

Abstract
Bulimia nervosa is characterized by consuming large amounts of food over a defined period with a loss of control over the eating. This is followed by a compensatory behavior directed at eliminating the consumed calories, usually vomiting. Current treatments include antidepressants and/or behavioral therapies. Consensus exists that these treatments are not very effective and are associated with high relapse rates. We review evidence from literature and present original data to evaluate the hypothesis that bulimia involves alterations in vago-vagal function. Evidence in support of this include (1) Laboratory studies consistently illustrate deficits in meal size, meal termination, and satiety in bulimia; (2) Basic science studies indicate that meal size and satiation are under vagal influences; (3) Anatomical, behavioral and physiological data suggest that achieving satiety and the initiation of emesis involve common neural substrates; (4) Abnormal vagal and vago-vagal reflexive functions extend to non-eating activational stimuli; and (5) Studies from our laboratory modulating vagal activation have shown significant effects on binge/vomit frequencies and suggest a return of normal satiation. We propose a model for the pathophysiology of bulimia based upon de-stabilization of a bi-stable positive vago-vagal feedback loop. This model is not meant to be complete, but rather to stimulate anatomical, psychobiological, and translational neuroscience experiments aimed at elucidating the pathophysiology of bulimia and developing novel treatment strategies.
Indexing terms: vagus nerves, destabilization of positive feedbacks, pathophysiology of bulimia nervosa
Classification of Eating Disorders
The Psychiatric Diagnostic and Statistical Manual IV-R [1] recognizes a total of four disorders with an additional one listed as worthy of for diagnostic use. These disorders include obesity, anorexia nervosa, bulimia nervosa, eating disorder-not otherwise specified (ED-NOS) and, under consideration, binge-eating disorder. This paper will concentrate primarily on bulimia nervosa, although the other disorders will be discussed when appropriate for comparative purposes. Bulimia Nervosa is an eating disorder characterized by eating a large amount of food over a defined period with a definite loss of control over the eating episode. Eating is then followed by some type of compensatory behavior directed at eliminating the consumed calories. Depending on the type of behavior, bulimia is further divided into two subtypes: (1) purging or (2) non-purging. In this paper we focus solely on the purging subtype and our studies only include vomiting which constitutes the major form of purging seen in this group. The binge-eating and vomiting must occur on average of two times per week for three months to make the diagnosis. Their self-evaluation must also include an over-concern about body shape and weight. Two distinguishing characteristics of bulimia nervosa have led use to view this disorder as resulting from primarily, if not entirely, disruption of normal meal patterning. This article contains both a review of the literature in support of this idea and original experiments from our laboratory designed to test the mechanism underlying the proposed alteration in meal patterning.
Several behavioral and phenotypic differences, along with correlated physiological abnormalities, distinguish bulimia nervosa from the other eating disorders. Firstly, the behaviors of binge eating and vomiting are episodic occurrences. Indeed the rate of occurrence of these behaviors is one of the diagnostic criteria. Conversely, anorexia and obesity are chronic conditions, although the extent of the illness may show a waxing and waning course. Secondly, the diagnostic importance of body weight standards (phenotypic variables) also differs for these three main eating disorders. That is, the phenotypic characteristics of weight or body mass index (BMI) are crucial criteria for the diagnoses of both anorexia and obesity, but not an integral part of the diagnosis of bulimia. Thirdly, and perhaps most clinically important, the hierarchical decision process of choosing treatment options for the respective disorders is determined, to some degree, by different methods. For example, in both anorexia and obesity, the degree of altered body weight is crucial in determining the type of intervention. Treatments are loosely based on a continuum of severity of altered body weight. Examples of phenotypic based treatment strategies for anorexia nervosa include in-patient treatment for severely underweight patients with possible life-threatening co-morbid complications, an outpatient partial-day program for those individuals with higher (but not normal) BMIs who do not display as severe morbidity and are not considered at risk of sudden death, and individual counseling sessions including nutritional counseling. Typically, these differences in body weight and general appearance are both mirrored and influenced by the degree of altered electrolyte status (including magnesium) and other nutritional laboratory values, such as presence of ketone bodies [2]. In obesity, phenotypic-based treatments also dominate therapeutics. The least heavy patients may receive structured guidance on diet and life style, such as exercise programs, whereas treatments for more severe cases, such as high moderate to morbid obesity, may include more invasive surgical interventions such as gastric banding, bypass, or gastric reduction [3].
No true consensus on severity-based treatments exists for bulimia nervosa. In the clinical population, the most widely accepted treatment options are: (1) manual based cognitive behavioral therapy (CBT), or (2) pharmacotherapy, predominately with a serotonin-selective (re)uptake inhibitors (SSRIs); the gold standard being high dose fluoxetine (Prozac) [4,5]. Often combinations of these, including different antidepressant drugs, are used either concomitantly or in a “stepped-care” fashion [4] until either the symptoms totally remit, become sub-diagnostic, or no effective treatment is found. In the latter case, the patient is essentially abandoned to either find another health care provider or to simply accept that there exists no new strategies for treatment. Not surprisingly, the bulimia sufferers who fall into the later “no hoper” category have the highest morbidity, the greatest frequencies of bulimic episodes, and the longest duration of illness. Some estimates range as high as nearly fifty-percent of bulimia patients may ultimately fall into this category [6].
Clinicians, as well as the general public, sometimes view bulimia nervosa as being less serious than anorexia or obesity simply because there is a lower mortality rate by comparison. However, bulimia has many associated serious morbidity and occasional mortality consequences [7]. The individual and societal costs of the illness are summarized in Figure 1 [8,9]. Sadly, it may have taken the Terri Schiavo case to bring the gravity of this illness into the public limelight [10]. In short, bulimia nervosa robs 3% of women over their lifetime from reaching their full potential. That percentage doubles during the college years, affecting 6% of women, who are unquestionably some of the best and brightest of our still-developing young women. Given the disappointing outcome of current treatments (see below), novel mechanistic perspectives on the disorder and corresponding innovative treatment approaches are essential.
Figure 1.

Complications and Costs of Bulimia Nervosa
Current Front-Line Treatments for Bulimia Nervosa
Currently, two therapies available for treating bulimia are considered to be the front-line options. First, the use of structured, manual based psychotherapies to treat bulimia nervosa has been well established over the last 20 years. Cognitive behavioral therapy (CBT) is generally viewed as the gold standard for the treatment of bulimia nervosa at this time, although there is some evidence that self-help (manual based treatment conducted individually or in a group) may hold promise for some individuals [4,5]. The reductions in binge-eating frequency following CBT have generally ranged from about 50% to 80%. Rates of complete abstinence following CBT average from 35-50%. The second major treatment strategy has been pharmacotherapy. Since 1983 many studies of reported that bulimia nervosa patients respond to antidepressants, independent of the presence or absence of co-morbid depression. The majority of antidepressants in current clinical usage have been examined and with few exceptions antidepressants have been more effective than pill placebo control [11]. The use of serotonin specific (re)uptake inhibitors as become the standard treatment and fluoxetine (in doses ranging from 60 mg -80 mg; typically higher than used for depression) has received FDA approval for this indication [12,13]. As with CBT, the effect on binge-eating frequency is significant, though not as great as those seen with psychotherapy, typically ranging from a 40-60% reduction with 10-35% of patients reaching abstinence. Several studies have examined the utility of combining pharmacotherapy and psychotherapy to treat bulimia nervosa [5,14,15]. Overall, these studies do not suggest that this combination results in a meaningful improvement in the response to treatment.
Studies such as those summarized above contribute little to unraveling the pathophysiology of the illness. The CBT studies in general are not designed to be hypothesis-testing experiments aimed at understanding the underlying therapeutic mechanisms, but rather are usually treatment-outcome studies. The anti-depressant studies also add little, if anything, to understanding factors maintaining or driving the illnesses. In fact, many are pharmaceutical company subsidized and are designed to statistically evaluate the effects of different doses of established pharmacotherapeutic treatments (e.g. 40 vs. 80 mg of Prozac) or new drugs within established drug families (e.g. the SSRIs Prozac versus Zoloft). These studies often rely on large sample sizes for increased statistical power such that small effects are statistically significant.
The pharmacological approach detailed above is fraught with problems. Foremost, the results have little predictive value to an individual, since the significance is only apparent statistically and since recruited patients may not reflect the general clinical population. While such studies may add additional choices and combinations of options to the individual clinician’s armamentarium of treatment options, long-term remission of symptoms remains illusive. Secondly, an incidental undesired outcome of the multitude of studies examining various antidepressants alone or in conjunction with varying combinations of psychotherapy, is that the patient is often left paying for multiple treatments with different drug/therapy combinations, of which most are ineffectual over time. Thirdly, there exists the temptation to use reverse logic when interpreting the results. For instance, if increasing serotonin (5-HT) reduces bulimic behaviors, then the cause of bulimia must be a central nervous system serotonin deficit [16]. However, given: (1) the array of neuroscience knowledge on the different neurotransmitters which co-exist with different serotonergic nuclear groups; (2) the ubiquitous distribution of serotonin within the nervous system; and (3) the array of serotonergic receptors; implicating central serotonin as the main pathophysiological factor in bulimia has little provocative value, equivalent to stating the intuitively obvious: that bulimia nervosa is a nervous system disorder. This point is further underscored by the fact that 80% of total body serotonin is synthesized in gut enterochromaffin cells and is the sole source for platelet 5-HT [17]; yet studies on serotonin platelet binding in bulimia are interpreted as reflecting central serotonergic function [16,18].
This interpretation ignores the likely contribution of peripheral influences such as the peripheral actions of enterochromaffin peripheral serotonin, the effects of other gastrointestinal (GI) hormones release during a meal, and the influence of peripheral afferents on central nervous system (CNS) function, including behavior. In short, a simple “central catecholaminergic model” is antiquated, even if the model has been updated to include specific receptor sub-types or specific brain nuclei (as these ignore the corresponding hodology). Continuing to primarily pursue this avenue of research is uninspiring especially when there are multitudes of novel approaches to be considered.
Etiology versus Pathophysiology of Bulimia Nervosa
Crucial to establishing new or enhanced methods for illness prevention versus treatment is emphasizing the distinction between the etiology (origin of bulimia behaviors) and the pathophysiology (the biological mechanism which promotes or continues the pathology of abnormal eating) of bulimia nervosa, respectively. The writings of K.R. Miller in “Finding Darwin’s God” [19] provide a useful framework for conceptualizing the importance of this distinction. In discussing whether evolution is a fact or theory, Miller contends that the occurrence of evolution is indeed a fact. Fossil records indisputably demonstrate changes in a species over time. That some species become extinct and new ones appear can also be clearly documented. These findings are fact--indisputable truths laid out in geological data. The processes by which these occur--namely natural selection and adaptation-- are theories laid out by Darwin. Similarly, the facts of altered eating behaviors and the behavior changes that occur over time (natural history of the disorder) in bulimia nervosa, are also clear and may be accepted as etiological fact. However, the proposed mechanisms by which changes in the disorder occur (the evolution of the disordered behaviors), such as loss of volitional control over the behaviors and the eventual worsening of the disease, should be viewed as pathophysiological theories, which can be tested by scientific experimentation. We will present a theory that de-stabilization of vagus nerve control of meal termination plays a significant role in the pathophysiology of bulimia nervosa. We do not claim to have solved, or by no means cured, the underlying pathology. Rather, proposing this theory is solely for provocation of new research ultimately aimed at achieving a treatment end-point of complete and maintained abstinence of the bulimic behaviors.
The Etiology of Bulimia Nervosa
Many facts are known with regard to the etiology or origin of the bulimic behaviors. In the vast majority of cases, the behaviors of binge eating followed by compensatory vomiting begin as voluntary acts aimed at reducing caloric intake. Western societies undeniably place an unrealistic value on an ultra-slim physique, especially for young women in an age group prime to develop this disorder. Typically, these elective behaviors can start in the pre-teen years and, more frequently, in response to the weight gain often seen during the early college years. Strict parental control and high ambitions (dance, gymnastics, athletics, etc.) for their young daughters provide a substrate for a young woman to rebel by emphasizing her own control over her eating. Some have also emphasized the importance of the family meal in preventing the initiation of binge eating. The above briefly summarizes only a few of many reasons that someone voluntarily chooses to begin to couple over-eating (the binge) with self-induced vomiting. Independent of whether or not one views bulimia nervosa as a true disease or as a collection of disorder symptoms, there is a consensus as to the facts above as reasons why some young people initiate experimentation with binge-eating and vomiting. Some estimates claim as high as 50% of college age students will try these behaviors for weight control. Importantly, in a sub-set of individuals (estimated at between 1-4 % of the female population; 0.1-1% of men), there is a loss of self-control to stop the binge-eating episodes [1]. To relieve the guilt and worry caused by over-consumption, manual (physical insertion of an object, such as their finger or a toothbrush deep into the throat) stimulation of vomiting occurs. Thus, binge-eating and vomiting become intimately coupled and when they occur together at an average frequency of two times a week for three months, the individual is then considered to have developed diagnosable bulimia nervosa. When treating these bulimic women, many researchers will consider a sub-diagnostic frequency of binge/purge episodes as the benchmark for a successful treatment program. However, the ultimate goal of any new treatment strategy aimed at correcting the underlying pathophysiology should be the establishment of complete abstinence without re-occurrence. Some of the key questions to addressing this goal are: What causes some individuals who experiment with the behaviors (at sub-diagnostic levels) to develop full-blown bulimia nervosa? What physiological mechanism(s), i.e. the underlying pathophysiology, drives the loss of control over the binge-eating/ vomiting cycle such that after a certain point an individual cannot just give up the behaviors and start eating normally?
The Pathophysiology of Bulimia Nervosa
Bulimia nervosa most often evolves from a mild condition to a severe debilitating disorder. Therefore, initial insight to the pathophysiology of bulimia may lie simply in the fact that an individual does not just become bulimic, as one might just wake up with a cold. An individual that has never electively coupled binge-eating with self-induced vomiting rarely will develop the disorder. Rather, the behaviors evolve or change over time to become a self-perpetuating illness with an associated high morbidity, where the loss of voluntary control over binge-eating coupled with vomiting apparently has been induced (reinforced) by the bulimic behaviors itself.
Repeatedly engaging in the bulimic behaviors leads to self-consuming urges to continue an episodic pattern of binge-eating followed by vomiting. At some point, engaging in the bulimic behaviors produce what patients often independently call a positive “after-glow,” a time of near euphoria, when all anxiety, tension, and depressive thoughts are relieved. This euphoric state is relatively short lived, and the urges to binge-eat and vomit return. Engaging in the bulimic behaviors provides a period of positive reinforcement of short-duration, followed by a period of uneasiness during which the urge to binge/vomit increases and is accompanied by feelings of loss of control over the urge to binge-eat. Feelings of self-doubt, dissatisfaction with many facets of life, body image disturbances, and mini-depressive episodes also accompany the urge to binge/eat. These unsettling feelings are relieved, and thereby positively reinforced, by engaging in the next episode of binge-eating and vomiting. The duration between the end of the positive feelings following the completion of a bulimic episode and the peak of the uncontrollable urge to repeat the behaviors would correspond to the approximate length of time between bulimic episodes, or the inter-binge interval (IBI). Thus, by definition, the more frequent the episodes are, the shorter the IBI, and the more severe the illness in terms of frequency of abnormal eating behaviors. At some point during this period of increasing frequencies of engaging in the behaviors, the need to manually stimulate the vomiting reflex becomes unnecessary and emesis becomes a natural consequence of the binge-eating episode. Succinctly stated, the abnormal behaviors are spinning out of control, with the probability of successful treatment decreasing accordingly.
Evidence for a Vagal Pathophysiology in Bulimia Nervosa
Multiple scientific approaches from independent research groups converge to suggest that the vagus nerves play a significant, if not primary, biological role responsible for causing the bulimic behaviors to become self-perpetuating, or self-reinforcing. These findings include: (1) Laboratory studies in bulimia and control patients consistently illustrate deficits in meal size, meal termination, and satiety; (2) Basic science studies in both animals and humans clearly indicate that meal size and the satiety accompanying meal termination are under vagal influences; (3) Anatomical, behavioral and physiological data in both rats and humans suggest that achieving satiety (the process of becoming satiated) and the initiation of emesis involve common neural substrates; (4) Abnormal vagal and vago-vagal reflexive functions occur when vagal activity is stimulated in bulimia patients using non-eating activational stimuli; and (5) Studies from our laboratory examining experimental methods for modulating vagal activation have shown significant effects on binge/vomit frequencies and suggest a return of normal satiation. Collectively, this information summates to strongly support a focus by multiple research groups on understanding abnormal vagal function in bulimia. Each of these five converging lines of inquiry are presented in detail below, including some summarized original data from our investigations (these summarized data will be presented in their entirety elsewhere).
(1) Meal size and meal termination are inadequately controlled in bulimia patients
A dampened satiety response to meal consumption in patients with bulimia nervosa has become a well-established biological finding [20,21,22]. When the development of satiety is studied in response to a fixed-volume test meal, the satiating potency of the meal is reduced in bulimia nervosa subjects [23]. Under a more naturalistic setting in which bulimia and control subjects were allowed to freely eat from a “banquet” of foods, the bulimia nervosa subjects required more food to achieve the same level of satiety as the free-feeding control subjects [21]. An extension of this study compared the development of intra-meal satiety in bulimia nervosa patients to normal controls [22]. Measuring subjective ratings of fullness, hunger, etc., after each 75ml increment of consumed liquid diet, showed a significantly greater amount of food was required to produce an equivalent change in both “hunger” and “fullness” ratings in the bulimia subjects. These findings make an important contribution in that they provide evidence that individuals with bulimia nervosa are capable of feeling full, it just requires a larger food bolus, or gastric load. Collectively, the findings summarized in this section suggest that the neural systems mediating meal termination and satiety are intact (i.e. bulimic patients can feel full); rather the threshold for their activation appears to have been shifted upward (i.e. a greater food volume is required). Therefore, identification of the neural systems involved in the control of meal termination will provide a foundation upon which to base further investigations into the underlying pathophysiology.
2. Neural control of meal size and meal termination involves vagal control
Activation of vagal afferents has long been recognized as sufficient for meal termination. Vagal afferent provide the major connection between the gastrointestinal tract and the brain. During feeding, vagal afferent fibers are activated by both physical distention of the stomach [24,25] and by select duodenal peptides and pancreatic hormones [26,27,28]. The ultimate goal of this paper is to present information relevant to possible abnormalities in vagally-induced satiety in bulimia. To accomplish this goal, specific theoretical constructs will be used to differentiate between various processes in eating and energy balance. While these theoretical constructs may over-compartmentalize the highly coordinated processes that ultimately enable a normal eating individual to regulate and maintain a relatively stable body weight over long periods of time, this paper will dissect this process into detailed components to aid in identification of where the process fails in individuals with bulimia.
In almost all mammals, eating occurs in defined episodes or bouts, commonly referred to as a meal. Cephalic factors such as taste, odor, and environmental circumstances exert the first influence on meal consumption, and in part function to prepare the gastrointestinal tract to accept and process nutrients. A variety of digestive enzymes and gastrointestinal motility change occur during this stage, and these factors are considered a ‘positive’ influence aimed at promoting meal consumption [29]. Once food passes through the pharynx and esophagus to enter the stomach atrium, the ensuing events are considered “negative” as they feedback to the central nervous system to inhibit or terminate the meal [30]. The vagus nerve is recognized as a major route for relaying information on food intake to the brain [31]. During the meal, the stomach fills with food and relaxes to allow for more food, through two different mechanisms [32]. Following acidification and emulsification of gastric contents, the process of gastric emptying moves food contents through the pyloric sphincter and into the small intestines, specifically the duodenum. It is here that intra-luminal contents, such as long-chain amino acids, fatty acids, and carbohydrates stimulate the release of specific hormones and gastric enzymes (e.g. amylase). Luminal factors are chiefly aimed at converting the consumed food into absorbable nutrients. The released luminal hormones can summate with gastric distention to increase vagal stimulation apparently directly through c-fibers and through a-fibers by augmenting gastric distention (e.g. cholecystokinin (CCK), serotonin, mucosal leptin [26,33,34,35,36,37]. At approximately this point in the digestive process, no actual calories have been absorbed into the local circulation for ultimate distribution to the rest of the body. Therefore, this portion of the meal is typically termed the pre-absorptive phase. Short-term satiety (intra-meal and early phase post-prandial satiety) occurs during this period.
As food continues through the intestines specific macronutrients are further processed and are either absorbed and function to supply energy or are stored, evolutionarily for the purpose of future use during times of limited food availability. This later, post-absorptive stage is quite complex, and includes (1) nutrient acquisition, namely measuring sugars, fats, and proteins, by specific receptors and (2) monitoring nutrients through specific hormones with the goal of signaling the amount of calories consumed during that feeding episode. This post-absorptive phase is not essential for short-term satiety, meaning meal termination, but rather is balanced with mechanisms monitoring energy expenditure and meal initiation to regulate body weight.
As mentioned above, women with bulimia do not reach the same level of satiety as control subjects when given a fixed volume liquid diet. This finding is highly replicable, and G.P. Smith suggested over a decade ago that this deficit likely accounted for the binge eating [38]. Subsequent research has demonstrated that satiety is not limited to cognitive perception but is also demonstrated by the abnormal gastrointestinal processing of a meal in bulimia patients. These abnormalities will be detailed below. First, however, it cannot be over-emphasized that unlike anorexia nervosa and obesity, the vast majority of women with bulimia are of normal body weight [39]. That is, despite the extreme physiological stress placed on their GI and inter-related systems by the repeated binge-eating and vomiting, these women with bulimia are capable of monitoring calories absorbed and balance this with energy expenditure, such that a relatively stable, and normal body weight is maintained. Thus, it would appear that the long-term energy balance phase of eating is functioning properly in bulimic individuals. Instead, pre-absorptive short-term factors underlying meal termination seem to be specifically altered. This distinction is particularly relevant given that eating disorder specialists often combine bulimia nervosa and binge-eating disorder, in which the latter are typically obese.
As we develop our presentation of evidence in support of vagal involvement in bulimia, it is crucial to always be cognizant of the fact that cortical processing of feelings surrounding food is also altered in bulimia [20]. Somehow, the mechanisms tying environmental influences and higher thought processing need to be connected to abnormal peripheral vagal physiology. Thus, we conducted a Positron Emission Tomography (PET) study in normal eating young women to better understand the CNS processing of a pure mechanical gastric load. This study was conducted at the VA Medical Center, Minneapolis, in collaboration with Jose Pardo and Elke Stephan [40]. Eighteen young healthy women were recruited into this VA Institutional Review Board approved study. After insertion of an arterial line, the subject moved into the PET suite where a balloon connected to an oro-gastric tube was inserted. After being placed into the scanner, the balloon was inflated with tepid water until the subject reported a sense of satiety and gastric fullness (“like after a Thanksgiving dinner”). Labeled water was injected through the intra-arterial line, and a 90s scan was conducted. The balloon was then deflated, and the scan was repeated. Scans were separated from each other by approximately 10 minutes. On average, 3 cycles of inflation/deflation scans were conducted in each subject. Analyses compared brain activation during the “inflated” (cognitive feeling of gastric fullness) and “deflated” conditions. A significant increase in regional cerebral blood flow was seen in the brain stem, and in primary and association cortices. While the nucleus of the solitary tract (NTS) lies too deep for visualization using this method, significant activity was apparent within the pontine parabrachial nucleus (PBN). The PBN is a major relay nucleus for ascending information from the NTS, sending projections to both hypothalamic and thalamic nuclei. Thus, this finding is very consistent with what would be expected to accompany vagal stimulation by a gastric load. Higher order activation was observed in the left inferior frontal gyrus and several subnuclei within the insular cortex. Lateral inferior frontal and orbitofrontal areas respond to food-associated stimuli, such as visual, olfactory and gustatory inputs. Affective components of feeding have also been attributed to these regions [Fig. 2].
Figure 2.
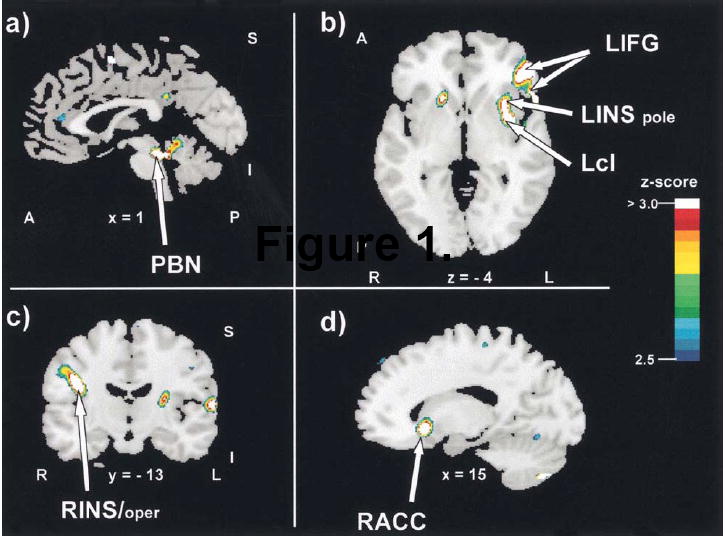
Increases in regional cerebral blood flow during stomach distension. Arrows point to significant changes in blood flow in the difference image (inflation minus deflation). Letters and numbers below the slices indicate their orientation and stereotactic coordinates. A = anterior; I = inferior; L = left; Lcl = left claustrum; LIFG = left inferior frontal gyrus; LINS = left insula; LINS pole = left insular pole; P = posterior; PBN = parabrachial nucleus; R = right; RACC = right anterior cingulate cortex; rCBF = regional cerebral blood flow; RINS/oper = right insula/operculum; S = superior; x = sagittal plane; y = coronal plane; z = horizontal plane.
3. Both satiety and emesis are dependent on vagal afferent stimulation
The mechanisms underlying meal size, meal termination and the production of satiety have been discussed above. While over-eating occurs in many situations, in bulimia a large volume of food (compared to that of a normal eating individual) is consumed rapidly, with an accompanying lose of control, over a relatively short period of time, or in other words, during a defined meal. That is, the bulimic behaviors are organized into well-defined bouts. Another feature which makes bulimia nervosa unique among other over-eating syndromes is the compensatory behavior which occurs following food consumption with the aim of reducing caloric intake. The most common compensatory behavior is vomiting. In more severe cases, vomiting can occur reflexively, without any manual stimulation of the gag reflex. Thus, it seems essential that any valid pathophysiological model of bulimia must incorporate a mechanism by which binge-eating, (viewed here as a large, unphysiological, gastric load) could become so intimately tied to the emetic reflex. In light of the above discussion of vagal control of short-term satiety, it seems relevant to examine the role of the vagus in the initiation of vomiting.
Vomiting can occur in response to several stimuli. For example, the vestibular system is well known to connect with medullary regions to initiate vomiting in response to motion. In some instances, especially in infants, vomiting can occur for specified reasons or be idiopathic and can resolve on its own or with limited treatment [39]. Vomiting may also occur in response to the presence of consumed or circulating toxins. The ultimate causation of this type of emetic reflex is to remove the toxic material before absorption into general circulation can take place [41]. Both ingested and systemic toxins can initiate this type of vomiting. For example, chemotherapeutic agents and radiation exposure leads to toxin exposures at levels high enough to serve as an activational stimulus of the emetic reflex. The mechanism underlying toxin induced vomiting has been delineated and, like meal termination, involves some of the same subdiaphragmatic vagal branches, most notably the gastric branches. The presence of a toxin elicits the release of serotonin from gastric enterochromaffin cells [41]. Serotonin of this origin acts in a paracrine fashion on excitatory, ligand gated ion channels on vagal afferents which reflexively activates dorsal motor nucleus vagal (parasympathetic preganglionics) efferents to cause vomiting and empty the gastric cavity. The act of binge-eating followed by manually stimulating vomiting would be expected to result in suprathreshold levels of activation of the vago-vagal reflexive pathway. Binge-eating would stimulate vagal afferents through application of a large gastric stimulus followed by further stimulation of vagal afferents through self-induced vomiting.
Direct experimental evidence also supports the premise that meal termination and vomiting involve activation of a vagally-dependent pathway. Some of this work has stemmed from the necessity to show that an administered agent decreases food intake specifically, and is not explained by making the animal sick or causing a generalized malaise. Two GI hormones serve as good examples that satiety versus emesis can be a dose-dependent function. For example, CCK is naturally released at relatively low levels in response to specific luminal nutrients and administration of low doses of CCK, within the physiological range, have been demonstrated to induce a true satiety [42,43], and antagonists specific to vagal CCK-A receptor increase food intake (FI). Doses in a relatively low range do not support a conditioned taste aversion, as would be expected if a malaise was being produced. However, higher doses of CCK do indeed appear to induce nausea and vomiting, release oxytocin or vasopressin, depending on the species, from the paraventricular nucleus (PVN) (an event associated with emetic agents), supporting the finding of a conditioned taste aversion [44].
The induction of both satiety and emesis has also been shown for peripherally administered serotonin acting on the 5-HT3 vagal receptor [41,45]. As mentioned above, serotonin liberated from enterochromaffin cells in the GI tract acts on vagal afferents to initiate vomiting. Clinically, 5-HT3 antagonists are widely used to combat the debilitating side-effect of chemotherapeutic agents. The emetic action of peripheral serotonin is mediated through activation of vagal afferent receptors. Additionally, peripheral serotonin suppresses food intake in a vagally-dependent manner. Whether the effect of the exogenous ligand is satiety or nausea/emesis, both are apparently mediated through higher order neurons in the NTS. In this regard, there are differences in the literature as to whether the same or different vagal fibers are involved. That is, it is possible that the two neurochemical phenotypes act synergistically through parallel pathways [37].
Given the importance of the intimate relationship of binge-eating followed by vomiting in characterizing bulimia nervosa, we have conducted two experiments to directly address this question. Others [46] have conducted studies analyzing the effects of CCK across a range of doses to determine if satiety or malaise resulted. We extended these studies (Hofbauer et al., in preparation), by examining the effect of doses of intraperitoneal (IP) CCK at both satiating and malaise producing doses. The number of cellular nuclei positively stained for c-fos proteins using immunohistochemistry were counted in the NTS and area postrema at four different levels along the anterior-posterior axis. C-fos nuclear proteins are induced upon cellular depolarization, and their localization provides a neural activity map. The doses of CCK investigated were 0, 1.5, 3, 6, 10, and 100 μg/kg. The behavioral shift from true satiety to beginning to cause malaise would be expected to start occurring no lower than between the 3 and 6 μg/kg dose [42]. The number of c-fos positive cells increased in a linear fashion until saturation occurred with no differences observed between the 10 and 100 μg/kg doses. While a detailed location of stained nuclei remains to be completed, the linear nature of the dose-response is suggestive of activation of a single pathway or independent pathways (through different fiber types, c vs. a, or wide-dynamic range neurons) acting in concert to activate NTS neurons [37] in a graded fashion. This finding establishes the existence of the neural substrates necessary for the establishment of a bi-stable system of low activity between meals and maximal activity during emesis (see below for significance).
In a second study aimed at determining the relationship between satiety and emesis, we reported the effect of an established anti-emetic dose of ondansetron, a 5-HT3 ion channel blocker in reducing the suppression of food intake by CCK [47] [Fig. 3,4]. CCK interactions with the 5-HT3 receptor blockade were monitored behaviorally by measuring liquid food intake in mildly deprived rats and also physiologically/anatomically by quantifying c-fos induction in the NTS. This study found ondansetron (2mn/kg) produced approximately a 50% reduction in both the suppression of food intake and the activation of C-fos by IP CCK (3μg/kg). Subsequently, Hayes, Savastano, and Covasa found that the enhancement of CCK-induced c-fos in the hindbrain by gastric distention was also dependent on activation of the 5-HT3 receptor [37].
Figure 3.
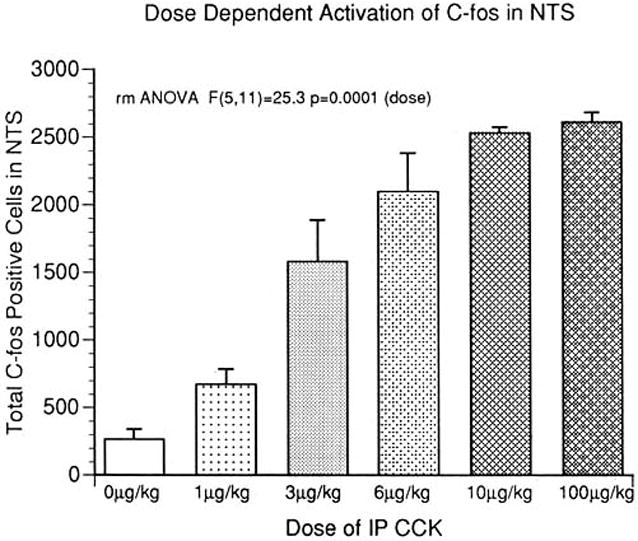
Administration of CCK-8 increased the average number of c-fos positive nuclei sampled at four levels of the NTS. The doses (0 μg/kg - 100 μg/kg) ranged from those that elicit true satiety to those associated with malaise. The results indicate a linear, or graded, response with saturation occurring at 10 μg/kg.
Figure 4.
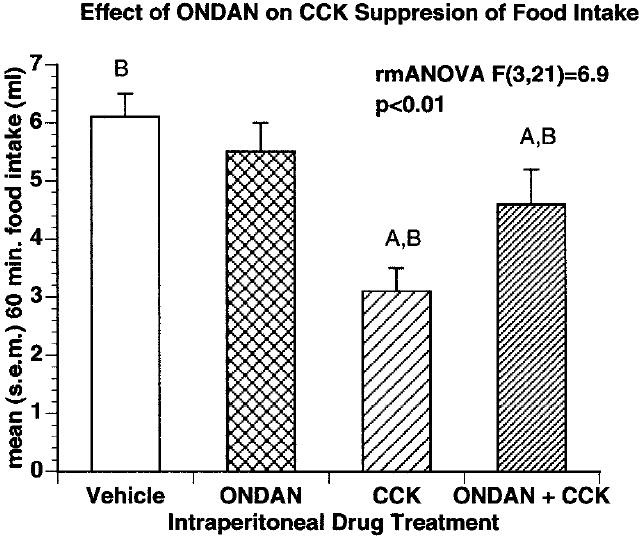
Effect of i.p. drug treatment on liquid food intake. Administration of 3 mg/kg CCK significantly reduced food intake over sixty minutes compared to vehicle. Injection of 1 mg/kg ONDAN in combination with 3 mg/kg CCK significantly attenuated the satiating effect of CCK on food intake. Groups with the same letters are significantly different from each other P , 0.05.
To summarize the above, whether parallel or the same vagal fibers carry satiety versus emetic information, these two functions are intimately connected, and provide strong support that the non-physiological, repeated coupling of binge-eating and vomiting would result in a stronger vagal activational stimulus than either behavior by itself. Given the facts that in bulimia, a stronger stimulus of food is required for eliciting satiety and that vomiting becomes automatically coupled with the binge, it seems possible that the repeated and frequent engaging in these behaviors induces a desensitization in the threshold for activation of the functionally corresponding vagal afferent fibers. Such a mechanism, explored in more detail below, would explain the malignant course of this illness, where first voluntary control over the binge is lost, then the frequency of the behaviors increases, and vomiting becomes an unstoppable consequence of the illness. In other words, an oscillatory shift in the threshold for activation of vagal afferents would provide a pathophysiological mechanism capable of explaining the evolution of this illness over time.
4. Vagal abnormalities extend to other non-satiety related functions in bulimia
If vagal afferent function has been altered in bulimia as proposed above, then it would be likely that other vagally-mediated processes not directly related to satiety or emesis would also be abnormal. Indeed, several other incidences of vagal dysfunction have been reported by our group and others. We first began to explore the possibility of a wider range of vagal abnormalities in bulimia back in the late 1980s [48]. At that time, there was evidence from other laboratories, primarily from Randich and colleagues [49,50], that vagal stimulation resulted in inhibition of somatic pain at the level of inhibition of pre-synaptic nociceptive input through dorsal root ganglia by stimulation of a poly-synaptic descending pain-inhibitory pathway. Thus, we reasoned that an increase in vagal activity (reflected the increase in the activational threshold) would result in a decrease in sensitivity to somatic pain in bulimia compared to control subjects. Therefore, we studied pain detection thresholds in these two subject populations using a constantly increasing pressure stimulus applied to the ventral surface of the most distal phalanx of fingers 2-5. Indeed, the pressure required to exert a painful stimulus was significantly elevated in the bulimia compared to control patients [Fig. 5]. This effect appeared specific to pain detection, as both the more cortically modulated ability to tolerate pain was not different between groups when the data were analyzed using the pain detection threshold as a co-variant (unpublished analysis) and the ability to detect a light touch stimulus also was not significantly different between groups [Fig. 6].
Figure 5.
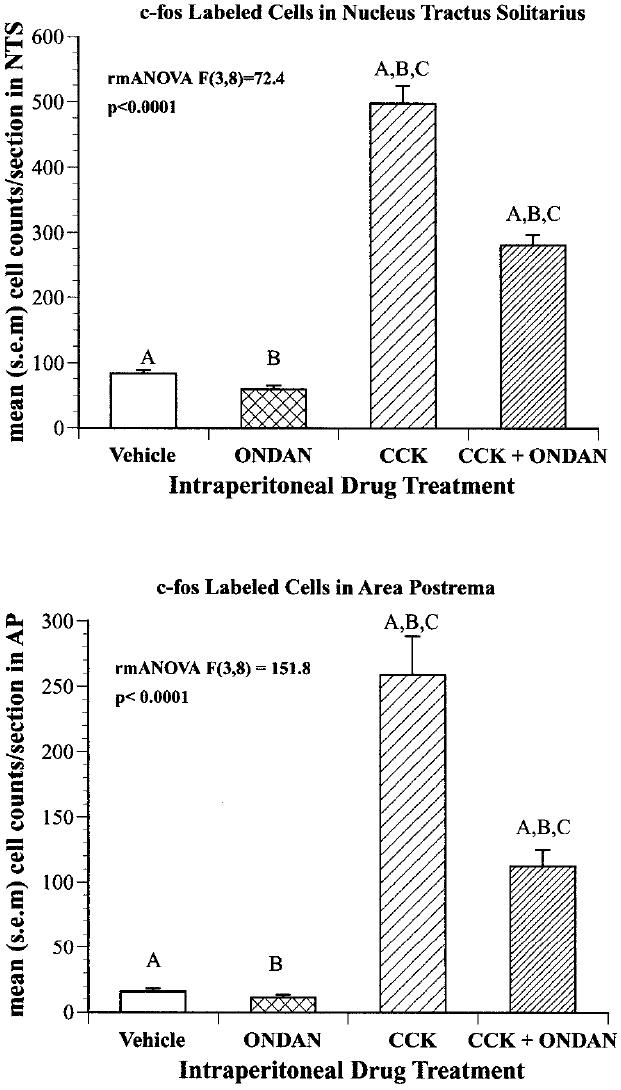
Effect of 3 g/kg CCK and/or 1 mg/kg ONDAN on the number of c-fos labeled nuclei in the nucleus of the solitary tract (TOP) and in the area postrema (BOTTOM). Values on the Y-axis represent the mean number of cells per animal sampled (n 3) from four anatomic levels along the caudal-rostral plane. Groups with the same letters are significantly different from each other P 0.05.
Figure 6.
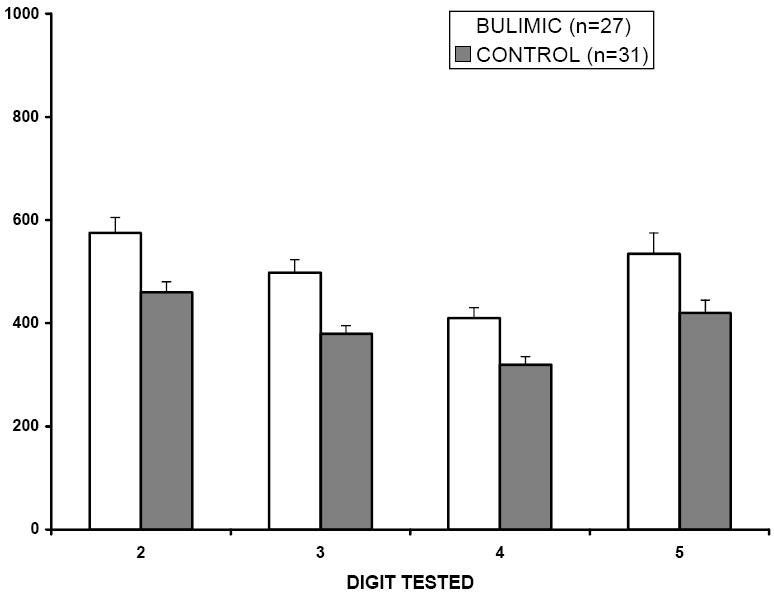
Pain thresholds (mean ± SEM) in normal subjects and in patients with bulimia nervosa. The ability to detect a noxious pressure stimulus was determined in 27 female patients with bulimia nervosa and in 31 normal, sex and age-matched controls. The force, in grams, being exerted at the time of the subjects’ verbal response was recorded. Each finger of the non-dominant hand was tested in succession, starting with the fifth digit. Pain detection thresholds were found to be significantly elevated in the bulimic population (p< 0.01; repeated measures ANOVA).
Other groups have both replicated and extended these findings using pressure stimuli as well as testing other nociceptive modalities such as thermal pain detection [48,51,52,53]. Several factors have been demonstrated not to be statistically related to pain thresholds. These include dietary restriction [51], depressive symptoms [48,52], alexithymia [52], anxiety levels [51], plasma cortisol [51], skin temperature as an index of abnormal thermoregulatory processes [51], body weight [48], and peripheral nerve function [51]. Collectively, these findings indicate a certain degree of specificity of the elevated pain detection thresholds to bulimia, as opposed to either an unrelated factor or to a consequence of the illness.
In addition to pain detection, others have studied vagal and vago-vagal responses in bulimia [54]. During their study of altered pain detection in bulimia, Girdler et al. [53] noted that only in the women with bulimia was blood pressure correlated to pain sensitivity. During this study, blood pressure regulation was also studied as this measure represents an autonomic function influenced in part by vagal efferents. From a mechanistic standpoint, these investigators mentioned that one explanation for their results would be maladaptive changes in afferent vagal function since systolic blood pressure regulation is modulated by peripheral vagal afferent stimulation (e.g. peripherally administered opioids). In a more recent study, these findings were nicely replicated and extended by Murialdo et al [55]. Their study used the tilt-table test to assess sympatho-vagal modulation of heart rate variability. They found that both bulimia and anorexia patients showed decreased sympathetic activation and prevalent vagal activity after tilting. Rissanen et al [56] reported that cardiac vagal tone was elevated in women with bulimia nervosa and that this elevation was reduced to control levels by fluoxetine (a SSRI) treatment. However, SSRI treatment did not significantly reduce bulimic behaviors, not atypical for this treatment method. Despite this lack of a demonstrated relationship between cardiac vagal tone and the primary disease variables, these authors noted that SSRIs can desensitize 5-HT3 receptors. Collectively, these findings suggest that the altered afferent activity, evidenced by changes in satiation, is accompanied by changes in both mono- and poly- synaptic vagal efferent activity.
Additional evidence for vago-vagal abnormalities perpetuating bulimic symptoms is provided by physiological studies on GI processing of a meal. Geliebter et al. [57] have reported an increase in gastric capacity in bulimia nervosa subjects when meal-induced gastric distention is experimentally simulated by inserting a balloon into the stomach. Additionally, Devlin et al. [58] measured both the postprandial release of CCK and gastric emptying in bulimia patients. They found that both measurements were blunted in bulimia patients, compared to controls. They concluded that the delayed emptying most likely resulted from increased gastric capacity [57] and thereby likely led to the blunted CCK release.
Since the behaviors of bulimia nervosa are episodic, i.e. occur in defined bouts, as opposed to being a constant feature as in obesity and anorexia, any pathophysiological model for bulimia should include a component responsible for controlling the defined bouts of binge/eating and vomiting. Therefore, we conducted an experiment to extend our earlier finding on pain thresholds in which we examined if there was a temporal relationship between the elevation in pain thresholds and the occurrence of a bulimic episode [59]. Our approach was to repeatedly measure pain detection thresholds (PDTs) in the same individuals at weekly intervals and to statistically determine if the time since the subject had last engaged in a bulimic episode was a significant predictor of pain thresholds. Fourteen subjects participating in ongoing clinical trials were enrolled in this study. PDTs were measured weekly over the period of time which the subject was not on active drug. For comparison, data collected by our laboratory from 67 age-, weight-, and sex- matched normal-eating control subjects indicate a mean pressure PDT of 387 grams (s.e.m. ± 19; unpublished data).
The resultant findings yielded valuable insights into the relationship between PDT and the bulimic behaviors. Mixed regression analysis indicates that the time elapsed since last engaging in a binge/vomit episode was a significant predictor of pain detection thresholds [59,60]. That is, pain detection thresholds change dynamically over the interval between binges reaching their zenith prior to a bulimic episode and their nadir by one hour after a vomiting episode. An additional analysis of this data was done to take into consideration individual severity as defined by frequency of bulimic behaviors. For each individual, an average weekly duration of time between bulimic episodes was calculated. This value simply amounted to dividing the number of hours in one week (168 hours) by the number of episodes that individual had during the week between PDT evaluations. The calculated value represents the average weekly IBI. Data from each testing session from all subjects were then portioned into two bins: (1) PDT data collected within their average IBI (this bin on average contained data prior to the overwhelming urge to binge-vomit); and (2) data collected at a time when a subject would have not had an episode within their average IBI (these data would on average be temporally closely associated with the occurrence of the next bulimic episode and thereby likely associated with higher urges to engage in the bulimic behaviors). When mixed regression analyses were conducted on the IBI data, only data collected at times exceeding the average IBI were significantly elevated compared to normal control data (unpublished comparison) and to data collected within the average IBI [Fig 7]. That is, not engaging in the behaviors (exceeding the normal IBI) is associated with a worsening of the underlying physiology whereas engaging in the behaviors is associated with a short-term normalization of the underlying physiology, thus reinforcing the abnormal eating behavior.
Figure 7.
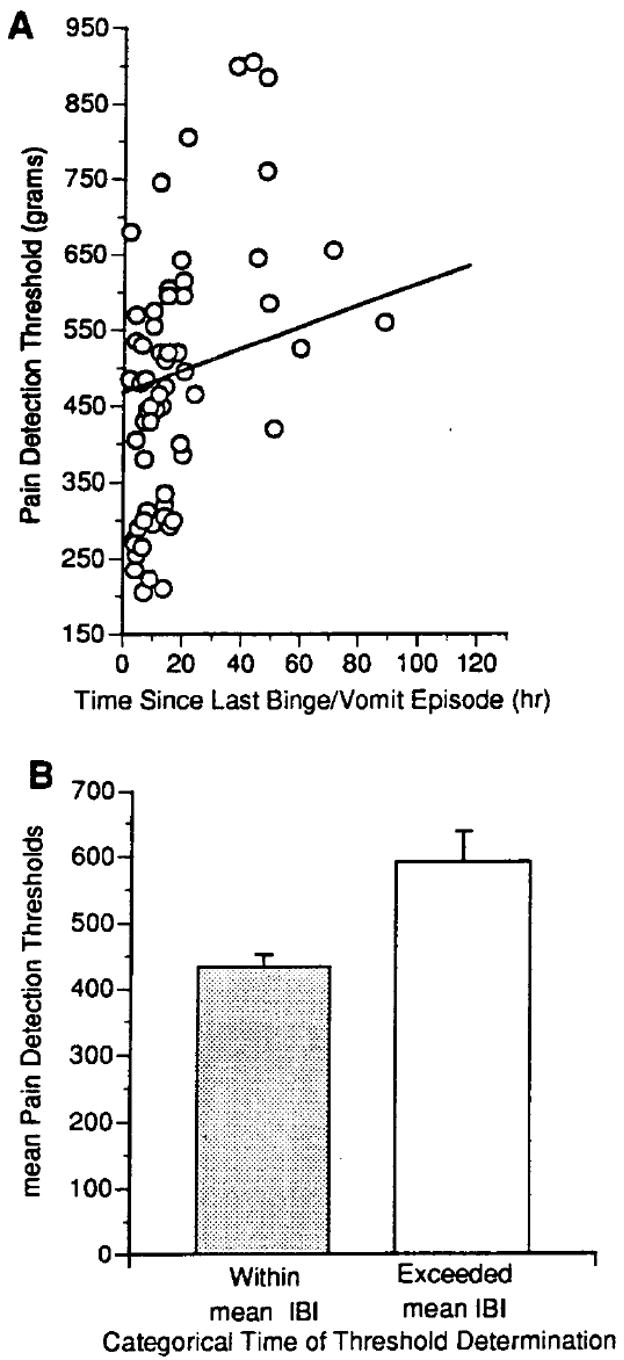
Relationship between pain detection thresholds and engaging in bulimic behaviors in 14 bulimia nervosa patients not concurrently receiving active treatment. (A) A scatterplot of 63 total observations illustrating pain detection thresholds measured at different times following an episode of binge-eating and vomiting. The line indicates the slope of the maximum likelihood regression obtained by random regression analysis (z = +2.42; P = 0.0140. (B) Categorical subdivision of pain detection thresholds based on whether the measurements were collected prior to (within category; 34 observations) or after (exceeded category; 20 observations) the expected duration of an inter-binge interval (z = +2.42; P = 0.03). IBI = average length of an inter-binge interval during the preceding week.
The specific patterning of the changes in pain detection is significant for three reasons. First, since the elevation in pain thresholds occurs prior to, not after, a bulimic episode, it is unlikely that this elevation is a direct effect of binge-eating or vomiting. Second, the elevation in PDT prior to a bulimic episode is consistent with the possibility that the mechanism giving rise to the increase in pain detection is also the factor driving the abnormal behaviors, in other words a mechanism representing the pathophysiology of the disorder. Third, the fact that pain thresholds return to normal in close temporal association with having preformed the bulimic behaviors suggests a cyclic oscillation in the underlying neural substrate(s) and a period of relative refractoriness during this time of “re-setting” neural activity to approximate normal levels.
5. Clinical interventions aimed at modulating vagal neurotransmission result in an improvement of bulimic symptoms
The above findings indicated that pain detection thresholds vary dynamically across the inter-binge interval and therefore appear physiologically connected to the abnormal eating patterns. The following set of experiments were aimed at determining the involvement of vagal afferent neurotransmission as a common mechanism in mediating the concordant relationship between cyclic changes in pain detection thresholds and the initiation of an episode of binge-eating and vomiting. To address this question, we relied on the use of ondansetron (ONDAN) (Zofran; Glaxo Wellcome) as a pharmacological tool to suppress activity in vagal afferent fibers.
ONDAN was chosen as the pharmacological agent to conduct a “proof-of-concept” inquiry (used here to describe studies aimed at testing underlying physiology with the purpose of distinguishing these experiments from a formal, definitive clinical trial aimed at devising a specific new treatment) into vagal involvement in perpetuating binge-eating and vomiting episodes in bulimia nervosa. It was chosen based on its functional action on vagal nerve activity, rather than to evaluate the neurochemical phenotype of vagal afferents. ONDAN is a potent and selective antagonist of the 5-HT3 receptor [61]. This serotonin receptor subtype is an excitatory, ligand-gated cation channel that mediates fast depolarizations [62]. In humans, ONDAN displays relatively poor penetrability into the CNS [63]. Steady state cerebrospinal fluid concentrations measured in six individuals after oral administration of 32 mg/day for 2.5 days were less than 15% of that in blood (ranges 5-15%). One reason for the poor brain permeability appears to be explained by the finding that ONDAN is a fair to good substrate for P-glycoproteins located in the endothelial calls of blood capillaries within the blood brain barrier which function to extrude specific drugs and toxins from entering the brain [64]. ONDAN is marketed for treatment of the nausea and vomiting induced by serotonin release from gastric enterochromaffin cells in response to chemotherapeutic agents and radiation treatment. Electrophysiological studies have shown that 5-HT3 antagonists block the activation of receptors on chemosensitive afferent vagal fibers [17] and functionally, this vagal blockade is responsible for their anti-emetic action [65,66]. Li and colleagues have demonstrated that either 5-HT3 antagonists or subdiaphragmatic vagotomy would block nodose ganglia firing elicited by maltose and glucose infused into the intestinal lumen[67,68]. We have also reported that ONDAN blocks vagally-mediated c-fos activation in response to intraperitoneal CCK [47]. Thus, ONDAN not only has an indisputable peripheral site of action on vagal afferents in general, but also is active on the subpopulation of vagal afferent fibers with functional relevance to bulimia nervosa-namely those mediating satiety and emesis.
Given that ONDAN clearly acts on vagal afferent fibers to suppress neural activity, if treatment with this drug did not moderate bulimic symptoms then we would accept the null hypothesis that elevations in vagal activity were not likely to play a major pathophysiological role in this disorder. We have reported the effect of ONDAN on bulimic symptoms in a group of severe patients under a randomized, placebo controlled, intention-to-treat design [61]. During the fourth week of double-blind treatment, mean binge/vomit frequencies for the Placebo and ONDAN groups were 13.2 (SD 11.6) versus 6.5 (SD 3.9), respectively [est. diff. 6.8 (CI 4.0-9.5); p<0.0001][Fig. 8]. The amount of time spent engaging in bulimic behaviors was decreased on average by 7.6 hours per week in the ONDAN group compared to 2.3 hours in the Placebo group [est. diff. 5.1 (CI 0.6-9.7] [Fig. 9]. Similarly, the number of “normal” meals and snacks increased on average by 4.3 normal eating episodes/week without vomiting in the ONDAN group compared to 0.2 in the Placebo group [est. diff. 4.1 (CI 1.0-7.2)] [Fig. 9]. Thus, ONDAN treatment was associated not only with a decrease in binge-eating and vomiting, but also with an apparent return towards normal meal termination and satiety.
Figure 8.

Effect of ondansetron treatment on the number of binge/vomit episodes per week. Points = means; Error bars = SE.
Figure 9.
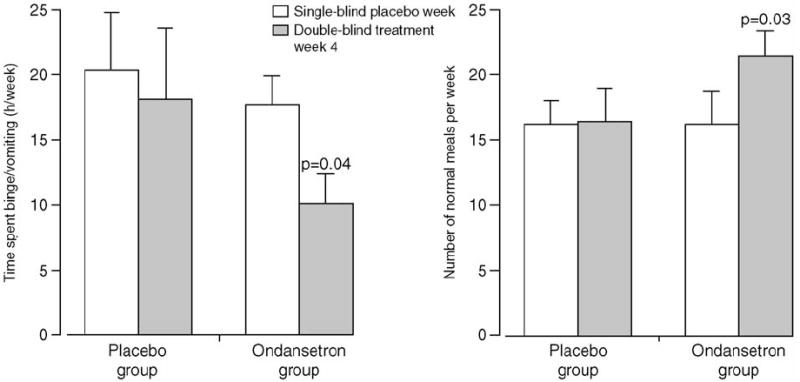
Effect of ondansetron treatment on secondary indicators of disease.
In addition to bulimic symptoms, the cyclic elevations in pain thresholds across the IBI also did not occur when subjects were being treated with ONDAN [61] [Fig. 10]. The abolition by ONDAN of the elevation in pain threshold, at times exceeding the normal IBI, is particularly significant given our primary clinical finding that ONDAN also reduced the number of binge/vomit episodes in these and other individuals treated as private patients (unpublished clinical data). Collectively, the findings summarized above suggest that afferent vagal fibers drive both the changes in pain detection and the occurrence of a bulimic episode.
Figure 10.
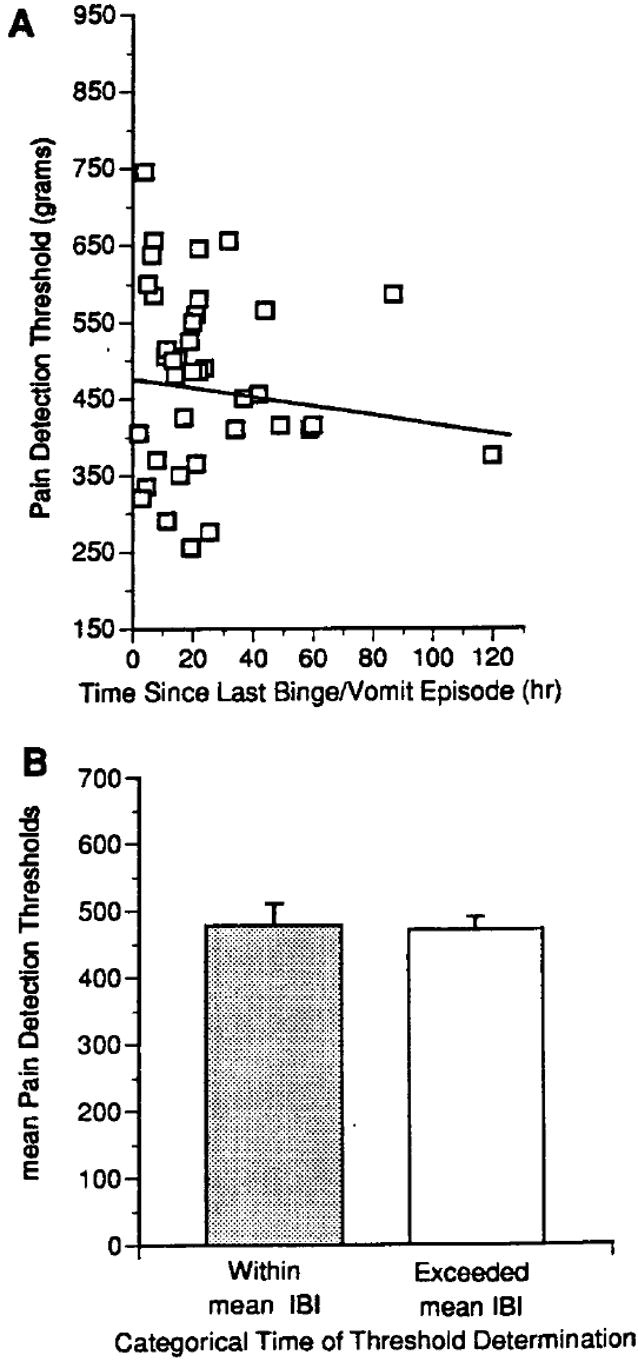
Effect of ondansetron treatment on the relationship between pain detection thresholds and engaging in bulimic behaviors in figure 7 of the bulimia nervosa patients presented in Fig. 1. (A) A scatterplot of 40 total observations illustrating that the time elapsed since the occurrence of a bulimic episode was no longer a significant predictor of pain detection threshold. The line indicates the slope of the maximum likelihood regression obtained by random regression analysis (z = -1.84; P = 0.066). (B) Categorical subdivision of pain detection thresholds based on whether the measurements were collected prior to (within category; 19 observations) or after (exceeded category; 21 observations) the expected duration of an inter-binge interval (z = -1.37; P = 0.172). IBI = average length of an inter-binge interval during the preceding week.
The above reviewed studies with ONDAN provide strong support for heightened vagal activity driving the abnormal behaviors of binge-eating and vomiting and that suppression of this abnormal activity will result consumption on normal meals and, and presumably, the re-establishment of short-term satiety. As interesting and thought provoking as these finding might be, there are some inherent weaknesses in this experiment. The number of subjects studied was small and all participants would be considered, by most clinicians, to have severe, unremitting bulimia nervosa and therefore not characteristic of the general clinical population. These factors obviously compromise the ability to generalize to the larger, more heterogeneous, population of women with this disorder. While ample evidence demonstrates a vagal site of action for ONDAN, and available data suggest that this drug does not cross the blood-brain barrier, other sites of action can’t be completely discounted. Due to the short half-life of ONDAN, a multiple daily dosing strategy was required. This may have decreased compliance to the dosing protocol, although such an effect would be expected to minimize (not increase) the observed effectiveness. As mentioned earlier, this was a “proof-of-concept” level inquiry designed for rejecting the null hypothesis, that vagal involvement was not involved in bulimia. That is, if a vagally active drug (ONDAN) known to act on afferents modulating both satiety and emesis did not affect the abnormal eating, then the results would have suggested that vagal involvement was unlikely to play a major role in perpetuating the disordered eating behaviors. Given these limitations, to further examine vagal involvement in bulimia, another approach was taken.
The NeuroCybernetic Prosthesis© (Cyberonics Inc.; Houston, TX) is an implantable device used to deliver Vagal Nerve Stimulation (VNS) Therapy© [69]. This device utilizes stimulating electrodes which are placed around the surgically dissected cervical branch (along the carotid sheath) of the left vagus. The electrodes are connected by a subcutaneous lead to a pulse generated surgically placed under the axial of the left arm. The generator can be programmed to deliver a set current at designed pulse rates and widths. This device has long been used for treatment of epilepsy and more recently approved by the FDA for treatment of Major Depressive Disorders [70]. This device has been often referred to as being a “neural pacemaker”. Thus, we reasoned that pacing the activity in the vagus might dampen neural oscillations in the vagus, analogous to a cardiac pacemaker stabilizing electrical activity of the heart. Furthermore, by testing the efficacy of this device, the experimental design would address two of the weaknesses of the ONDAN trial, namely the dosing/compliance issue and the anatomical site of the delivered treatment. The full results of this study will be published elsewhere; however, the summarized findings below provide another complementary approach to determine if modulation of vagal activity has a positive action on bulimic behaviors.
Briefly, ten chronic unremitting patients with binge eating and vomiting sub-type of bulimia nervosa participated in this study. This study was conducted under FDA IDE GO10289 and with approval of our Institutional Review Board. All participants had previously failed multiple attempts of treatment by conventional front-line methods. After a lead in period during which baseline behaviors were quantified, subjects were implanted with the NeuroCybernetic Prosthesis©, a relatively minor surgery. After a two week recovery period, the two-week period of stimulation adjustment occurred. The goal of this period was to increase the current up to the maximally tolerable level as rapidly as possible (2 mA). The subjects then were followed for a six-week period during which no further adjustment to the pulse generator was made. Subjects kept daily records on a meal-patterning form, where all bulimic behaviors as well as normal meals and bowel movements were recorded on a 24 hour time line. In addition, subjects made weekly visits to see their assigned psychiatrist, to complete both self- and semi-structured interviews, and on some visits, to have blood electrolytes measured. Data from two of the subjects are not presented here because: (1) one subject’s behavioral reports of a decrease in binge/vomit episodes were not congruent with to her lab values of low potassium. When directly questioned about this discrepancy, she admitted that she had been under reporting her bulimic behaviors; (2) when the second patient underwent explantation surgery for device removal, the stimulating electrodes were no longer coiled around the vagus nerve. This situation apparently resulted from failure of the tie-downs used to immobilize the electrode coil, and there was no way to determine when exactly this occurred, or if the position of the coil remained sufficient to continue to provide ample stimulation via current spread through interstitial fluids. Therefore, the analysis presented below does not follow Intent-to-Treat guidelines [71]. It should be noted that the full report of this study will be based on an Intent-to-Treat design and significant results were obtained under these more rigorous conditions.
The mean number of binge/vomit episodes averaged over the baseline lead in period was 27.75 ± 5.46. By the end of the eight week acute phase, during which the stimulation parameters were not altered, the mean binge/vomit episodes had been reduced to 1.38 ± 0.73 [Fig. 11]. Importantly five of the eight subjects had achieved complete abstinence. During four of the study visits, participants also were given a meal challenge test to determine the effect of VNS on a liquid meal induced satiety. Comparing baseline values to those collected at the exit visit following the acute phase of the study in the five complete responders, there was a significant and complete normalization of in the satiating potency of the meal [Fig. 12]. This finding is extremely important in that it indicates that achieving abstinence is accompanied by a return of normal satiation.
Figure 11.
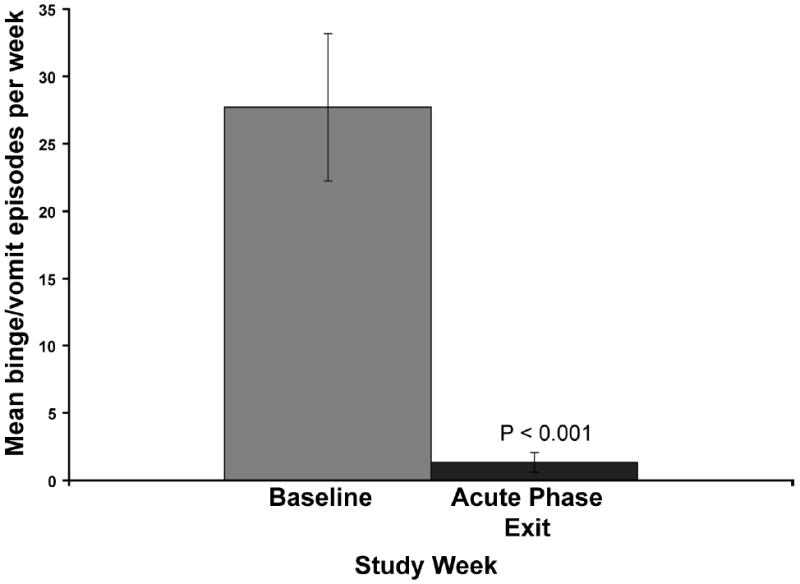
Effect of VNS treatment on the number of binge/vomit episodes per week. Baseline values are average of greater then two week baseline observation period. Bars = means; Error bars = SE.
Figure 12.
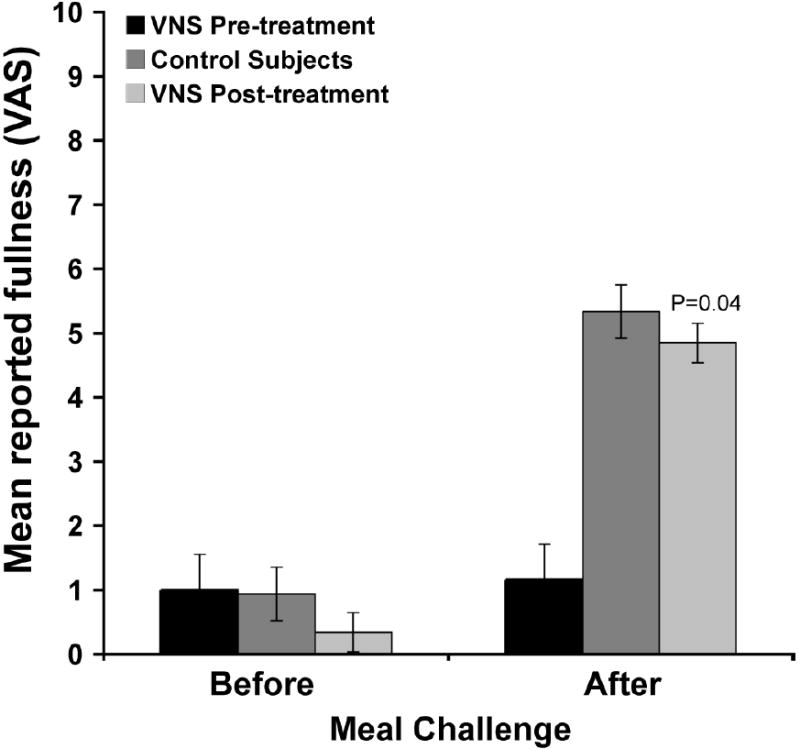
Effect of VNS treatment on reported Satiety after meal challenge. Subject self-report feelings of fullness before 450ml meal challenge and 60 minutes after. Means represent max satiety for control subjects or 5 responders at baseline and acute phase study exit.
Summary of Vagal Afferent Involvement in Bulimia Nervosa
Several abnormalities seen in bulimia nervosa have been discussed above. This section outlines this information integrated with that discussed above on the normal control of food intake. Collectively, this information will be used to lay a logical foundation for a theoretical model for the pathophysiology of bulimia:
Women with bulimia are typically of normal weight despite eating abnormally large meals followed by vomiting. This fact distinguishes them from sufferers of other eating disorders. Their ability to feel full during a meal is decreased compared to appropriate controls. These findings indicate that bulimia most likely involves dysregulation of short-term, pre-absorptive satiety mechanisms.
Intra-meal satiety and early post-prandial satiety involves activation of vagal afferent fibers. Fibers in the afferent gastric branches of the vagus also mediate vomiting. Experimental evidence supports a functional interaction between these two behavioral processes. This interaction may explain why binge-eating and vomiting are so tightly coupled in bulimia nervosa. In the studies from our laboratory summarized above, the bulimia participants would have vomited following food consumption on average around 1000 times per year.
Vagal activation is first processed in the CNS within higher order neurons within the dorsal vagal complex (DVC). In this brain area two divisions, ascending and descending, of neural pathways can be activated. An ascending poly-synaptic pathway which includes relays in many brain areas where additional nutrient, taste, physiological, and environmental factors can modulate the cortical process of satiation. Other non-food related responses also occur, such as activation of the descending pain inhibitory pathway. In bulimia, this pathway also does not function normally, but rather demonstrates oscillations temporally suggestive of an oscillatory neural pattern within the vagal afferent neural pathways. Second, within the DVC, a number of reflexes are activated involving vagal efferent stimulation [72]. Most pertinent to this discussion is the accommodation reflex, which demonstrates heightened activation in bulimic women and accompanying delays in gastric emptying. Collectively, these finding suggest concomitant abnormalities in vagal afferent-efferent reflexive functions.
De-stabilization of the Positive Vago-Vagal Feedback Loop as an Integrative Model for the Psychobiology of Bulimia Nervosa
We propose that one possible mechanism that may underlie the behavioral effects of binge-eating and vomiting on nervous system function is de-stabilization of a positive feedback loop, which results in an uncontrollable perpetuation of the behaviors. Positive feedback loops can always be de-stabilized if the strength of the activational stimulus significantly increased [73]. Supra-threshold stimulation causes a positive feed-back system, be it an electric circuit, mechanical or biological, to oscillate at greater frequencies and amplitudes. When the application of the supra-threshold activation of the system continues, the resultant function spins out of control resulting in the induction of a vicious cycle (circle) which can be debilitating and catastrophic [74,75] and has been implicated in causing apoptosis in cultured heart cells and neonatal rat myocytes [76]. Today, an active area of research is investigating the positive feedbacks involved in intra-cellular processes. This research has lead to the description of several types of destabilization patterns [77]. However, some general statements are typically true for the stability of feedback mechanisms, namely (1) negative feedback loops lead to a steady-state condition and resistant to change making the system robust against alterations, (2) any system can be stabilized by adding negative feedback, and alternatively adding positive feedback can destabilize any system, and (3) positive feedback control is always responsible for graded interpretations, as presented above for graded satiety at sub-maximal levels of afferent stimulation [Fig. 3] [73,74,75].
Based on these mathematical models of electronic and biochemical mechanisms, we suggest that during the voluntary period of the illness, the binge-eating and vomiting behaviors result in the application of supra-high levels of stimulation. These levels of stimulation are sufficient for destabilization of positive vago-vagal reflexive processes. These alterations would cause perturbations in higher order neurons, altering psychological aspects such as pain detection, body image, and mood [78]. Available evidence for bulimia is suggestive of a bi-stable system. Bi-stability may apply in particular to biological systems that switch between discrete states, generate oscillatory responses or sensitize to transitory (episodic) stimuli [77]. Applying this bi-stable system to bulimia, the low activity stable-state would correspond to basal firing rates at the time of meal initiation and the high activity stable-state would represent the threshold for emesis. In this model, the middle state corresponds to the stimulation (input) required for eliciting satiety. The middle state exists in an unstable condition and is subject to changes in sensitivity and destabilization which would result in oscillations in the system. In support of this, vagal afferents are known to demonstrate a wide dynamic range in their thresholds for activation [31], indicating that multiple fibers can be recruited by various stimuli. Figure 13 shows a graphical presentation of how environmental stimuli and societal pressure to maintain a normal body weight could lead to an abnormal, oscillating, bi-stable positive feedback system and the development of full-blown bulimia nervosa.
Figure 13.
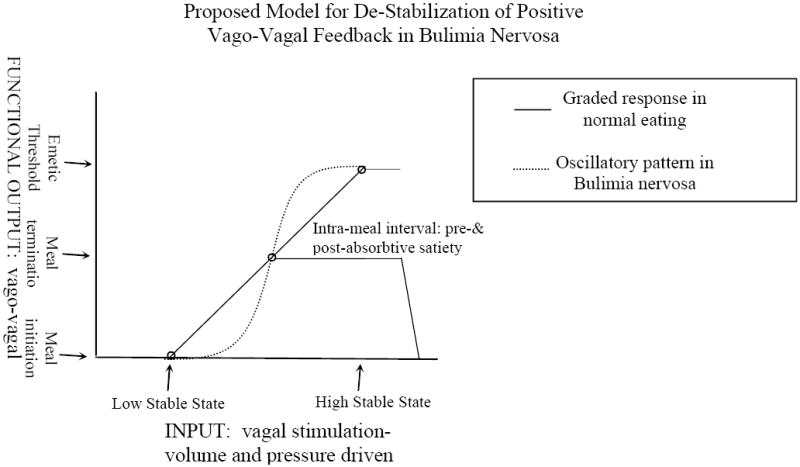
A schematic representation of the proposed de-stabilization of positive vago-vagal feedback illustrates how an induced oscillation in vagal afferent responsiveness would result in the uncontrollable coupling of binge-eating and vomiting. The x-axis depicts relative inputs of increasing vagal stimulation. The y-axis presents the functional outcome of different stimulation intensities. The solid line represents the strength of input as a function of vagal afferent activity. The top and bottom circles on this linear response pattern represent the two stable steady states. Theoretically these would correspond to vagal afferent activity at meal initiation and activity at maximum at which emesis occurs. The middle point represents the stimulation level at which satiety occurs and is the one unstable state. The dashed line depicts oscillations induced by destabilizing the positive feedback loop by binge-eating and vomiting. In this case, a stronger stimulus, approximating maximal, is required to reach satiety, with emesis becoming a natural consequence of maximal stimulation. Modified from [77].
The model proposed here would explain at least two functions that would be altered by the voluntary consumption of a large gastric load along with the coupled vomiting. First, initiation of a gastric vagal emetic reflex becomes involuntary; and second, the CNS becomes “desensitized” such that subsequent episodes become associated with stronger urges and further loss of control, accompanied by greater difficulty in achieving satiety. Positive feedbacks are designed as control systems with the purpose of driving or accomplishing a given process. However, they can lead to “instability and even catastrophe” events [73,74].
Implications for Future Treatment of Bulimia Nervosa and Other Disorders
The presented model stems from the early principles of American Psychobiology [79], which emphasized that act of performing behaviors can feedback to affect nervous system function, which in turn affects behavior. That is, binge-eating and vomiting desensitizes vago-vagal projections and this leads to a further increase in bulimic behaviors. This model of behavior affecting neural function, which in turn affects behavior, has several important implications. First, the neurochemical anatomy of the vago-vagal reflex and higher order projections suggests several new pharmacological approaches. Drugs used to block serotonin release from enterochromaffin cells, such as the somatostatin analogs, may provide a mechanism to dampen the strength of the activational stimulus. Other new age anti-emetics which act on either vagal afferents or in the dorsal vagal complex also warrant consideration. Given that the efferents are intimately involved in the vicious cycle, treatments affecting both pre- and post-ganglionic parasympathetic function would also be of interest. Since the effects of the vago-vagal circuitry affects nervous system function at both distal and caudal sites, the motility of the GI tract and enteric nervous system function may also require corrective action to completely return eating behaviors to normal. Prokinetic agents should be useful in this regard. The antidepressants currently used may prove useful in conjunction with other vagally directed methods to modify cortical effects of ascending perturbations in areas in the higher neural axis. We have previously provided some evidence that the depression frequently accompanying bulimia may result from vagal activation of higher brain areas, and that this activity is interpreted as specific emotions, rather than peripheral events, a process we termed “referred emotions” [78].
Second, psychobiology principles from animal research act as a reminder that the cognitive issues and environmental pressures that initially lead to the voluntary bulimic behaviors may still exist and that the brain’s responsiveness to these external influences may have resulted in neuronal or cognitive restructuring [78,80]. Thus, CBT may also be a very useful adjunctive treatment. With regard to the mention of combining treatment options, it may be that the current first line treatments are partially affective by exerting a corrective action of neural sites upstream from the main vagal origin responsible for perpetuating the illness. An analogy would be the use of diet, exercise, stress reduction techniques and discouraging drug and alcohol use in a diabetic without providing insulin replacement. Depending on the severity of the diabetes, these other treatment modalities would on average result in an amelioration or marginal reduction in diabetic symptoms in some patients. Such is true with the current treatment strategies for bulimia.
Third, the involvement of destabilization of positive feedback control systems may also play some causative role in other disorders. Likely candidates would include other episodic illnesses such as bipolar disease, obsessive-compulsive disorder, restless leg syndromes and other cyclically occurring movement disorders, along with impulse control or “urge” driven syndromes such as gambling, compulsive shopping, self-mutilation, and trichotillomania [81,82]. Cinquin and Demongeot [73] have previously argued that if memories are stored as strengths of connections between neurons, then activated connections would be reinforced by the positive feedback resulting from membrane depolarization. Finally, since it is likely that most neural systems involved in positive feedback control use glutamate as the transmitter in the afferent limb and GABA as an inhibitory interneuron (a governor in essence) [83], appropriate ligands for these neurotransmitters may have general value for many disorders and diseases.
Conclusions
In the eating disorder literature, there is clear and wide-spread recognition that the two front line treatments, along with the multitude of their combinations evaluated to date, are inadequate [5,6,84]. Yet, there has been a strong resistance and non-valid critiques of efforts to integrate new conceptual approaches and novel second line treatments into clinical research [5]. If progress is to be made toward prevention and treatment of bulimia nervosa, a consensus should be established to emphasize and fund investigations into new ways of conceptualizing and treating this debilitating disorder. New treatment options should be viewed openly as being in the “proof-of-concept” phase. Elegant studies on the role of the endogenous opioids had been conducted by Marrazzi and colleagues [85] [Subsequent to the publication of this work, Dr. Marrazzi passed away. This section of our paper is dedicated to her tireless efforts, even during her illness, to draw attention to the need for translational research in improving clinical treatments for bulimia nervosa]. Given the reported orexogenic effects of this class of peptides, this group carefully examined the effects of naltrexone on bulimic symptoms. They reported a significant reduction in binge/vomit frequencies with high doses. This thought-provoking work continues to receive negative comments in the literature, because of the potential hepatotoxic side-effects of naltrexone. Yet, the main point was not to encourage all clinicians to start using naltrexone as a main line treatment, but rather to emphasize that other neurotransmitters besides the classic catecholamines have a proven role in food intake and energy balance. The hepato-toxic criticism is actually without merit as similar doses have been used to successfully treat gambling and other impulse-control disorders with careful monitoring of liver enzyme levels [86]. Similarly, our double-blind study with ondansetron was also a proof-in-concept level analysis aimed at determining likely vagal involvement in this disorder. This work was not well received by the scientific community due to the expense of the drug and the need for multiple daily dosing (e.g. 5). Ondansetron and alternative 5-HT3 antagonists are now available in both generic and long-acting forms, illustrating the short-sightedness of the previous reviews. Furthermore, similar dosing conditions are commonly encountered by physicians treating diseases such as diabetes and AIDS, yet studies continue using these medications to obtain insight into the illnesses. Such close-mindedness regarding novel approaches with strong basic-science rationales detract from clinically relevant progress being made toward developing new treatments for this disorder.
The importance of treatment choice based on the frequency of the behaviors requires more scientific consideration. Published treatment studies rarely use frequency of bulimic behaviors or other determinates of severity as a factor in the statistical analysis of their results, although it is well recognized both clinically and from empirical evidence that successful treatment outcomes are most often associated with early-stage treatment and with lower frequencies of binge-eating and vomiting. Conversely, long illness durations and high frequencies of vomiting are associated with the poorest prognosis [87]. Failure to analyze data using severity as a variable in efficacy analysis lumps all patients into a homogenous group and ignores the natural progression of the illness along with the accompanying likelihood of potential changes in the relative contributions of the underlying physiology. Treatment choices also may depend upon distinguishing if the clinical presentation of bulimia nervosa is due to a relapse of a previous bout or the presentation of a new episode occurring after complete recovery had been achieved. A good starting point might be devising criteria based upon duration of abstinence to delineate between an existing or new condition. Incorporating laboratory tests on the return of normal meal-induced (short-term) satiety into clinical trials may provide an excellent physiological indicator of recovery and aid in making this distinction.
Our approach to studying and ultimately formulating a model for the pathophysiology of bulimia has been to use multiple avenues of inquiry. This route of investigation was chosen because each avenue of research has its own inherent and unique weaknesses. Through employing different approaches, the results compliment each other and summate to provide strong support for vagal involvement in bulimia nervosa. Future translational neuroscience studies aimed at evaluating the function of vagally-activated pathways through a variety of means, including (but not limited to) pharmacological challenges, neuroimaging, neural stimulation/inhibition through either implantable or non-invasive techniques (e.g. acupuncture), and neuropsychological methods are necessary to evaluate vago-vagal function. The proposed pathophysiology and accompanying model presented here is not intended to be complete nor without error; this cannot be overemphasized. Many limitations in experimental design exist and great liberty has been taken with making theoretical connections. However, the goal of this paper was to offer a new perspective based on hodology (nuclear organization and neural projections) rather than neurochemical anatomy (i.e. central catecholamines). Our intent is merely to stimulate conversation and experimentation using hodology, psychobiology, and translational neuroscience to lead to the development of more effective treatments for this baffling disorder.
Acknowledgments
Portions of this work were conducted by Randall D. Hofbauer, M.D. in partial fulfillment of the requirements for a Ph.D. in Neuroscience from the University of Minnesota. His current address is: Singing River Hospital 2809 Denny Avenue, Pascagoula, MS 39581. This work was supported by R01 DK5229103, R03 DK065167, a gift of Vagal Nerve Stimulators from Cyberonics, Inc., and a monetary gift from the Mark A. Nugent Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, D.C.: American Psychiatric Association; 1994. [Google Scholar]
- 2.Fairburn CG. Evidence-based treatment of anorexia nervosa. Int J Eat Disord. 2005;37:S26–S30. doi: 10.1002/eat.20112. [DOI] [PubMed] [Google Scholar]
- 3.Fernandes M, Atallah AN, Soares BGO, Humberto S, Guimaraes S, Matos D, Monteiro L, Richter B. Intragastric balloon for obesity. Cochrane Database Syst Rev. 2007;24:CD004931. doi: 10.1002/14651858.CD004931.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefano SC, Bacaltchuk J, Blay SL, Hay P. Self-help treatments for disorders of recurrent binge eating: A systematic review. Acta Psychiatr Scand. 2006;113:452–459. doi: 10.1111/j.1600-0447.2005.00735.x. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell JE, Agras S, Wonderlich S. Treatment of bulimia nervosa: Where are we and where are we going? Int J Eat Disord. 2007;40:95–101. doi: 10.1002/eat.20343. [DOI] [PubMed] [Google Scholar]
- 6.Halmi KA, Agras WS, Mitchell J, Wilson GT, Crow S, Bryson SW, Kraemer H. Relapse predictors of patients with bulimia nervosa who achieved abstinence through cognitive behavioral therapy. Arch Gen Psychiatry. 2002;59:1105–1109. doi: 10.1001/archpsyc.59.12.1105. [DOI] [PubMed] [Google Scholar]
- 7.Mcgilley BM, Pryor TL. Assessment and treatment of bulimia nervosa. Am Fam Physician. 1998;57:2743–2750. [PubMed] [Google Scholar]
- 8.Striegel-Moore RH, Leslie D, Petrill SA, Garvin V, Rosenheck RA. One-year use and cost of inpatient and outpatient services among female and male patients with an eating disorder: Evidence from a national database of health insurance claims. Int J Eat Disord. 2000;27:381–389. doi: 10.1002/(sici)1098-108x(200005)27:4<381::aid-eat2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 9.Spoor TP, Stice E, Burton E, Bohon C. Relations of bulimic symptom frequency and intensity to psychosocial impairment and health care utilization: Results from a community-recruited sample. Int J Eat Disord. 2007;40:505–514. doi: 10.1002/eat.20410. [DOI] [PubMed] [Google Scholar]
- 10.Lynne D, editor. Life and death tug of war: The whole Terri Schiavo story. World Net Daily. 2005 March 24; www.worldnetdaily.com.
- 11.Mitchell JE, Groat R. A placebo-controlled, double-blind trial of amitriptyline in bulimia. J Clin Psychopharmacol. 1984;4:186–193. [PubMed] [Google Scholar]
- 12.Goldstein DJ, Wilson MG, Thompson VL, Potvin JH, Rampey AH., Jr Long-term fluoxetine treatment of bulimia nervosa. Fluoxetine Bulimia Nervosa Research Group. Br J Psychiatry. 1995;166:660–666. doi: 10.1192/bjp.166.5.660. [DOI] [PubMed] [Google Scholar]
- 13.Fichter MM, Leibl C, Kruger R, Rief W. Effects of fluvoxamine on depression, anxiety, and other areas of general psychopathology in bulimia nervosa. Pharmacopsychiatry. 1997;30:85–92. doi: 10.1055/s-2007-979488. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell JE, Halmi K, Wilson GT, Agras WS, Kraemer H, Crow S. A randomized secondary treatment study of women with bulimia nervosa who fail to respond to CBT. Int J Eat Disord. 2002;32:271–281. doi: 10.1002/eat.10092. [DOI] [PubMed] [Google Scholar]
- 15.Walsh BT, Fairburn CG, Mickley D, Sysko R, Parids MK. Treatment of bulimia nervosa in a primary care setting. Am J Psychiatry. 2004;161:556–561. doi: 10.1176/appi.ajp.161.3.556. [DOI] [PubMed] [Google Scholar]
- 16.Brewerton TD. Toward a unified theory of serotonin dysregulation in eating and related disorders. Psychoneuroendocrinology. 1995;20:561–590. doi: 10.1016/0306-4530(95)00001-5. [DOI] [PubMed] [Google Scholar]
- 17.Blackshaw LA, Grundy D. Effects of 5-hydroxytryptamine on discharge of vagal mucosal afferent fibres from the upper gastrointestinal tract of the ferret. J Auton Nerv Syst. 1993;45:41–50. doi: 10.1016/0165-1838(93)90360-7. [DOI] [PubMed] [Google Scholar]
- 18.Steiger H, Gauvin L, Engelberg MJ, Ying Kin NM, Israel M, Wonderlich SA, Richardson J. Mood- and retraint-based antecedents to binge episodes in bulimia nervosa: Possible influences of the serotonin system. Psychol Med. 2005;35:1553–1562. doi: 10.1017/S0033291705005817. [DOI] [PubMed] [Google Scholar]
- 19.Miller KR. Finding Darwin’s God: A scientist’s search for common ground between God and Evolution. Harper Perennial. 2007 [Google Scholar]
- 20.Halmi KA, Sunday SR. Temporal patterns of hunger and fullness ratings and related cognitions in anorexia and bulimia. Appetite. 1991;16:219–37. doi: 10.1016/0195-6663(91)90060-6. [DOI] [PubMed] [Google Scholar]
- 21.Hadigan CM, Walsh BT, Devlin MJ, LaChaussee JL, Kissileff HR. Behavioral assessment of satiety in bulimia nervosa. Appetite. 1992;18:233–241. doi: 10.1016/0195-6663(92)90200-p. [DOI] [PubMed] [Google Scholar]
- 22.Kissileff HR, Wentzlaff TH, Guss JL, Walsh BT, Devlin MJ, Thornton JC. A direct measure of satiety disturbance in patients with bulimia nervosa. Physiol Behav. 1996;60:1077–1085. doi: 10.1016/0031-9384(96)00086-8. [DOI] [PubMed] [Google Scholar]
- 23.Geracioti TD, Jr, Liddle RA. Impaired cholecystokinin secretion in bulimia nervosa. N Engl J Med. 1988;319:683–688. doi: 10.1056/NEJM198809153191105. [DOI] [PubMed] [Google Scholar]
- 24.Weigert N, Li YY, Schick RR, Coy DH, Classen M, Schusdziarra V. Role of vagal fibers and bombesin/gastrin-releasing peptide-neurons in distention-induced gastrin release in rats. Regul Pept. 1997;69:33–40. doi: 10.1016/s0167-0115(97)02127-7. [DOI] [PubMed] [Google Scholar]
- 25.Phillips RJ, Powley TL. Gastric volume detection after selective vagotomies in rats. Am J Physiol. 1998;274:R1626–R1638. doi: 10.1152/ajpregu.1998.274.6.R1626. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz GJ, Netterville LA, McHugh PR, Moran TH. Gastric loads potentiate inhibition of food intake produced by a cholecystokinin analogue. Am J Physiol. 1991;261:R1141–R1146. doi: 10.1152/ajpregu.1991.261.5.R1141. [DOI] [PubMed] [Google Scholar]
- 27.Grundy D. Signalling the state of the digestive tract. Auton Neurosci. 2006;125:76–80. doi: 10.1016/j.autneu.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Kissileff HR, Carretta JC, Geliebter A, Pi-Sunyer FX. Cholecystokinin and stomach distension combine to reduce food intake in humans. Am J Physiol. 2003;285:R992–R998. doi: 10.1152/ajpregu.00272.2003. [DOI] [PubMed] [Google Scholar]
- 29.Zafra MA, Molina F, Puerto A. The neural/cephalic phase reflexes in the physiology of nutrition. Neurosci Biobehav Rev. 2006;30:1032–1044. doi: 10.1016/j.neubiorev.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Ritter RC. Gastrointestinal mechanisms of satiation for food. Physiol Behav. 2004;81:249–273. doi: 10.1016/j.physbeh.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 31.Moran TH, Ladenheim EE, Schwartz GJ. Within-meal gut feedback signaling. Int J Obes Relat Metab Disord. 2001;25:539–541. doi: 10.1038/sj.ijo.0801910. [DOI] [PubMed] [Google Scholar]
- 32.Arakawa T, Uno H, Fukuda T, Higuchi K, Kuroki T. New aspects of gastric adaptive relaxation, reflex after food intake for more food: involvement of capsaicin-sensitive sensory nerves and nitric oxide. J Smooth Muscle Res. 1997;33:81–88. doi: 10.1540/jsmr.33.81. [DOI] [PubMed] [Google Scholar]
- 33.Berthoud HR, Patterson LM. Anatomical relationship between vagal afferent fibers and CCK-immunoreactive entero-endocrine cells in the rat small intestinal mucosa. Acta Anat (Basel) 1996;156:123–131. doi: 10.1159/000147837. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz GJ, Moran TH. Sub-diaphragmatic vagal afferent integration of meal-related gastrointestinal signals. Neurosci Biobehav Rev. 1996;20:47–56. doi: 10.1016/0149-7634(95)00039-h. [DOI] [PubMed] [Google Scholar]
- 35.Van de Wall EH, Duffy P, Ritter RC. CCK enhances response to gastric distension by acting on capsaicin-insensitive vagal afferent. Am J Physiol Regul Integr Comp Physiol. 2005;389:R695–R703. doi: 10.1152/ajpregu.00809.2004. [DOI] [PubMed] [Google Scholar]
- 36.Peters JH, McKay BM, Simasko SM, Ritter RC. Leptin-induced satiation mediated by abdominal vagal afferents. Am J Physiol Regul Integr Comp Physiol. 2005;288:R879–R884. doi: 10.1152/ajpregu.00716.2004. [DOI] [PubMed] [Google Scholar]
- 37.Hayes MR, Savastano DM, Covasa M. Cholecystokinin-induced satiety is mediated through interdependent cooperation of CCK-A and 5-HT3 receptors. Physiol Behav. 2004;82:663–669. doi: 10.1016/j.physbeh.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Smith GP. Animal models of human eating disorders. Ann N Y Acad Sci. 1989;575:63–72. doi: 10.1111/j.1749-6632.1989.tb53233.x. [DOI] [PubMed] [Google Scholar]
- 39.Kondo DG, Sokol MS. Eating disorders in primary care. Postgrad Med. 2006;119:59–65. doi: 10.1080/00325481.2006.11446052. [DOI] [PubMed] [Google Scholar]
- 40.Stephan E, Pardo JV, Faris PL, Hartman BK, Kim SW, Ivanov EH, Daughters RS, Costello PA, Goodale RL. Functional neuroimaging of gastric distention. J Gastrointest Surg. 2003;7:740–749. doi: 10.1016/s1091-255x(03)00071-4. [DOI] [PubMed] [Google Scholar]
- 41.Freeman AJ, Cunningham KT, Tyers MB. Selectivity of 5-HT3 receptor antagonists and anti-emetic mechanisms of action. Anticancer Drugs. 1992;3:79–85. [PubMed] [Google Scholar]
- 42.Gibbs J, Young RC, Smith GP. Cholecyctokinin decreases food intake in rats. J Comp Physiol Psychol. 1973;84:488–495. doi: 10.1037/h0034870. [DOI] [PubMed] [Google Scholar]
- 43.Woods SC. Gastrointestinal satiety signals I. An overview of gastrointestinal signals that influence food intake. Am J Physiol Gastrointest Liver Physiol. 2004;286:G7–G13. doi: 10.1152/ajpgi.00448.2003. [DOI] [PubMed] [Google Scholar]
- 44.Verbalis JG, Richardson DW, Stricker EM. Vasopressin release in response to nausea-producing agents and cholecystokinin in monkeys. Am J Physiol. 1987;252:R749–R753. doi: 10.1152/ajpregu.1987.252.4.R749. [DOI] [PubMed] [Google Scholar]
- 45.Mazda T, Yamamoto H, Fujimura M, Fujimiya M. Gastric distention-induced release of serotonin stimulates c-fos expression in specific brain nuclei via 5-HT3 receptors in conscious rats. Am J Physiol Gastrointest Liver Physiol. 2003;287:G228–G235. doi: 10.1152/ajpgi.00373.2003. [DOI] [PubMed] [Google Scholar]
- 46.Gulley S, Sharma SK, Moran TH, Sayegh AI. Cholecystokinin-8 increases Fos-like immunoreactivity in the brainstem and myenteric neurons of rats through the CCK(1) receptors. Peptides. 2005;26:1617–22. doi: 10.1016/j.peptides.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 47.Daughters RS, Hartman BK, Grossman AW, Marshall AM, Faris PL. Ondansetron attenuates CCK-induced satiety and c-fos labeling in the dorsal medulla. Peptides. 2001;22:1331–1338. doi: 10.1016/s0196-9781(01)00460-0. [DOI] [PubMed] [Google Scholar]
- 48.Faris PL, Raymond NC, De Zwaan M, Howard LA, Eckert ED, Mitchell JE. Nociceptive, but not tactile, thresholds are elevated in bulimia nervosa. Biol Psychiatry. 1992;32:462–466. doi: 10.1016/0006-3223(92)90134-l. [DOI] [PubMed] [Google Scholar]
- 49.Randich A, Maixner W. [D-Ala]methionine enkephalin-amide reflexively induces antiociception by activating vagal afferents. Pharmacol Biochem Behav. 1984;21:441–448. doi: 10.1016/s0091-3057(84)80107-0. [DOI] [PubMed] [Google Scholar]
- 50.Ren K, Randich A, Gebhart GF. Electrical stimulation of cervical vagal afferents. I. Central relays for modulation of spinal nociceptive transmission. J Neurophysiol. 1990;64:1098–1114. doi: 10.1152/jn.1990.64.4.1098. [DOI] [PubMed] [Google Scholar]
- 51.Lautenbacher S, Pauls AM, Strian F, Pirke KM, Krieg JC. Pain sensitivity in anorexia nervosa and bulimia nervosa. Biol Psychiatry. 1991;29:1–6. doi: 10.1016/0006-3223(91)90249-l. [DOI] [PubMed] [Google Scholar]
- 52.de Zwaan M, Biener D, Schneider C, Stacher G. Relationship between thresholds to thermally and to mechanically induced pain in patients with eating disorders and healthy subjects. Pain. 1996;67:511–512. doi: 10.1016/0304-3959(96)03143-0. [DOI] [PubMed] [Google Scholar]
- 53.Girdler SS, Koo-Loeb J, Pedersen CA, Brown HJ, Maixner W. Blood pressure-related hypoalgesia in bulimia nervosa. Psychosomatic Med. 1998;60:736–743. doi: 10.1097/00006842-199811000-00016. [DOI] [PubMed] [Google Scholar]
- 54.Moran TH, McHugh PR. Cholecystokinin suppresses food intake by inhibiting gastric emptying. Am J Physiol. 1982;242:R491–R497. doi: 10.1152/ajpregu.1982.242.5.R491. [DOI] [PubMed] [Google Scholar]
- 55.Murialdo G, Casu M, Falchero M, Brugnolo A, Patrone V, Cerro PF, Ameri P, Andraghetti G, Briatore L, Copello F, Cordera R, Rodriguez G, Ferro AM. Alterations in the autonomic control of heart rate variability in patients with anorexia or bulimia nervosa: Correlations between sympathovagal activity, clinical features, and leptin level. J Endocrinol Invest. 2007;30:356–362. doi: 10.1007/BF03346310. [DOI] [PubMed] [Google Scholar]
- 56.Rissanen A, Naukkarinen H, Virkkunen M, Rawlings RR, Linnoila M. Fluoxetine normalizes increased cardiac vagal tone in bulimia nervosa. J Clin Psychopharmacol. 1998;18:26–32. doi: 10.1097/00004714-199802000-00005. [DOI] [PubMed] [Google Scholar]
- 57.Geliebter A, Melton PM, McCray RS, Gallagher DR, Gage D, Hashim SA. Gastric capacity, gastric emptying, and test-meal intake in normal and bulimic women. Am J Clin Nutr. 1992;56:656–661. doi: 10.1093/ajcn/56.4.656. [DOI] [PubMed] [Google Scholar]
- 58.Devlin MJ, Walsh BT, Guss JL, Kissileff HR, Liddle RA, Petkova E. Postprandial cholecystokinin release and gastric emptying in patients with bulimia nervosa. Am J Clin Nutr. 1997;65:114–120. doi: 10.1093/ajcn/65.1.114. [DOI] [PubMed] [Google Scholar]
- 59.Faris PL, Kim SW, Meller WH, Goodale RL, Hofbauer RD, Oakman SA, Howard LA, Stevens ER, Eckert ED, Hartman BK. Effect of ondansetron, a 5 HT3 receptor antagonist, on the dynamic association between bulimic behaviors and pain thresholds. Pain. 1998;77:297–303. doi: 10.1016/S0304-3959(98)00108-0. [DOI] [PubMed] [Google Scholar]
- 60.Hedeker D. MIXREG: A Fortran Program for Mixed-Effects Linear Regression Models. Chicago: Prevention Research Center, University of Illinois at Chicago; 1993. [Google Scholar]
- 61.Faris PL, Kim SW, Meller WH, Goodale RL, Oakman SA, Hofbauer RD, Eckert ED, Hartman BK. Effect of decreasing afferent vagal activity with ondansetron on symptoms of bulimia nervosa: A randomised, double-blind trial. Lancet. 2000;355:792–797. doi: 10.1016/S0140-6736(99)09062-5. [DOI] [PubMed] [Google Scholar]
- 62.Galligan JJ. Electrophysiological studies of 5-hydroxytryptamine receptors on enteric neurons. Behav Brain Res. 1996;73:199–201. doi: 10.1016/0166-4328(96)00096-4. [DOI] [PubMed] [Google Scholar]
- 63.Simpson KH, Murphy P, Colthup PV, Whelan P. Concentration of ondansetron in cerebrospinal fluid following oral dosing in volunteers. Psychopharmacology (Berl) 1992;109:497–8. doi: 10.1007/BF02247730. [DOI] [PubMed] [Google Scholar]
- 64.Schinkel AH, Wagenaar E, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996:972517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reynolds DJ, Barber NA, Grahame SD, Leslie RA. Cisplatin-evoked induction of c-fos protein in the brainstem of the ferret: the effect of cervical vagotomy and the anti-emetic 5-HT3 receptor antagonist granisetron (BRL 43694) Brain Res. 1991;565:231–236. doi: 10.1016/0006-8993(91)91654-j. [DOI] [PubMed] [Google Scholar]
- 66.Fukui H, Yamamoto M, Sato S. Vagal afferent fibers and peripheral 5-HT3 receptors mediate cisplatin-induced emesis in dogs. Jpn J Pharmacol. 1992;59:221–6. doi: 10.1254/jjp.59.221. [DOI] [PubMed] [Google Scholar]
- 67.Li Y, Hao Y, Zhu J, Owyang C. Serotonin released from intestinal enterochromaffin cells mediates luminal non-cholecystokinin-stimulated pancreatic secretion in rats. Gastroenterology. 2000;118:1997–2207. doi: 10.1016/s0016-5085(00)70373-8. [DOI] [PubMed] [Google Scholar]
- 68.Zhu JX, Zhu XY, Owyang C, Li Y. Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat. J Physiol. 2001;530:431–442. doi: 10.1111/j.1469-7793.2001.0431k.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Hatton KW, McLarney JT, Pittman T, Fahy BG. Vagal nerve stimulation: Overview and implications for anesthesiologists. Anesth Analg. 2006;103:1241–1249. doi: 10.1213/01.ane.0000244532.71743.c6. [DOI] [PubMed] [Google Scholar]
- 70.Rush AJ, George MS, Sackeim HA, Marangell LB, Husain MM, Giller C, Nahas Z, Haines S, Simpson RK, Jr, Goodman R. Vagus nerve stimulation (VNS) for treatment-resistant depressions: A multi-center study. Biol Psychiatry. 2000;47:276–286. doi: 10.1016/s0006-3223(99)00304-2. [DOI] [PubMed] [Google Scholar]
- 71.Rennie D. How to report randomised controlled trials: the CONSORT statement. JAMA. 1996;276:649. [PubMed] [Google Scholar]
- 72.Rinaman L, Card JP, Schwaber JS, Miselis RR. Ultrastructural demonstration of a gastric monosynaptic vagal circuit in the nucleus of the solitary tract in rat. J Neurosci. 1989;9:1985–1996. doi: 10.1523/JNEUROSCI.09-06-01985.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cinquin O, Demongeot J. Roles of positive and negative feedback in biological systems. C R Biol. 2002;325:1085–1095. doi: 10.1016/s1631-0691(02)01533-0. [DOI] [PubMed] [Google Scholar]
- 74.Brunton LL. A positive feedback loop contributes to the deleterious effects of angiotensin. Proc Natl Acad Sci U S A. 2005;102:14483–14484. doi: 10.1073/pnas.0507070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robertson DS. Feedback theory and Darwinian evolution. J Theor Biol. 1991;152:469–484. doi: 10.1016/s0022-5193(05)80393-5. [DOI] [PubMed] [Google Scholar]
- 76.Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, Liu W, Molina CA, Sadoshima J, Blaxall BC, Berk BC, Yan C. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A. 2005;102:14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Angeli D, Ferrell JE, Jr, Sontag ED. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback Systems. Proc Natl Acad Sci U S A. 2004;101:1822–1827. doi: 10.1073/pnas.0308265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Faris PL, Eckert ED, Kim SW, Meller WH, Pardo JV, Goodale RL, Hartman BK. Evidence for a vagal pathophysiology for bulimia nervosa and the accompanying depressive symptoms. J Affect Disorders. 2006;92:79–90. doi: 10.1016/j.jad.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 79.Lehrman DS. Ethology and psychology. Recent Adv Biol Psychiatry. 1961;4:86–94. doi: 10.1007/978-1-4684-8306-2_11. [DOI] [PubMed] [Google Scholar]
- 80.Lehrman DS, Wortis RP. Previous breeding experience and hormone-induced incubation behavior in the ring dove. Science. 1960;132:1667–1668. doi: 10.1126/science.132.3440.1667. [DOI] [PubMed] [Google Scholar]
- 81.Kim SW, Grant JE, Eckert ED, Faris PF, Hartman BK. Pathological gambling and mood disorders: Clinical associations and treatment implications. J Affect Disorders. 2006;92:109–116. doi: 10.1016/j.jad.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 82.Kim SW. Opioid antagonists in the treatment of impulse-control disorders. J Clin Psychiatry. 1998;59:159–164. [PubMed] [Google Scholar]
- 83.Zheng H, Kelly L, Patterson SM, Berthoud HR. Effect of brain stem NMDA-receptor blockade by MK-801 on behavioral and fos responses to vagal satiety signals. Am J Physiol. 1999;277:R1104–R1111. doi: 10.1152/ajpregu.1999.277.4.R1104. [DOI] [PubMed] [Google Scholar]
- 84.Mitchell JE, de Zwann M, Roerig JL. Drug therapy for patients with eating disorders. Curr Drug Targets CNS Neurol Disord. 2003;2:17–29. doi: 10.2174/1568007033338850. [DOI] [PubMed] [Google Scholar]
- 85.Marrazzi MA, Kinzie J, Luby ED. A detailed longitudinal analysis on the use of naltrexone in the treatment of bulimia. Int Clin Psychopharmacol. 1995;10:173–176. doi: 10.1097/00004850-199510030-00006. [DOI] [PubMed] [Google Scholar]
- 86.Kim SW, Grant JE, Yoon G, Williams KA, Remmel RP. Safety of high-dose naltrexone treatment: Hepatic transaminase profiles among outpatient. Clin Neuropharmacol. 2006;29:77–79. doi: 10.1097/00002826-200603000-00004. [DOI] [PubMed] [Google Scholar]
- 87.Wilson GT, Loeb KL, Walsh BT, Labouvie E, Petkova L, Liu X, Waternaux C. Psychological versus pharmacological treatments of bulimia nervosa: Predictors and processes of change. J Consult Clin Psychol. 1999;67:451–459. doi: 10.1037//0022-006x.67.4.451. [DOI] [PubMed] [Google Scholar]