

Abstract
Acute secretory diarrhea induced by infection with enterotoxigenic strains of Escherichia coli involves binding of stable toxin (STa) to its receptor on the intestinal brush border, guanylyl cyclase type C (GC-C). Intracellular cGMP is elevated, inducing increase in chloride efflux and subsequent accumulation of fluid in the intestinal lumen. We have screened a library of compounds and identified a pyridopyrimidine derivatives {5-(3-bromophenyl)-1,3-dimethyl-5,11-dihydro-1H-indeno[2′,1′:5,6]pyrido[2,3-d]pyrimidine-2,4,6-trione; BPIPP} as an inhibitor of GC-C that can suppress STa-stimulated cGMP accumulation by decreasing GC-C activation in intact T84 human colorectal carcinoma cells. BPIPP inhibited stimulation of guanylyl cyclases, including types A and B and soluble isoform in various cells. BPIPP suppressed stimulation of adenylyl cyclase and significantly decreased the activities of adenylyl cyclase toxin of Bordetella pertussis and edema toxin of Bacillus anthracis. The effects of BPIPP on cyclic nucleotide synthesis were observed only in intact cells. The mechanism of BPIPP-dependent inhibition appears to be complex and indirect, possibly associated with phospholipase C and tyrosine-specific phosphorylation. BPIPP inhibited chloride-ion transport stimulated by activation of guanylyl or adenylyl cyclases and suppressed STa-induced fluid accumulation in an in vivo rabbit intestinal loop model. Thus, BPIPP may be a promising lead compound for treatment of diarrhea and other diseases.
Keywords: adenylyl cyclase, cGMP, guanylyl cyclase, cAMP, toxin-induced diarrhea
Diarrhea is one of the principal causes of mortality in children in the developing world, being responsible for 2.5 million deaths per year (1, 2), and about 60% of these deaths are caused by enterotoxigenic strains of bacteria, including Escherichia coli (3). One of the toxins, heat-stable enterotoxin (STa), binds to intestinal epithelial cell membrane receptor, guanylyl cyclase type C (GC-C), and activates the enzyme to increase the synthesis of cyclic guanosine 3′,5′-monophosphate (cGMP) (4–6). GC-C is a transmembrane protein with an extracellular receptor domain, a short membrane span, and an intracellular catalytic domain (7). Natural endogenous ligands of GC-C are peptide hormones guanylin and uroguanylin, which regulate secretion of salt and fluids by the intestine (8). Upon stimulation of GC-C, elevated levels of cGMP induce activation of a cGMP-dependent protein kinase and a chloride-ion channel, cystic fibrosis transmembrane conductance regulator (CFTR). Activation of CFTR increases transport of chloride into the intestinal lumen and accumulation of water and sodium ions, thus causing diarrhea (6). Since defining the cell signaling pathways involved in pathogenesis of enterotoxigenic diarrhea, it had become apparent to us that this pathway represents a molecular target to interrupt the signaling as a therapeutic approach to STa-induced diarrhea. The goal of the present study was to develop an innovative approach to therapy of acute diarrhea based on an inhibitor of stimulated cyclic nucleotide synthesis. We have identified a promising lead compound, 5-(3-bromophenyl)-1,3-dimethyl-5,11-dihydro-1H-indeno[2′,1′:5,6]pyrido[2,3-d]pyrimidine-2,4,6-trione (BPIPP), which can suppress cyclic nucleotide synthesis and is active in vivo in an animal model of acute diarrhea.
Results and Discussion
Screening a compound library allowed us to identify a class of pyridopyrimidine derivatives that can suppress STa-dependent cGMP accumulation in cultured human colorectal carcinoma T84 cells (Table 1). Accumulation of cGMP was suppressed by ≈80% by BPIPP (IIa). Other derivatives with a 4-hydroxyl group in the phenyl moiety or with an acylated N-11 atom and 4-chloride in the phenyl moiety had considerably lower activity. However, the 5-(3-fluorophenyl) derivative IIIa, being essentially isoelectronic to BPIPP, had similar potency. Oxidation products of BPIPP and compounds Ia and IIIa (Ib, IIb, and IIIb, respectively) were prepared by exposing the solutions to air for extended periods of time, and they did not inhibit STa-stimulated cGMP accumulation. All compounds had no influence on baseline intracellular or extracellular cGMP levels (8.8 ± 2.3 and 1.6 ± 0.6 pmol/mg of protein in the presence of vehicle and 9.0 ± 1.0 and 2.6 ± 1.6 pmol/mg of protein in the presence of 50 μM BPIPP, respectively; n = 6; P > 0.1). The most potent derivative BPIPP was further investigated in T84 cells.
Table 1.
Inhibition of STa-stimulated cGMP accumulation in T84 cells
| Compound Inhibition, % | Formula | Compound Inhibition, % | Formula |
|---|---|---|---|
| Ia | 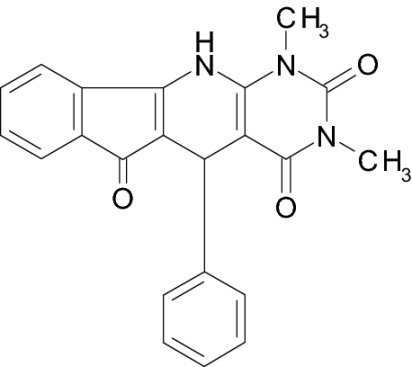 |
IVa |  |
| 19 ± 4 | 26 ± 7 | ||
| Ib |  |
Va |  |
| 7 ± 1 | 29 ± 6 | ||
| IIa (BPIPP) | 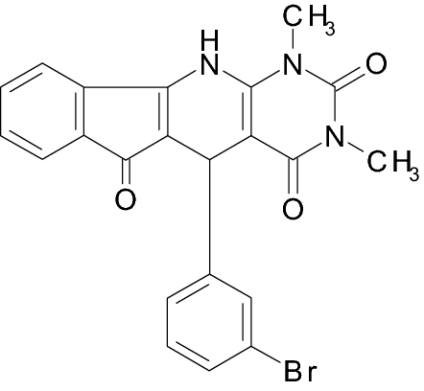 |
VIa | 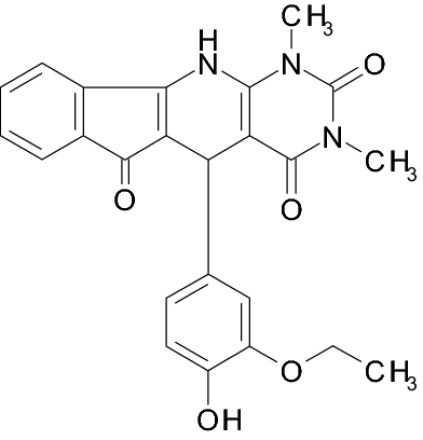 |
| 86 ± 4 | 42 ± 5 | ||
| IIb | 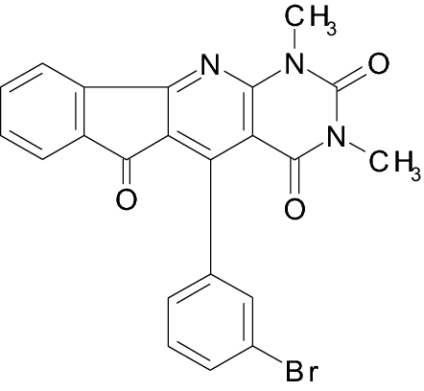 |
VII | |
| 2 ± 1 | 38 ± 6 | ||
| IIIa | 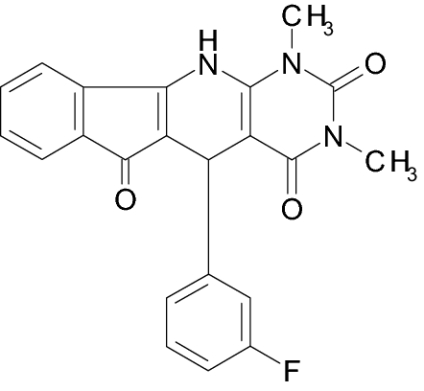 |
VIII | |
| 83 ± 16 | 22 ± 4 | ||
| IIIb | 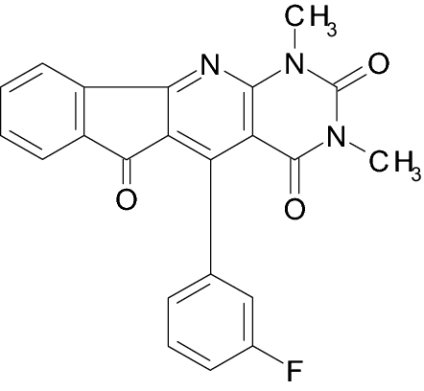 |
IXa | 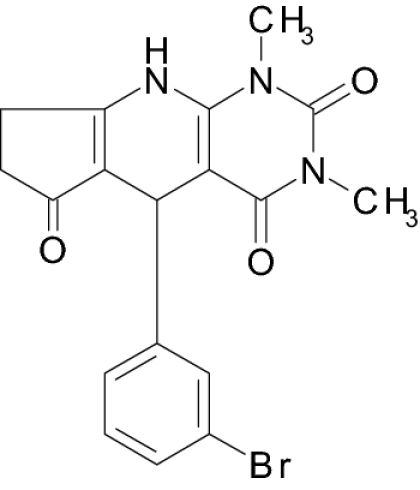 |
| 17 ± 2 | 56 ± 11 |
T84 cells were pretreated with the indicated compounds at 50 μM or vehicle (0.1% DMSO) and 1 mM 3-isobutyl-1-methylxanthine (IBMX) for 10 min and then treated with 100 nM STa for 10 min, and cGMP accumulation in the cells was measured. Accumulation of cGMP in the presence of vehicle was 221 ± 34 pmol of cGMP per mg of protein. Data are mean ± SEM of six independent experiments assayed in duplicate.
Intracellular cGMP accumulation is a balance of three major processes: cGMP synthesis by guanylyl cyclases (9, 10), cGMP degradation by phosphodiesterases (11), and cGMP extrusion into the medium (12). Thus, suppression of cGMP accumulation by BPIPP can be due to inhibition of guanylyl cyclase activation or to stimulation of cGMP degradation or extrusion. To rule out enhanced degradation, all experiments were performed in the presence of the nonspecific phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; 1 mM). Cells were also treated with vehicle or BPIPP, and rates of cGMP degradation were assayed in the homogenate (21.8 ± 0.8 and 25.7 ± 2.3 nmol of cGMP per min per mg of protein in cells pretreated with vehicle and BPIPP, respectively; n = 4; P > 0.1). Treatment of T84 cells with BPIPP in the presence or in the absence of IBMX had no influence on the efflux of cGMP in cells stimulated with STa (Fig. 1A). STa-dependent increase in extracellular cGMP was inhibited by BPIPP to the same extent as intracellular cGMP accumulation, suggesting that the effect of BPIPP on cGMP levels cannot be due to enhanced cGMP degradation or extrusion.
Fig. 1.

Effect of BPIPP on cGMP accumulation in cultured cells. (A) T84 cells were pretreated with 50 μM BPIPP or vehicle (0.1% DMSO) with or without 1 mM IBMX for 10 min and then treated with 0.1 μM STa (ST0.1) or 1 μM STa (ST1) for 10 min; intracellular (i) and extracellular (e) cGMP were assayed in the extract or in the treatment medium, respectively. n = 3–6. (B) Membranes were isolated from T84 cells and GC-C activity was assayed without stimulation (basal) or with 0.1 μM STa (STa 0.1), 1 μM STa (STa 1), or 3 mM MnCl2 (Mn) in the presence of 50 μM BPIPP or vehicle. Similar data were obtained in membranes isolated in buffer containing protein phosphatase inhibitors from cells pretreated for 10 min with BPIPP. n = 3–4. (C) T84 cells were pretreated with 50 μM BPIPP or vehicle for 10 min and then with indicated concentrations of STa for 10 min, and cGMP accumulation was measured. n = 3. (D) T84 cells were pretreated with indicated concentrations of BPIPP or vehicle (Con) for 10 min and then with 0.5 μM guanylin or 100 nM STa for 10 min, and cGMP accumulation was measured. n = 6. (E) BE-2 cells were pretreated with indicated concentrations of BPIPP or vehicle (Con) for 10 min and then treated with 1 μM atrial natriuretic peptide (ANP) or 0.5 μM C-type natriuretic peptide (CNP) for 10 min, and intracellular cGMP was measured. n = 4. (F) RFL-6 cells were pretreated with 50 μM BPIPP or vehicle for 10 min and then with 10 μM nitric oxide donor benzotrifuroxan (NO donor), 1 μM ANP, or 1 μM CNP for 4 min. *, P < 0.01; n = 3.
The effect of BPIPP can result from decreased stimulation of GC-C with STa because of diminished binding of STa to GC-C in BPIPP-treated cells or to other mechanisms. BPIPP did not induce detectable changes in 125I-STa binding to the intact T84 cells or to the membranes of cells pretreated with vehicle or BPIPP (1.92 ± 0.23 and 1.87 ± 0.18 fmol/mg of protein in intact cells and 48.8 ± 0.5 and 46.1 ± 0.4 fmol/mg of protein in membranes, respectively; n = 5–7; P > 0.1). These data (plus data shown in Fig. 1C) also rule out possible STa scavenging by direct competitive binding to BPIPP because in this case, specific receptor binding should be decreased, whereas it remains unaffected. The effect of BPIPP can be of a nonspecific nature and might result from cytotoxic activity or induction of ATP depletion. No cell death was observed with BPIPP treatment according to trypan blue exclusion or lactate dehydrogenase (LDH) release into the medium (LDH release was 0.42 ± 0.03% and 0.49 ± 0.04% in vehicle- and 50 μM BPIPP-treated cells, respectively; n = 8; P > 0.1). Assay of intracellular ATP did not reveal significant changes (18.3 ± 1.5 and 16.4 ± 1.7 nmol of ATP per mg of protein in cells treated with vehicle and BPIPP, respectively; n = 8; P > 0.1). Thus, BPIPP specifically decreases activation of GC-C by STa.
In membranes isolated from T84 cells, BPIPP did not cause inhibition of basal GC-C activity or activity with 0.1 μM and 1 μM STa or with Mn2+ (Fig. 1B). Similar data were obtained with homogenates and membranes isolated in the presence of protease and phosphatase inhibitors from cells pretreated with BPIPP, indicating that the influence of BPIPP is indirect and can be observed only in the intact cells because after disruption of the cells, the effects are lost. The mechanism of BPIPP inhibition appears to be noncompetitive, because extent of inhibition was similar at low and high concentrations of STa, and even 5 μM STa (Fig. 1C) could not overcome the effect of BPIPP.
BPIPP action was reversed by washing of BPIPP-treated T84 cells with Dulbecco's PBS (DPBS) containing BSA. In the cells that had not been washed or in the cells washed with DBPS, the STa-stimulated accumulation of cGMP was inhibited by 50 μM BPIPP by 87.4 ± 7.7% and 59.2 ± 10.2%, respectively (n = 6; P > 0.1). However, in cells washed with DPBS containing 1% BSA, inhibition was only 22.6 ± 7.7% (n = 6; P < 0.01). In addition to this observation, both BPIPP-induced inhibition of 250 nM STa-stimulated cGMP accumulation and BPIPP binding to the cells were suppressed in DPBS containing BSA (but not ovalbumin). In control incubations without BSA, inhibition was 87.4 ± 7.7% (32.7 ± 5.6 nmol of BPIPP bound per mg of protein) and in the presence of 1 mg/ml or 5 mg/ml BSA, inhibition was 48.0 ± 5.8% or 12.7 ± 3.5% (2.7 ± 2.3 nmol of BPIPP bound per mg of protein), respectively (cGMP accumulation in cells treated with 250 nM STa was 307 ± 31 pmol of cGMP per mg assumed as 100%; n = 3). This can be explained by BPIPP binding to BSA with relatively high affinity and indeed was confirmed by ultrafiltration of the BSA–BPIPP complex (supporting information (SI) Materials and Methods). The apparent equilibrium binding constant was 8.3 ± 0.8 μM and the stoichiometry was 5.6 molecules of BPIPP per molecule of BSA. No binding of BPIPP to ovalbumin was detected under similar conditions.
BPIPP suppressed cGMP accumulation in T84 cells stimulated with STa or the endogenous GC-C activator guanylin (8) (Fig. 1D) with IC50 3.4 ± 1.6 and 7.2 ± 1.7 μM, respectively. In the human neuroblastoma cell line BE-2 (Fig. 1E), BPIPP suppressed cGMP accumulation induced by atrial natriuretic peptide (stimulation of GC-A) or C-type natriuretic peptide (stimulation of GC-B); IC50 values were 8.4 ± 1.6 and 11.2 ± 1.5 μM, respectively. Similar effects were observed in rat fetal lung fibroblasts RFL-6 (Fig. 1F). In addition to inhibiting particulate guanylyl cyclases (GC-A, GC-B, and GC-C), BPIPP also suppressed activation of soluble guanylyl cyclase by a nitric oxide donor (benzotrifuroxan) in intact RFL-6 cells (Fig. 1F). However, similar to the inability of BPIPP to influence enzymatic activity of GC-C in membranes of T84 cells (Fig. 1B), BPIPP had no inhibitory effect on basal or nitric oxide-stimulated activity of purified soluble guanylyl cyclase. Basal activity of the purified enzyme was 0.16 ± 0.01 μmol/min per mg, and with 100 μM spermine/NONOate the activity increased to 2.96 ± 0.22 μmol/min per mg; corresponding values in the presence of 50 μM BPIPP were 0.10 ± 0.01 and 3.76 ± 0.17 μmol/min per mg (n = 6; P > 0.05).
BPIPP also inhibited cGMP accumulation stimulated with STa in COS-7 cells (which lack endogenous functional GC-C) transfected with full-length recombinant human GC-C. In cells transiently transfected with an empty vector, the basal level of cGMP was 2.8 ± 0.3 pmol/mg and it was not enhanced with STa; in cells transfected with GC-C and stimulated with 100 nM STa, cGMP increased to 22.2 ± 1.5 pmol/mg, and this stimulation was inhibited by BPIPP by 81 ± 3% (n = 3).
Additionally, BPIPP can suppress activation of adenylyl cyclases. Pretreatment of RFL-6 cells with BPIPP suppressed activation of cAMP accumulation induced by treatment with direct adenylyl cyclase activator forskolin, receptor-dependent stimulation by β-adrenergic agonist isoproterenol, and GTP-binding protein-dependent stimulation with cholera toxin (Fig. 2A). Because of poor water solubility of BPIPP, the maximal concentration of the drug tested was 50 μM, and it is unclear whether further increases in BPIPP concentrations can produce greater inhibition. It seems that BPIPP preferentially inhibits activation of guanylyl cyclase compared with adenylyl cyclase. Similar data were obtained in T84 cells. For example, basal cAMP accumulation in T84 cells was not influenced by treatment with 50 μM BPIPP (31.4 ± 3.0 and 32.6 ± 2.7 pmol of cAMP per mg with vehicle and BPIPP, respectively; n = 6; P > 0.1), whereas in cells stimulated with 100 μM isoproterenol, BPIPP inhibited cAMP accumulation by 58.2% (284 ± 22 and 119 ± 7 pmol of cAMP per mg with vehicle and BPIPP, respectively; n = 3).
Fig. 2.

Effect of BPIPP on cAMP accumulation in cultured cells. (A) RFL-6 cells were pretreated with 50 μM BPIPP or vehicle for 10 min and then with 50 μM forskolin (For) or 100 μM isoproterenol (Iso); cells were pretreated with 1 μg/ml cholera toxin for 30 min (CT30) or 60 min (CT60) and then, after a 10-min incubation with IBMX and BPIPP, cAMP accumulation was measured. *, P < 0.01; n = 4. (B) T84 cells were pretreated in serum-free growth medium in the presence of 1 μg/ml adenylyl cyclase toxin from Bordetella pertussis (BAC), a combination of 2 μg/ml protective antigen and 0.1 μg/ml edema factor from Bacillus anthracis (edema toxin; ET), or 1 μg/ml Vibrio cholera toxin (CT) for 1 h (pretreatment phase; pretreat); cells were washed and further incubated in DBPS containing 1 mM IBMX for 10 min to measure cAMP accumulation (incubation phase; incubate); cells were treated with vehicle or with 50 μM BPIPP during the corresponding phase as indicated. Control (100% values) for BAC, ET, and CT were 9.98 ± 0.72, 0.14 ± 0.01, and 7.85 ± 0.82 nmol of cAMP per mg of protein. *, P < 0.05; **, P < 0.01; n = 3. (C) T84 cells were pretreated with 50 μM BPIPP or vehicle and 1 mM IBMX for 10 min and then treated with indicated concentrations of forskolin for 10 min. n = 4.
BPIPP can suppress accumulation of cAMP in cells pretreated with various bacterial toxins, including adenylyl cyclase toxin from Bordetella pertussis (13), edema toxin from Bacillus anthracis (14), and cholera toxin. BPIPP effects on the efficacy of pretreatment were also assessed. The data of Fig. 2B indicate that BPIPP does not significantly inhibit apparent efficacy of pretreatment of pertussis adenylyl cyclase or edema factor but markedly suppressed efficacy of pretreatment with cholera toxin. However, cAMP accumulation induced by pertussis cyclase and edema factor and activation of endogenous adenylyl cyclase by cholera toxin were efficiently inhibited by BPIPP. Thus, BPIPP is capable of inhibiting not only eukaryotic adenylyl cyclase but also bacterial adenylyl cyclase toxins. Similar to inhibition of guanylyl cyclase (Fig. 1C), suppression of adenylyl cyclase appears to be noncompetitive because increasing forskolin concentrations to 25 μM did not overcome the inhibition by BPIPP (Fig. 2C). Also, BPIPP had no influence on basal, isoproterenol-, fluoride-, or forskolin-stimulated adenylyl cyclase activity in membranes prepared from T84 cells pretreated with BPIPP or when BPIPP was added to the incubation medium for assay of adenylyl cyclase activity in the membranes (Fig. S1). Thus, the effects of BPIPP on adenylyl cyclase activity are indirect and cannot be seen after destruction of the cells.
In cultured T84 cells, BPIPP inhibited the downstream effects of cGMP, which involve activation of chloride efflux from the cells. The data shown in Fig. 3A indicate that increasing concentrations of STa enhanced chloride ion efflux from the cells, and this increase was effectively blocked by pretreatment with BPIPP. BPIPP also suppressed increase in chloride efflux induced by treatment of cells with forskolin, cholera toxin, or isoproterenol (Fig. 3B). However, BPIPP did not influence basal chloride efflux (not shown), efflux stimulated with 8-bromo-cAMP (0.1 and 1 mM), or calcium-dependent efflux stimulated with 10 μM ionomycin (Fig. 3B), suggesting that the effects of BPIPP are mediated by inhibition of cyclic nucleotide synthesis and BPIPP does not affect the downstream signaling or chloride transport directly. Monolayers of T84 cells were subjected to a short-circuit current (ISC) measurements in an Ussing chamber. Treatment of cells with 0.1 μM STa increased ISC to 9.6 ± 1.3 μA/cm2, whereas in cells pretreated with 50 μM BPIPP, the STa effect was greatly diminished (2.7 ± 0.8 μA/cm2; n = 6). Similar data were obtained when ISC was stimulated with 5 μM forskolin (increase in ISC to 18.7 ± 1.6 and 2.9 ± 2.0 μA/cm2 in cells pretreated with vehicle and 50 μM BPIPP, respectively; n = 4). BPIPP had no influence on basal current or resistance of the monolayer.
Fig. 3.

Antisecretory activity of BPIPP in cell culture and in vivo. (A) T84 cells were pretreated with 50 μM BPIPP or vehicle in solution 1 containing NaCl, and chloride transport was measured in the presence of indicated concentrations of STa in solution 2 containing NaNO3 by using 6-methoxy-1-(3-sulfonatopropyl)quinolinium (SPQ) fluorescence as a Cl− sensor. n = 8. (B) Effect of 50 μM BPIPP on Cl− transport in T84 cells stimulated by various agents in solution 1 containing NaCl; 100 μM isoproterenol (Iso), 10 μg/ml cholera toxin (CT; CT-ptr, BPIPP was present during pretreatment similar to Fig. 2B; CT-inc, BPIPP was present only during incubation and assay phase; CT-both, BPIPP was present during pretreatment and incubation-assay phase), 25 μM forskolin (For), 1 mM or 0.1 mM 8-bromo-cAMP (BcA-1 or BcA-0.1), or 10 μM ionomycin (Iono). Fluorescence was measured by using SPQ or N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE) as indicator. Absolute Ft − Fc fluorescence values for vehicle control were 138 ± 6 for isoproterenol, 74 ± 5 for cholera toxin, 1,031 ± 36 for forskolin, 52 ± 2 and 780 ± 84 for 0.1 and 1 mM 8-bromo-cAMP in the presence of SPQ, and 635 ± 75 and 1,945 ± 102 for 0.1 and 1 mM 8-bromo-cAMP and 720 ± 81 for ionomycin in the presence of MQAE. *, P < 0.05 BPIPP vs. vehicle; n = 8. (C) Effect of compounds on secretion in rabbit intestinal segments (ileal loops) injected with 1 ml of PBS containing indicated concentrations of STa [none (Control), 0.1, 0.2, or 1.0 μM] with vehicle, 10 or 50 μM BPIPP, and 50 μM compound IVa. Incubations were continued for 5 h, and then volume/length ratios of the segments were determined. *, P < 0.05; n = 4–8.
We then hypothesized that BPIPP can suppress STa-mediated diarrhea in an animal model. The data of Fig. 3C indicate that in an in vivo rabbit intestinal loop model, 50 μM BPIPP slightly decreased basal secretion and completely suppressed secretion of fluid into the intestinal lumen stimulated with 0.1 μM or 0.2 μM STa but had no effect with 1 μM STa, and 10 μM BPIPP was also effective; 50 μM compound IVa (a weaker inhibitor of STa-stimulated cGMP accumulation in T84 cells; Table 1) had little or no effect on STa-stimulated secretion. Thus, BPIPP concentration-dependently suppressed STa-stimulated liquid secretion in an in vivo rabbit intestinal loop model and can potentially be used for therapy of toxin-induced secretory diarrhea.
The class of compounds studied in the present manuscript has not been described in the biochemical or pharmacological literature previously. Thus, we did not know or anticipate a specific direct target of BPIPP and therefore, investigation of molecular mechanism of action of this compound on synthesis of cyclic nucleotides was very challenging. As a first step, we have performed a pharmacological screening of various drugs acting on certain key intracellular signaling pathways known to be associated with regulation of cyclic nucleotide synthesis in cells. Our approach included treatment of T84 cells with various pharmacological agents, and then we evaluated the extent of inhibition of STa-mediated cGMP accumulation by BPIPP (Table S1).
Our results indicate that Ca2+-dependent signaling pathways are not directly involved in the effects of BPIPP on STa-mediated stimulation of cGMP accumulation in T84 cells. A number of extracellular and intracellular calcium chelators and antagonists as well as inhibitors and activators of calcium-dependent proteolysis and protein phosphorylation fail to interfere with BPIPP effect. The same is apparently true in the case of general inhibitors of serine/threonine-specific and tyrosine-specific protein kinases. However, an inhibitor of tyrosine-specific phosphatases, activated sodium orthovanadate, partially protected cells from the effects of BPIPP at high concentration (2 mM), significantly decreasing BPIPP inhibition by 30.8 ± 2.6%. Because orthovanadate is known to have considerable nonspecific effects unrelated to inhibition of phosphatases, we have sought to confirm whether BPIPP can influence the level of tyrosine-specific phosphorylation in T84 cells. The results shown in Fig. 4A indicate that treatment of T84 cells with 50 μM BPIPP significantly stimulates the apparent activity of tyrosine-specific protein kinases that can use various synthetic tyrosine-containing polypeptides as the substrates by 77–200%, depending on the substrate. Moreover, fractionation of the cells followed by a similar tyrosine kinase activity assay indicates that stimulation of protein kinase is more pronounced in the cytosol (85.0 ± 5.5%) versus membrane fraction (29.8 ± 3.2%; Fig. 4B).
Fig. 4.

Stimulation of activity of a tyrosine-specific protein kinase in T84 cells by BPIPP. (A) Cells were treated with 50 μM BPIPP or vehicle for 20 min and protein kinase activity was measured in the extract by using various tyrosine-containing protein kinase substrates. E4Y, (Glu)4-Tyr; EAY, Glu-Ala-Tyr; EY, Glu-Tyr. n = 3. (B) Cells were pretreated in a similar manner and then homogenized with a glass–glass homogenizer in a hypotonic buffer and fractionated to separate crude membrane fraction from the cytosol. Protein kinase activity was measured by using EAY as the substrate. *, P < 0.05; n = 6.
The effect of BPIPP can be attenuated by two inhibitors of phospholipase C, U73122 (54.0 ± 15.8% and 60.5 ± 12.8% suppression of BPIPP inhibition at 20 and 50 μM, respectively) and D609 (52.6 ± 5.7% suppression at 50 μM). However, inhibitors of phosphatidylinositol 3-kinase or phospholipase A2 had no detectable effect (Table S1). We then tested whether BPIPP can influence the activities of various phospholipases in intact T84 cells. BPIPP has no significant influence on inositol phosphate accumulation or on apparent phosphatidylcholine-specific phospholipase C or phospholipase D activities (Fig. S2). Thus, even though the inhibitors of phospholipase C interfere with BPIPP-dependent effects on cyclic nucleotide synthesis, BPIPP does not directly stimulate or inhibit phospholipases.
Because BPIPP can be oxidized in aqueous solutions in the presence of oxygen to form the corresponding compound IIb, we have tested two enzymes capable of metabolizing reactive oxygen species, catalase and superoxide dismutase. We found that these enzymes have an insignificant effect on BPIPP-mediated inhibition of STa-stimulated cGMP accumulation (Table S1).
We suggest that BPIPP can act on an unknown target, which then influences stimulation of synthesis of cyclic nucleotides in the cell, and the processes associated with this can involve cytosolic tyrosine-specific protein phosphorylation and might be affected by phospholipase C-dependent pathways.
Thus, BPIPP is a promising lead compound that suppresses stimulated synthesis of cyclic nucleotides, cAMP and cGMP, in intact cells. BPIPP acting via an unidentified target suppresses the effects of various stimulatory agents on membrane-bound and soluble guanylyl cyclases and membrane-bound adenylyl cyclases.
The interaction of BPIPP with BSA and inactivation of BPIPP during the course of this interaction might be an important pharmacokinetic property of this compound that might block systemic effects of BPIPP. Thus, BPIPP can be used for localized tissue treatment of disorders with excessive production of cyclic nucleotides without undesirable systemic effects. This property could make BPIPP an especially promising candidate for therapy of diarrheal diseases with oral administration. BPIPP could suppress activation of adenylyl and guanylyl cyclases by toxins produced by infectious agents in the intestine but should have minimal or no systemic adverse effects. The relatively easy synthesis (single step) and a presumed low cost should make it very attractive for therapy of diarrhea in developing countries, which is one of the major causes of mortality.
Materials and Methods
The chemical library was provided by Neuronautics. Compounds IIa, IVa, Va, VIa, VII, and VIII were purchased from ChemDiv. Other compounds (Ia, Ib, IIa, IIb, IIIa, IIIb, and IXa) were synthesized as described (15–17); see SI Materials and Methods for details.
Cell Culture.
Human colorectal carcinoma T84, rat fetal lung fibroblasts RFL-6, human neuroblastoma BE-2(C), and monkey kidney COS-7 cells were from the American Type Culture Collection and were grown according to the supplier's guidelines in the presence of 10% FBS (20% serum for RFL-6 cells) in a humidified atmosphere of 95% air/5% CO2 at 37°C. Cell viability was determined by trypan blue exclusion and with an lactate dehydrogenase assay kit (Cayman Chemical). Intracellular ATP was assayed with a luciferase-based ATPlite-M kit (Perkin–Elmer). For expression of human GC-C in COS-7 cells, the pZeo plasmid encoding the full-length human GC-C (from S. Waldman, Thomas Jefferson University, Philadelphia, PA) was mutated to the original sequence listed in the National Center for Biotechnology Information database (S139P) and then recloned into the pcDNA3.1 vector. COS-7 cells were transfected with 1 μg of purified plasmid per well of a 12-well plate by using Lipofectamine 2000 reagent (Invitrogen), and 24 h later cGMP accumulation in the cells was measured. Expression of GC-C was verified by Western blotting with an anti-GC-C antibody (18) (from S. Waldman).
Assay of cGMP and cAMP in Intact Cells.
Cells were grown to confluence in 12-well plates and washed three times with DPBS. Cells were treated with vehicle (DMSO, 0.1%) or BPIPP or other substances as indicated in DPBS containing 1 mM IBMX for 10 min, and then cyclic nucleotide accumulation was stimulated with activators of guanylyl or adenylyl cyclase as indicated for 4–10 min. Medium was aspirated and cGMP was extracted by rapid freezing of the plates at −80°C in the presence of 50 mM sodium acetate (pH 4.0; 0.3 ml per well) and measured by ELISA (19). cAMP was extracted with 0.1 M HCl (0.3 ml per well) and measured with a cAMP assay kit (acetylation protocol; Cayman Chemical). Cells were pretreated with bacterial toxins [cholera toxin (Calbiochem), pertussis adenylyl cyclase (E. Hewlett, University of Virginia, Charlottesville, VA), and anthrax edema factor (E. Hewlett)] for 30–60 min in the corresponding growth medium without serum.
STa Binding.
Whole-length STa (R. Giannella, University of Cincinnati, Cincinnati, OH) was iodinated and purified as described (20) to yield specific activity of 95 kBq/pmol. Binding of 25 pM 125I-STa to intact T84 cells was performed in F-12 medium for 1 h at 37°C. Cells were washed and solubilized with 0.1 M NaOH, and bound radioactivity was counted. Binding of 20–25 pM labeled STa to membranes was assayed as described (21), replacing 1 mg/ml BSA with 1 mg/ml ovalbumin. Nonspecific binding for the intact cell and membrane assays was determined in the presence of 100 nM STa and was subtracted to calculate the amount of bound toxin.
Assay of Guanylyl Cyclase Activity.
Activity of guanylyl cyclase was determined by RIA (22). Activity of purified human recombinant soluble guanylyl cyclase (23) (from E. Martin, University of Texas, Institute of Molecular Medicine, Houston, TX) was measured as described in ref. 24. For detailed description see SI Materials and Methods.
Effects of BPIPP on Ion Transport.
Transport of chloride in T84 cells was assayed as described in ref. 25. In some experiments, 6-methoxy-1-(3-sulfonatopropyl)quinolinium (SPQ) was used as a chloride ion fluorescent sensor instead of N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE) as specified in figure legends; for details see SI Materials and Methods.
Short-circuit current was assayed in T84 cells grown on 12 mm Millipore nitrocellulose inserts by using an Ussing chamber protocol (26). Cells were pretreated with 50 μM BPIPP or vehicle for 10 min in both hemichambers before addition of STa (100 nM) or forskolin (5 μM).
Rabbit intestinal ileal loop experiments were performed as described in ref. 27. Loops were injected with PBS containing indicated concentrations of BPIPP and STa or corresponding vehicles, and volume to length ratio was measured after 5 h.
Assay of tyrosine-specific phosphorylation in T84 cell extracts was performed with a K-LISA PTK screening kit (Calbiochem); for details see SI Materials and Methods.
Data are shown as mean ± SEM and statistical significance was calculated by using a Student's t test assuming P < 0.05 as significant. All experiments were repeated at least three times in duplicate, and either representative data or mean pooled values of multiple experiments are shown.
Supplementary Material
Acknowledgments.
We thank S. Waldman for pZeo plasmid encoding human GC-C and for anti-GC-C antibody; R. Giannella for STa; A. Morris for help with Ussing chamber experiments; E. Martin for a gift of purified human recombinant soluble guanylyl cyclase; S. Khatami (Neuronautics, Chicago, IL) for a chemical library; E. Hewlett for pertussis adenylyl cyclase, edema factor, and protective antigen of B. anthracis; and members of F.M.'s laboratory for helpful discussions. The work was supported by the Welch Foundation (AU-1437), the Dunn Foundation, the National Institute of Health (GM-061731), the Department of Defense (T5 grant), and the University of Texas at Houston Health Science Center.
Footnotes
Author contributions: A.Y.K. and B.-K.C. contributed equally to this work; A.Y.K. and F.M. designed research; A.Y.K., B.-K.C., M.E.E.-J., C.A.W., and R.L.G. performed research; A.Y.K., B.-K.C., M.E.E.-J., C.A.W., S.R.G., and R.L.G. analyzed data; and A.Y.K., S.R.G., and F.M. wrote the paper.
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0803096105/DCSupplemental.
References
- 1.Guerrant RL, et al. Magnitude and impact of diarrheal diseases. Arch Med Res. 2002;33:351–355. doi: 10.1016/s0188-4409(02)00379-x. [DOI] [PubMed] [Google Scholar]
- 2.Kosek M, Bern C, Guerrant RL. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull WHO. 2003;81:197–204. [PMC free article] [PubMed] [Google Scholar]
- 3.Qadri F, Svennerholm AM, Faruque AS, Sack RB. Enterotoxigenic Escherichia coli in developing countries: Epidemiology, microbiology, clinical features, treatment, and prevention. Clin Microbiol Rev. 2005;18:465–483. doi: 10.1128/CMR.18.3.465-483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Field M, Graf LH, Jr, Laird WJ, Smith PL. Heat-stable enterotoxin of Escherichia coli: In vitro effects on guanylate cyclase activity, cyclic GMP concentration, and ion transport in small intestine. Proc Natl Acad Sci USA. 1978;75:2800–2804. doi: 10.1073/pnas.75.6.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes JM, Murad F, Chang B, Guerrant RL. Role of cyclic GMP in the action of heat-stable enterotoxin of Escherichia coli. Nature. 1978;271:755–756. doi: 10.1038/271755a0. [DOI] [PubMed] [Google Scholar]
- 6.Vaandrager AB. Structure and function of the heat-stable enterotoxin receptor/guanylyl cyclase C. Mol Cell Biochem. 2002;230:73–83. [PubMed] [Google Scholar]
- 7.Schulz S, Green CK, Yuen PS, Garbers DL. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell. 1990;63:941–948. doi: 10.1016/0092-8674(90)90497-3. [DOI] [PubMed] [Google Scholar]
- 8.Forte LR., Jr Uroguanylin and guanylin peptides: Pharmacology and experimental therapeutics. Pharmacol Ther. 2004;104:137–162. doi: 10.1016/j.pharmthera.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Murad F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355:2003–2011. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- 10.Lucas KA, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- 11.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 12.Sager G. Cyclic GMP transporters. Neurochem Int. 2004;45:865–873. doi: 10.1016/j.neuint.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Hewlett EL, Gordon VM, McCaffery JD, Sutherland WM, Gray MC. Adenylate cyclase toxin from Bordetella pertussis. Identification and purification of the holotoxin molecule. J Biol Chem. 1989;264:19379–19384. [PubMed] [Google Scholar]
- 14.Gordon VM, et al. Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J Biol Chem. 1989;264:14792–14796. [PubMed] [Google Scholar]
- 15.Agarwal A, Chauhan PMS. First report on the abnormal dearylation/alkylation reaction in one-pot Hantzsch synthesis with 6-amino-1,3-dimethyl uracil. Synth Commun. 2004;34:4447–4461. [Google Scholar]
- 16.Agarwal A, Chauhan PMS. Solid supported synthesis of structurally diverse dihydropyrido[2,3-d]pyrimidines using microwave irradiation. Tetrahedron Lett. 2005;46:1345–1348. [Google Scholar]
- 17.Agarwal A, et al. Dihydropyrido[2,3-d]pyrimidines as a new class of antileishmanial agents. Bioorg Med Chem. 2005;13:6678–6684. doi: 10.1016/j.bmc.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 18.Birbe R, et al. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathol. 2005;36:170–179. doi: 10.1016/j.humpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Horton JK, et al. Enzyme immunoassays for the estimation of adenosine 3′,5′ cyclic monophosphate and guanosine 3′,5′ cyclic monophosphate in biological fluids. J Immunol Methods. 1992;155:31–40. doi: 10.1016/0022-1759(92)90268-x. [DOI] [PubMed] [Google Scholar]
- 20.Thompson MR, Luttrell M, Overmann G, Giannella RA. Biological and immunological characteristics of 125I-4Tyr and -18Tyr Escherichia coli heat-stable enterotoxin species purified by high-performance liquid chromatography. Anal Biochem. 1985;148:26–36. doi: 10.1016/0003-2697(85)90623-2. [DOI] [PubMed] [Google Scholar]
- 21.Crane JK, et al. Regulation of intestinal guanylate cyclase by the heat-stable enterotoxin of Escherichia coli (STa) and protein kinase C. Infect Immun. 1992;60:5004–5012. doi: 10.1128/iai.60.12.5004-5012.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krumenacker JS, Katsuki S, Kots A, Murad F. Differential expression of genes involved in cGMP-dependent nitric oxide signaling in murine embryonic stem (ES) cells and ES cell-derived cardiomyocytes. Nitric Oxide. 2006;14:1–11. doi: 10.1016/j.niox.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Martin E, Sharina I, Kots A, Murad F. A constitutively activated mutant of human soluble guanylyl cyclase (sGC): Implication for the mechanism of sGC activation. Proc Natl Acad Sci USA. 2003;100:9208–9213. doi: 10.1073/pnas.1633590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gocmen C, et al. The relaxant activity of 4,7-dimethyl-1,2,5-oxadiazolo[3,4-d]pyridazine 1,5,6-trioxide in the mouse corpus cavernosum. J Pharmacol Exp Ther. 2006;316:753–761. doi: 10.1124/jpet.105.094250. [DOI] [PubMed] [Google Scholar]
- 25.West MR, Molloy CR. A microplate assay measuring chloride ion channel activity. Anal Biochem. 1996;241:51–58. doi: 10.1006/abio.1996.0377. [DOI] [PubMed] [Google Scholar]
- 26.Broughman JR, et al. Chronic PKC-β activation in HT-29 Cl.19a colonocytes prevents cAMP-mediated ion secretion by inhibiting apical membrane current generation. Am J Physiol. 2006;291:G318–G330. doi: 10.1152/ajpgi.00355.2005. [DOI] [PubMed] [Google Scholar]
- 27.Alcantara CS, et al. Angiotensin II subtype 1 receptor blockade inhibits Clostridium difficile toxin A-induced intestinal secretion in a rabbit model. J Infect Dis. 2005;191:2090–2096. doi: 10.1086/430316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.