, however, true kcat and Km values obtained for both the Trx reductant and several hydroperoxides for E. coli Tpx showed H2O2 to be a much worse substrate than cumene hydroperoxide for this enzyme, with kcat/Km values of ≈4 × 104 M−1·s−1 and 7.7 × 106 M−1·s−1, respectively (26). This specificity is exactly the opposite relative to our findings for AhpC, showing that specificity can strongly vary among Prxs. We expect that these differences in specificity are traceable to differences in the active site shapes of the enzyme, but this hypothesis cannot yet be confirmed as no crystal structure is available for the peroxide binding conformation of Tpx (3). With such dramatic differences in substrate specificity as observed in these two examples of Prxs, both present in E. coli, this issue of better defining the active site pocket by solving structures of Prxs with substrates or products bound in a productive way becomes all the more interesting as an avenue of investigation for future studies.
We note that a better understanding of true differences in substrate specificities also provides important information on which we can base our understanding of the distinct biological roles that may be played by given Prx enzymes. In the organisms of relevance to the present findings, E. coli and S. typhimurium, Tpx would appear to play the major role as the reductant of organic hydroperoxides, whereas AhpC would be more targeted toward hydrogen peroxide detoxification. Each may also reduce peroxynitrite if exposed to this oxidant given the report that StAhpC can reduce this species (with a second-order rate constant of ≈106 M−1·s−1) (53), and a range of other Prxs have also been noted to possess this activity to similar or greater degrees (54–57). One might suspect that any of these reactive oxygen and nitrogen species may be encountered by pathogenic bacteria during times of stress and/or when encountering a host's immune system during infection, and that Tpx and AhpC provide overlapping and complementary protection against them. Indeed, results from Seaver and Imlay (12) have emphasized the importance of E. coli AhpC in maintaining low, endogenous levels of hydrogen peroxide; the other major player in hydrogen peroxide scavenging, catalase, provided an important biological antioxidant activity once levels of hydrogen peroxide reached 10 μM or more caused by saturation of the AhpC activity, consistent with the high reactivity and low Km value observed in vitro for AhpC in these studies.
Finally, our overoxidation studies, although carried out on a relatively robust enzyme that requires mM concentrations of peroxides to observe modest inactivation, provide evidence that the propensity of a given substrate to cause overoxidation at the active site is a property that parallels catalytic efficiency, that is, the higher the Km for a hydroperoxide substrate, the less likely it is to effect inactivation during turnover. Some evidence that better substrates are also linked with a tendency to overoxidize the protein has been presented previously for Prxs (22, 26); to thoroughly investigate this linkage, we present here a full panel of four substrates with widely differing specificity constants compared side by side for both attributes, catalysis and inactivation. Prxs, including StAhpC, are well known to undergo local unfolding in the vicinity of the active site, e.g., when engaging in disulfide bond formation after peroxide reduction (3), and one possibility would be that the overoxidation reaction occurs when the sulfenic acid is outside of the putative protective environment of the active site. Our data suggest, however, that binding interactions within the active site pocket are indeed of importance in the overoxidation reaction, and that this inactivation occurs predominantly within the fully folded active site. Given the limitations of quantitatively studying overoxidation in this relatively robust enzyme, however, it will be of great interest to obtain such information, and perhaps even Ki values for overoxidation, for more sensitive enzymes that exhibit a range of specificities for particular peroxide substrates.
In closing, we note that earlier studies of Prx enzymology have led to the general views that (i) Prxs are relatively poor peroxidases, being 100- to 1,000-fold less efficient than the catalase and GSH peroxidase enzymes, and (ii) Prxs are relatively broad spectrum peroxidases for the most part, with activities varying only up to ≈10-fold for such different substrates as H2O2, cumene hydroperoxide, and/or t-butyl hydroperoxides (4, 17, 29, 58). The problem is that these generalizations are based on a body of literature that we now know is flawed because in most cases limitations in the assays carried out masked the true kinetic parameters. The issue of poor catalysis by Prxs is indeed undergoing revision as evidence accumulates that this misconception has resulted from limitations of the assays being used (10, 26, 27, 36, 56, 59). Our data reveal that substrate specificity issues also need to be revisited. Using AhpC as a case in point, our earlier work (44) suggested that AhpC had a catalytic turnover rate of ≈10 s−1 and had no significant discrimination between hydrogen peroxide and cumene hydroperoxide. Now, we know that the true turnover rate is 53 s−1, the efficiency is near 4 × 107 M−1·s−1, and there is a ≈100-fold preference for hydrogen peroxide over cumene hydroperoxide. Given this dramatic shift for AhpC, it may be that a reassessment of the kinetic properties of diverse Prxs will similarly shift our understanding of Prx activity and specificity in general and will allow us to better assess the true scope of the physiological roles of these ubiquitous peroxidases.
Experimental Procedures
Materials.
NADH, NADPH, cumene hydroperoxide, and t-butyl hydroperoxide were purchased from Sigma. Ethyl hydroperoxide (5%) was purchased from Polysciences. Hydrogen peroxide (30%), DMSO, and most buffer components were from Fisher. DTT was obtained from Anatrace.
Methods.
Expression and purification of enzymes.
StAhpC (44), S128W NTD fragment (10), and E. coli redox proteins Grx1 (60), Trx1 (61, 62), and Trx reductase (34) were expressed and purified as described. The extinction coefficients previously reported for each of those proteins at 280 nm were used for determining protein concentrations.
Enzyme assays.
For peroxide-dependent assays, our previously published, highly sensitive assay was used wherein the loss of fluorescence from the mutated electron donating domain of AhpF, S128W NTD, was monitored as it is oxidized in the presence of AhpC and hydroperoxide substrates. Briefly, S128W NTD was prereduced by DTT for 1 h, excess DTT was removed by gel filtration chromatography, and the prereduced S128W NTD and wild-type StAhpC in one syringe were mixed with reaction buffer (50 mM potassium phosphate at pH 7.0, 0.5 mM EDTA, and 100 mM ammonium sulfate, present in both syringes) containing the peroxide in the second syringe of an Applied Photophysics SX.18MV stopped-flow spectrophotometer. Because of solubility limitations, cumene hydroperoxide was first diluted into DMSO to a concentration of 121 or 12.1 mM, then into reaction buffer to the final concentrations. Fluorescence changes were observed, with excitation at 280 nm and emission >320 nm (using an emission filter). Temperature was maintained at 25°C. All of the rates reported here are averages of at least three rate measurements in at least two independent assays. The fluorescence changes were calibrated by measuring the total change in fluorescence upon oxidation of a known concentration of reduced S128W NTD by excess peroxide. The initial rate of fluorescence decrease was determined by linear regression of the data from about the first 0.5–2 s of the reaction. True values for kcat and Km for both peroxide and S128W substrates were calculated directly from global fits of all of the data for a single peroxide substrate by using the multiple-function nonlinear regression capability of SigmaPlot (Jandel Scientific), using Eq. 1.
To assess peroxide-dependent inactivation rates of the various AhpC mutants, assays were conducted in the presence of Trx reductase and Trx and mM concentrations of the hydroperoxides as described (13). The high concentrations of DMSO present while testing overoxidation by cumene hydroperoxide required a separate control analysis with DMSO alone, which acts as an inhibitor of turnover by AhpC at amounts in excess of 1.5% in this assay.
Determination of midpoint reduction (redox) potential.
The redox potential of wild-type AhpC was determined by direct protein–protein redox equilibration (30). Briefly, oxidized and reduced forms of both AhpC and E. coli Grx1 were separated by RP-HPLC (as established in preliminary experiments) for analysis of equilibrium mixtures of the two proteins. DTT-reduced AhpC and oxidized Grx1, or oxidized AhpC and reduced Grx1, were mixed anaerobically and allowed to equilibrate at room temperature. Both proteins were present at 50 μM in 100 mM potassium phosphate, pH 7.0, with 1 mM EDTA. Fifty-microliter samples were removed at various times from 4 to 7 h, quenched by adding 50% volume of 1 M phosphoric acid, and loaded immediately onto a Vydac C4 4.6 × 250-mm HPLC column. The protein components were quantified by running an acetonitrile gradient (31.5–40.6% acetonitrile for 30 min followed by a gradient to 70% acetonitrile for an additional 50 min, with 0.08–0.1% trifluoroacetic acid in both solvents, at a flow rate of 0.5 ml/min and room temperature), which was optimized to resolve reduced and oxidized AhpC and Grx1 species. Quantitation of the proteins was based on calibration standards of each of the proteins and peak area from the chromatogram.
Acknowledgments.
We thank Yuji Yamamoto (Kitasato University, Towada, Japan) for providing the purified E. coli Grx1 protein and Kim Nelson and Chananat Klomsiri for helpful discussions. This work was supported by National Institutes of Health Grant RO1 GM050389 (to L.B.P. with P.A.K. as the principal investigator of the subcontract).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Schröder E, Ponting CP. Protein Sci. 1998;7:2465–2468. doi: 10.1002/pro.5560071125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Copley SD, Novak WR, Babbitt PC. Biochemistry. 2004;43:13981–13995. doi: 10.1021/bi048947r. [DOI] [PubMed] [Google Scholar]
- 3.Karplus PA, Hall A. In: Peroxiredoxin Systems. Flohé L, Harris JR, editors. New York: Springer; 2007. pp. 41–60. [Google Scholar]
- 4.Hofmann B, Hecht H-J, Flohé L. Biol Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 5.Wood ZA, Schröder E, Harris JR, Poole LB. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 6.Noguera-Mazon V, Krimm I, Walker O, Lancelin JM. Photosynth Res. 2006;89:277–290. doi: 10.1007/s11120-006-9106-4. [DOI] [PubMed] [Google Scholar]
- 7.Dietz KJ, Jacob S, Oelze ML, Laxa M, Tognetti V, de Miranda SM, Baier M, Finkemeier I. J Exp Bot. 2006;57:1697–1709. doi: 10.1093/jxb/erj160. [DOI] [PubMed] [Google Scholar]
- 8.Veal EA, Day AM, Morgan BA. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 10.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guimaraes BG, Souchon H, Honore N, Saint-Joanis B, Brosch R, Shepard W, Cole ST, Alzari PM. J Biol Chem. 2005;280:25735–25742. doi: 10.1074/jbc.M503076200. [DOI] [PubMed] [Google Scholar]
- 12.Seaver LC, Imlay JA. J Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood ZA, Poole LB, Karplus PA. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 14.Poole LB, Karplus PA, Claiborne A. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 15.Rhee SG. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 16.Declercq JP, Evrard C, Clippe A, Stricht DV, Bernard A, Knoops B. J Mol Biol. 2001;311:751–759. doi: 10.1006/jmbi.2001.4853. [DOI] [PubMed] [Google Scholar]
- 17.Dietz KJ. Annu Rev Plant Biol. 2003;54:93–107. doi: 10.1146/annurev.arplant.54.031902.134934. [DOI] [PubMed] [Google Scholar]
- 18.Nogoceke E, Gommel DU, Kiess M, Kalisz HM, Flohe L. Biol Chem. 1997;378:827–836. doi: 10.1515/bchm.1997.378.8.827. [DOI] [PubMed] [Google Scholar]
- 19.Choi YO, Cheong NE, Lee KO, Jung BG, Hong CH, Jeong JH, Chi YH, Kim K, Cho MJ, Lee SY. Biochem Biophys Res Commun. 1999;258:768–771. doi: 10.1006/bbrc.1999.0714. [DOI] [PubMed] [Google Scholar]
- 20.Jeong W, Cha MK, Kim IH. J Biol Chem. 2000;275:2924–2930. doi: 10.1074/jbc.275.4.2924. [DOI] [PubMed] [Google Scholar]
- 21.Pedrajas JR, Miranda-Vizuete A, Javanmardy N, Gustafsson JA, Spyrou G. J Biol Chem. 2000;275:16296–16301. doi: 10.1074/jbc.275.21.16296. [DOI] [PubMed] [Google Scholar]
- 22.Sayed AA, Williams DL. J Biol Chem. 2004;279:26159–26166. doi: 10.1074/jbc.M401748200. [DOI] [PubMed] [Google Scholar]
- 23.Rouhier N, Gelhaye E, Gualberto JM, Jordy MN, De Fay E, Hirasawa M, Duplessis S, Lemaire SD, Frey P, Martin F, et al. Plant Physiol. 2004;134:1027–1038. doi: 10.1104/pp.103.035865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munhoz DC, Netto LE. J Biol Chem. 2004;279:35219–35227. doi: 10.1074/jbc.M313773200. [DOI] [PubMed] [Google Scholar]
- 25.Boucher IW, McMillan PJ, Gabrielsen M, Akerman SE, Brannigan JA, Schnick C, Brzozowski AM, Wilkinson AJ, Muller S. Mol Microbiol. 2006;61:948–959. doi: 10.1111/j.1365-2958.2006.05303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker LM, Poole LB. J Biol Chem. 2003;278:9203–9211. doi: 10.1074/jbc.M209888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akerman SE, Müller S. J Biol Chem. 2005;280:564–570. doi: 10.1074/jbc.M406367200. [DOI] [PubMed] [Google Scholar]
- 28.Poole LB. In: Peroxiredoxin Systems. Flohé L, Harris JR, editors. New York: Springer; 2007. pp. 61–81. [Google Scholar]
- 29.Rouhier N, Jacquot JP. Free Radical Biol Med. 2005;38:1413–1421. doi: 10.1016/j.freeradbiomed.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 30.Åslund F, Berndt KD, Holmgren A. J Biol Chem. 1997;272:30780–30786. doi: 10.1074/jbc.272.49.30780. [DOI] [PubMed] [Google Scholar]
- 31.Reckenfelderbaumer N, Krauth-Siegel RL. J Biol Chem. 2002;277:17548–17555. doi: 10.1074/jbc.M112115200. [DOI] [PubMed] [Google Scholar]
- 32.Poole LB. Arch Biochem Biophys. 2005;433:240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Poole LB, Higuchi M, Shimada M, Calzi ML, Kamio Y. Free Radical Biol Med. 2000;28:108–120. doi: 10.1016/s0891-5849(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 34.Poole LB, Godzik A, Nayeem A, Schmitt JD. Biochemistry. 2000;39:6602–6615. doi: 10.1021/bi000405w. [DOI] [PubMed] [Google Scholar]
- 35.Baker LM, Raudonikiene A, Hoffman PS, Poole LB. J Bacteriol. 2001;183:1961–1973. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Budde H, Flohé L, Hecht HJ, Hofmann B, Stehr M, Wissing J, Lünsdorf H. Biol Chem. 2003;384:619–633. doi: 10.1515/BC.2003.069. [DOI] [PubMed] [Google Scholar]
- 37.Prinz WA, Aslund F, Holmgren A, Beckwith J. J Biol Chem. 1997;272:15661–15667. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 38.König J, Baier M, Horling F, Kahmann U, Harris G, Schurmann P, Dietz KJ. Proc Natl Acad Sci USA. 2002;99:5738–5743. doi: 10.1073/pnas.072644999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones DP. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 40.Åslund F, Zheng M, Beckwith J, Storz G. Proc Natl Acad Sci USA. 1999;96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmgren A, Fagerstedt M. J Biol Chem. 1982;257:6926–6930. [PubMed] [Google Scholar]
- 42.Ritz D, Beckwith J. Methods Enzymol. 2002;347:360–370. doi: 10.1016/s0076-6879(02)47036-x. [DOI] [PubMed] [Google Scholar]
- 43.Latimer WM. Oxidation Potentials. New York: Prentice-Hall; 1938. [Google Scholar]
- 44.Poole LB, Ellis HR. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 45.Cha MK, Kim WC, Lim CJ, Kim K, Kim IH. J Biol Chem. 2004;279:8769–8778. doi: 10.1074/jbc.M312388200. [DOI] [PubMed] [Google Scholar]
- 46.Levick MP, Tetaud E, Fairlamb AH, Blackwell JM. Mol Biochem Parasitol. 1998;96:125–137. doi: 10.1016/s0166-6851(98)00122-4. [DOI] [PubMed] [Google Scholar]
- 47.Jeong JS, Kwon SJ, Kang SW, Rhee SG, Kim K. Biochemistry. 1999;38:776–783. doi: 10.1021/bi9817818. [DOI] [PubMed] [Google Scholar]
- 48.Park SG, Cha MK, Jeong W, Kim IH. J Biol Chem. 2000;275:5723–5732. doi: 10.1074/jbc.275.8.5723. [DOI] [PubMed] [Google Scholar]
- 49.Flohé L, Budde H, Bruns K, Castro H, Clos J, Hofmann B, Kansal-Kalavar S, Krumme D, Menge U, Plank-Schumacher K, et al. Arch Biochem Biophys. 2002;397:324–335. doi: 10.1006/abbi.2001.2688. [DOI] [PubMed] [Google Scholar]
- 50.Castro H, Budde H, Flohe L, Hofmann B, Lunsdorf H, Wissing J, Tomas AM. Free Radical Biol Med. 2002;33:1563–1574. doi: 10.1016/s0891-5849(02)01088-2. [DOI] [PubMed] [Google Scholar]
- 51.König J, Lotte K, Plessow R, Brockhinke A, Baier M, Dietz KJ. J Biol Chem. 2003;278:24409–24420. doi: 10.1074/jbc.M301145200. [DOI] [PubMed] [Google Scholar]
- 52.Jaeger T, Budde H, Flohe L, Menge U, Singh M, Trujillo M, Radi R. Arch Biochem Biophys. 2004;423:182–191. doi: 10.1016/j.abb.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 53.Bryk R, Griffin P, Nathan C. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 54.Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, Kissner R, Koppenol WH, Rees JF, Knoops B. FEBS Lett. 2004;571:161–165. doi: 10.1016/j.febslet.2004.06.080. [DOI] [PubMed] [Google Scholar]
- 55.Nickel C, Rahlfs S, Deponte M, Koncarevic S, Becker K. Antioxid Redox Signal. 2006;8:1227–1239. doi: 10.1089/ars.2006.8.1227. [DOI] [PubMed] [Google Scholar]
- 56.Ogusucu R, Rettori D, Munhoz DC, Soares Netto LE, Augusto O. Free Radical Biol Med. 2007;42:326–334. doi: 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 57.Trujillo M, Budde H, Pineyro MD, Stehr M, Robello C, Flohe L, Radi R. J Biol Chem. 2004;279:34175–34182. doi: 10.1074/jbc.M404317200. [DOI] [PubMed] [Google Scholar]
- 58.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 59.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 60.Yamamoto Y, Ritz D, Planson AG, Jönsson TJ, Faulkner MJ, Boyd D, Beckwith J, Poole LB. Mol Cell. 2008 doi: 10.1016/j.molcel.2007.11.029. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lennon BW, Williams CH., Jr Biochemistry. 1995;34:3670–3677. doi: 10.1021/bi00011a023. [DOI] [PubMed] [Google Scholar]
- 62.Veine DM, Mulrooney SB, Wang PF, Williams CH., Jr Protein Sci. 1998;7:1441–1450. doi: 10.1002/pro.5560070621. [DOI] [PMC free article] [PubMed] [Google Scholar]