

Abstract
Matrix-assisted laser desorption ionization–time-of-flight mass spectrometry was used to identify peptic fragments from protein complexes that retained deuterium under hydrogen exchange conditions due to decreased solvent accessibility at the interface of the complex. Short deuteration times allowed preferential labeling of rapidly exchanging surface amides so that primarily solvent accessibility changes and not conformational changes were detected. A single mass spectrum of the peptic digest mixture was analyzed to determine the deuterium content of all proteolytic fragments of the protein. The protein–protein interface was reliably indicated by those peptides that retained more deuterons in the complex compared with control experiments in which only one protein was present. The method was used to identify the kinase inhibitor [PKI(5–24)] and ATP-binding sites in the cyclic-AMP-dependent protein kinase. Three overlapping peptides identified the ATP-binding site, three overlapping peptides identified the glycine-rich loop, and two peptides identified the PKI(5–24)-binding site. A complex of unknown structure also was analyzed, human α-thrombin bound to an 83-aa fragment of human thrombomodulin [TMEGF(4–5)]. Five peptides from thrombin showed significantly decreased solvent accessibility in the complex. Three peptides identified the anion-binding exosite I, confirming ligand competition experiments. Two peptides identified a new region of thrombin near the active site providing a potential mechanism of how thrombomodulin alters thrombin substrate specificity.
Keywords: cyclic AMP-dependent protein kinase/thrombin/thrombomodulin/mass spectrometry/proton exchange
Amide hydrogens provide individualized probes along the entire protein sequence and measurements of amide proton [hydrogen/deuterium (H/D)] exchange rates have been used to analyze protein conformational changes, protein folding, and protein–protein interactions (1–3). NMR has been the method most used to study protein–protein interactions by amide H/D exchange since the first report in 1990 by Paterson et al. (4). NMR studies of other protein–protein complexes have not yielded unambiguous identification of the interface because amide H/D exchange rate variations also resulted from conformational changes (5–11). We propose that the key to separating conformational changes from interface information is to consider only those amides with rapid exchange rates in the uncomplexed state because these amides should be near the protein surface. An observed decrease in the exchange rate of a surface amide should primarily result from solvent accessibility changes, not from conformational changes. Information on the rapidly exchanging surface amides is nearly inaccessible by NMR because the surface amides exchange faster than the time required for measurement. In contrast, mass spectrometry is an ideal method to acquire information about decreased solvent accessibility of rapidly exchanging amides (12).
Two protein–protein complexes were analyzed. The first was a complex of known structure, the catalytic subunit of murine cAMP-dependent protein kinase (PKA) complexed to the kinase inhibitor, PKI(5–24) (13). A complex of unknown structure was also analyzed, human α-thrombin bound to an 83-aa fragment of human thrombomodulin [TMEGF(4–5)]. The surfaces of PKA that interact with PKI(5–24) are consistent with the crystal structure of the complex. The results of the thrombin-TMEGF(4–5) complex are consistent with the structure of the complex between thrombin and a 19-aa peptide from TM (14) and provide new information about where this important anticoagulant protein directly contacts thrombin.
METHODS
Proteins.
PKA was prepared as described previously (15), and experiments were carried out in the presence of either ATP/Mg2+ or ATP/Mg2+ and a 2-fold M excess of PKI(5–24). PKI(5–24), which has a Kd of 2 nM in the presence of 1 mM ATP and 5 mM Mg2+, was prepared as described (13). PKI(5–24) samples were dissolved with 6 μl of D2O containing 20 mM ATP and 100 mM MgCl2, pD 7.4.
Human α-thrombin was a generous gift of John Fenton (NY State Dept. of Health, Albany, NY) and was stored lyophilized at −20°C. Thrombin experiments were carried out with a 2.5-fold M excess of TMEGF(4–5). TMEGF(4–5), which has a Kd of 120 nM, was prepared as described (16), stored lyophilized at −20°C, and dissolved by addition of 6 μl of 25 mM TRIS in D2O, pD 7.9.
Mass Spectrometry.
Matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) mass spectra were acquired on a PerSeptive Biosystems Voyager DE STR. Data were acquired at a sampling rate of 2 GHz into 250,000 channels. Accelerating voltage was 20 kV, grid voltage 70%, and guide wire 0.01%. Delayed extraction was used with a nominal pulse delay of 100 ns. Typically, 256 scans were averaged in ≈3 min. All reported masses are monoisotopic MH+ masses unless otherwise noted. The matrix was 5 mg/ml α-cyano-4-hydroxycinnamic acid, in a solution containing 1:1:1 acetonitrile, ethanol, and 0.1% TFA, and was adjusted to pH 2.5 with 2% TFA. The MALDI targets were chilled on ice in a plastic case before use.
Identification of Each Peptic Fragment Present in the Mass Spectrum of the Mixture.
The analysis of each complex required the identification of the peptides produced when the protein of interest was cleaved with pepsin. This experiment was required because the pepsin cleavage sites cannot be predicted in advance, although a particular protein will be cleaved reproducibly each time. Either PKA (400 pmol/200 mM KCl/40 mM KH2PO4, pH 6.85, at a protein concentration of 33 μM in 12 μl) or thrombin (500 pmol/1 mM CaCl2/65 mM NaCl/33 mM KH2PO4, pH 6.5, at a protein concentration of 40 μM in 12 μl) was diluted to 132 μl with 0.1% TFA, pH 2.5, and digested with a 1:1 molar ratio of immobilized pepsin for 10 min at 0°C. Peptides were identified by a combination of sequence searching for accurate masses, post-source decay sequencing, and C-terminal ladder sequencing (15, 17, 18). Some peptides in the spectrum were not sequenced if the possible identities based on accurate mass determinations did not yield new information (i.e., they overlapped with already identified sequences).
Identification of Protein–Protein Complex Interfaces.
To detect regions of the protein experiencing slowed off-exchange caused by decreased solvent accessibility upon complexation, the rapidly exchanging surface amides were preferentially deuterated. The complexing proteins were therefore incubated in buffered D2O for short times before mixing them together and then off-exchanging the surface deuterons by 1:10 dilution in H2O. The starting protein concentration was 30–100 pmol/μl or 30–100 μM. The protein concentration during off exchange by 1:10 dilution was therefore 3–10 μM. For these high protein concentrations, which are above the Kd of the complex, the percentage bound can be calculated if the concentration of receptor (R) bound to ligand (L) and the Kd are known:
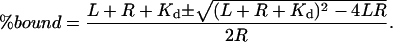 |
Consistent with previous work, we found that the percentage of the protein that is complexed is a function of the Kd and the protein concentrations (data not shown) (4). PKA (400 pmol) and PKI(5–24) (750 pmol) have a Kd of 2 nM and were 99.93% bound at these concentrations. Thrombin (500 pmol) and TMEGF(4–5) (1,200 pmol) have a Kd of 120 nM and are >97.8% bound. Amide H/D exchange rates at solvent accessible sites are highly variable for structured proteins but should be on the order of 0.05–4 sec−1 at pH 7 (ref. 15 and references therein). Based on the Kd and expected H/D exchange rates, off-exchange times of 0–10 min were chosen.
Experiments were carried out at 25°C unless otherwise noted and typically required 10 samples of the protein of interest. Five samples were deuterated for 10 min and then off-exchanged for varying times (0–10 min) by 1:10 dilution with H2O. Five equivalent samples were deuterated for 8 min, complexed with ligand for 2 min, and then off-exchanged for varying times (0–10 min) by 1:10 dilution with H2O. A short deuteration time was used so that primarily solvent accessible amides were labeled (15). Each off-exchange reaction was quenched at 0°C to pH 2.5 by addition of 1% TFA (≈5 μl). Each sample was digested with a 1:1 molar ratio of immobilized pepsin for 10 min at 0°C. A portion (10 μl of 132 μl total) was diluted 1:3 with cold 0.1% TFA, pH 2.5, and both the diluted and undiluted samples were stored at −40°C. Frozen samples were quickly defrosted to 0°C, mixed with 0°C matrix, and 1 μl was spotted onto the chilled MALDI target. The target was immediately placed in a dessicator under a moderate vacuum such that the spots would dry in 1–2 min. Slow drying under moderate vacuum was found to improve sample analysis, presumably because of improved crystal growth. The chilled, dried plate was transferred as quickly as possible from the dessicator to the mass spectrometer. Care was taken to collect data on corresponding control and complex samples after the same length of time in the mass spectrometer to avoid the necessity of correcting for back exchange in the mass spectrometer (15). Control experiments on an unstructured peptide showed that sample handling after quenching resulted in ≈50% loss of the maximal expected amount of deuteration (15). Previous work showed that for deuteration of structured proteins such as PKA, a 10 min deuteration time resulted in lower than expected and variable deuterium incorporation levels consistent with deuteration of primarily surface accessible sites (15).
Data Analysis.
The centroid of the isotopic envelope of each PKA or thrombin peptide from the mass spectrum of the digest mixture of the protein–protein complex was compared with the centroid for the same peptide from control reactions in which only PKA or thrombin was present. The control reactions were off-exchanged for the same amounts of time as the complex. Control peptides showed ≈5% residual deuteration, consistent with the expected 50% loss of the 10% residual expected from a 1:10 dilution (15). If the centroid of the peptide mass envelope was >0.5 deuterons greater than the control for all of the off-exchange times (1–10 min), this peptide was taken to be at the interface in the complex.
The program grasp (19) was used to calculate the solvent accessible surface area of PKA. Subsets of this surface were then created to determine the surface area of PKA spanning each proteolytic fragment in the presence and absence of PKI(5–24). The change in solvent accessible surface area was defined as SAunliganded − SAliganded, where SA is the solvent accessible surface area. For the PKA–PKI(5–24) interaction, the structure with Protein Data Bank ID 1ATP was used (13).
RESULTS
Measurement of Amide H/D Exchange by MALDI-TOF MS.
We recently reported a method by which amide H/D exchange could be monitored by MALDI-TOF MS (15). All of the data for each protein analysis could be obtained from a single mass spectrum of the peptic digest of the protein (Fig. 1). For PKA, ≈50 peptides were observed in the MALDI mass spectrum of 0.4 pmol of the mixture of peptic fragments, and 42 of these were identified by a combination of sequence searching for accurate masses, post-source decay sequencing, and C-terminal ladder sequencing (15). Thus, a single MALDI mass spectrum covered 65% of the PKA sequence and 80% of the solvent accessible protein surface (Fig. 2A). For thrombin, 29 peptides were identified out of the 40 in the spectrum, and these covered 50% of the sequence and 60% of the solvent accessible surface area (Fig. 2B). The lower level of coverage of the thrombin sequence is undoubtedly due to the fact that the four disulfide bonds were not reduced before pepsin cleavage.
Figure 1.

MALDI mass spectrum of a peptic digest of thrombin. (Inset) The region surrounding the isotopic mass envelope for the peptide with mass 2202.2809 (MH+, monoisotopic). All of the data required to completely identify the protein–protein interface can be obtained from four such spectra.
Figure 2.

(A) Sequence of PKA showing the 42 identified peptides that were observed in a single MALDI-TOF mass spectrum. The peptides cover 65% of the PKA sequence including two of the phosphorylation sites, which were identified as phosphopeptides in the mass spectrum. (B) Sequence of human thrombin showing the 29 identified peptides that were observed in a single MALDI-TOF mass spectrum. The sequence is numbered sequentially, although in the text the chymotrypsin-numbering system notation is given as well. The peptides cover 50% of the thrombin sequence. The cysteines that form the disulfide bonds between C9 and C155, between C64 and C80, between C209 and C223, and between C237 and C267 are shown in bold letters. The disulfide bonds were not reduced, and coverage of the sequence near the disulfide bonds appears to be inefficient.
The Interface of the PKA-PKI(5–24) Complex.
Amide H/D exchange rates were measured for the catalytic subunit of PKA complexed to the 20 residue inhibitor PKI(5–24). The solvent accessibility of amide protons on PKA alone was determined first. The maximum deuteration for each peptide varied depending on its surface accessibility (15). Analyses were then carried out on PKA complexed to ATP and on PKA complexed to ATP and PKI(5–24). Fig. 3 shows mass spectra expanded around the peptide of mass 1194.6463, one of the peptides from PKA that retained a significant number of deuterons in the PKI(5–24) complex compared with the controls even after 10 min of off-exchange. Much less deuteration was retained by this region of PKA in the presence of only Mg2+/ATP. Of the 42 peptic fragments identified in the mass spectrum of PKA peptides, eight showed marked slowing of deuteron off-exchange in the complex (Table 1), whereas 34 did not retain a significant excess of deuterium. Three overlapping peptides identified the ATP-binding site and catalytic loop (residues 164–172) including the essential residues K168, E170, and N171 (13). Three overlapping peptides identified the glycine-rich loop (residues 44–54) including S53, F54, and G55 that anchor the ATP phosphates as well as L49, which binds the adenine ring (13). One peptide corresponded to the substrate-binding shelf (residues 237–250) including residues 234–242 corresponding to the P11 pocket which binds PKI(5–24) (20, 21). One peptide corresponded to a second contact with the PKI(5–24) (residues 133–145) including R133 which is essential for PKI(5–24) binding (21) (Fig. 4).
Figure 3.

MALDI-TOF mass spectra of one of the eight peptides from the PKA-PKI(5–24) analysis that experienced slowed exchange in the complex. The spectra are expanded so as to show the isotopic distribution for the ion of interest (MH+ = 1194.6463). (A) The undeuterated peptide. The higher mass peaks in the envelope are caused by naturally occurring isotopes. (B) The isotopic envelope for the same peptide obtained from the PKA sample that was deuterated for 10 min before pepsin digestion. The additional peaks are due to the incorporation of deuterium in the peptide. (C) The peptide obtained from the deuterated PKA sample that was allowed to off-exchange for 10 min. (D) The peptide from the PKA-Mg2+ATP complex that was allowed to off-exchange for 10 min. (E) The peptide from the PKA-Mg2+ATP/PKI(5–24) complex that was allowed to off-exchange for 10 min.
Table 1.
Peptides that showed decreased solvent accessibility upon protein–protein complexation
| Peptide MH+ | Residues | k(complex)*, min−1 | No. of Ds slowly exchanging | Surface area protected, Å2 |
|---|---|---|---|---|
| PKA complexed to PKI(5–24) | ||||
| ATP-binding site | ||||
| 1147.6052 | 164–172 | 0.004 | 1.3 | 148 |
| 1260.6933 | 163–172 | 0.028 | 1.6 | 148 |
| 1373.7837 | 164–174 | 0.007 | 1.4 | 180 |
| Glycine-rich loop | ||||
| 1194.6463 | 44–54 | 0.037 | 2.4 | 223 |
| 1341.7118 | 43–54 | 0.024 | 2.0 | 224 |
| 1584.8087 | 41–54 | 0.014 | 2.1 | 224 |
| PKI(5–24)-binding site | ||||
| 1628.8925 | 133–145 | 0.016 | 1.0 | 80 |
| 1708.8816 | 237–250 | 0.0016 | 1.1 | 179 |
| Thrombin complexed to TMEGF(4–5) | ||||
| Anion-binding exosite I | ||||
| 2127.2265 | 96–112 | 0.36 | 2.1 | NA† |
| 2473.3970 | 97–116 | 0.13 | 3.7 | NA |
| 2586.4805 | 97–117 | 0.23 | 3.2 | NA |
| Potential connection to active site | ||||
| 2144.1661 | 117–132 | 0.74 | 2.4 | NA |
| 2530.3606 | 118–136 | ND‡ | ND | NA |
The value of the slowed off-exchange rate in the complex k(PKA-PKI(5–24) or k(thrombin-TM) corresponds to the slow ensemble off-exchange rate calculated from the bi-exponential fit of the data (PKA data shown in Fig. 5, thrombin data not shown).
The complex studied here is of TMEGF(4–5) for which a structure is not known. Only the structure of a 19 residue peptide from TM bound to thrombin is known.
Although the 2530.3606 peak shifted significantly, the signal was weak and quantitative kinetics were not determined.
Figure 4.

Structure of the PKA-Mg2+ATP/PKI(5–24) complex [PKA in blue, PKI(5–24) in black, and ATP in ball-and-stick, black] (13) showing the location of sequences that experienced slowed off-exchange in the complex. Three peptides identified the ATP-binding site and catalytic loop (residues 164–172, red). Three peptides identified the glycine-rich loop (residues 44–54, green). One peptide corresponded to the substrate-binding shelf (residues 237–250, purple) and one peptide corresponded to a second contact with the PKI(5–24) (residues 133–145, cyan).
Variation of the off-exchange time allowed the rate of off-exchange of deuterons from the interfacial peptic fragments to be determined. Fig. 5 shows representative plots of the rates of off-exchange of deuterons from peptides located in three different regions of PKA that contact either ATP or PKI(5–24) in the ternary complex. Kinetic data were fit to a bi-exponential because each peptide was expected to be only partly at the interface (only some of the amides would experience slowed exchange). The kinetic data gave four parameters: the number of deuterons that off-exchanged quickly, the corresponding rapid ensemble off-exchange rate, the number of deuterons experiencing slowed exchange, and the corresponding slow ensemble off-exchange rate (22). Comparison of the kinetic plots for off-exchange of deuterons from surface amides of PKA alone with those for the PKA-ATP complex show that some decrease in H/D exchange is observed upon ATP binding. In particular, the peptides corresponding to the ATP-binding site (residues 164–174; Fig. 5A) and the helix 6 Å away that alters conformation upon ATP-binding (residues 133–145; Fig. 5C) show some retention of deuterium upon ATP binding at the shorter time points. The levels of deuterium retained upon binding of ATP alone, however, are barely significant (confidence limits: 0.5 deuterons) whereas a highly significant decrease in solvent accessibility is observed for the ternary complex. The lost solvent accessible surface area of each peptide upon formation of the ternary complex was found to correlate with the number of deuterons experiencing slowed exchange in the complex (r = 0.81).
Figure 5.

Kinetic plots of the off-exchange data obtained for various peptides from pepsin digestion of PKA. (a) The peptide of mass 1373.7837 (ATP-binding site). (b) The peptide of mass 1341.7118 (glycine-rich loop). (c) The peptide of mass 1628.8925 (residues 133–145). (d) The peptide of mass 1708.8816 (residues 237–250). In each graph, data for the PKA-Mg2+ATP/PKI(5–24) complex (⧫), the PKA-Mg2+ATP complex (■), and the PKA alone (•) are shown.
Thrombin-TMEGF(4–5) Complex.
A complex of unknown structure also was analyzed, human α-thrombin bound to an 83-aa fragment of human thrombomodulin, TMEGF(4–5). This is the smallest fragment of TM that retains anticoagulant activity and has a thrombin-binding constant of ≈120 nM (16). Of the 29 peptides identified from the MALDI-TOF mass spectrum of the thrombin-TMEGF(4–5) complex, five showed marked slowing of off-exchange compared with thrombin alone (Table 1). Mass spectral data from one of the peptides is shown in Fig. 6. Three overlapping peptides corresponded to the anion-binding exosite I (residues 97–117). This region contains residues R103 (73), T104 (74), R105 (75), Y106 (76), and R108 (77a) that have been shown by site-directed mutagenesis to be important for TM binding or that make contacts to a 19-aa peptide from TM in the structure of the peptide-thrombin complex (refs. 14, 23, and 24; E. Di Cera, unpublished data). Two other overlapping peptides, which showed decreased solvent accessibility upon binding of TMEGF(4–5), corresponded to residues 117–132 (Fig. 7). Two peptides corresponding to residues 117–125 and 117–126 were not significantly shifted, which therefore narrowed the interacting region of thrombin to residues 127–132. This is a region that was not previously known to interact with TM.
Figure 6.

Expansion of the isotopic envelope for the thrombin peptide of mass 2586.4805, one of the five peptides that experienced slowed exchange in the thrombin:TMEGF(4–5) complex. The order of spectra is the same as for PKA but without the PKA:Mg2+ATP complex.
Figure 7.

Structure of human α-thrombin (blue) complexed to a 19-aa residue fragment from TM (black) and to an active site inhibitor (blue) (14) showing two sequences that experienced slowed off-exchange in the complex. The sequence that corresponds to the anion-binding exosite I (residues 97–117) is colored green. An additional sequence of decreased solvent accessibility upon TMEGF(4–5) binding (residues 127–132), is shown in red.
DISCUSSION
Mass spectrometry has made possible the rapid analysis of solvent accessibility changes upon protein–protein complexation. Data for an entire protein sample was obtained from a single spectrum of the mixture of peptides resulting from peptic digestion after H/D exchange. For PKA, this single spectrum gave information on 80% of the solvent accessible surface area. All of the regions represented by the peptic fragments that retained deuterium were indeed at the interface of the ATP- or PKI(5–24)-binding site, and together, they comprehensively indicate the surface of interaction with PKA.
The results on the thrombin-TMEGF(4–5) complex show that H/D exchange can be used to identify protein–protein interfaces when no prior structural information is available. The data show that TMEGF(4–5) binds to anion-binding exosite I, confirming site-directed mutagenesis and ligand competition studies. A new area of thrombin that interacts with TMEGF(4–5) also was discovered. This region corresponds to a β-turn that is within 5 Å of the active site and indicates a possible mechanism of TM alteration of thrombin substrate specificity. The region identified could be narrowed to residues 127–132 because two other peptides corresponding to residues 117–125 and 117–126 did not retain deuterium in the complex. If binding at anion-binding exosite I had transmitted a conformational change to the 127–132 loop, one would certainly have expected to see a decrease in solvent accessibility of residues 117–126. The observed deuteration of residues 127–132 and not 117–126 suggests two distinct regions of thrombin may bind to TM. A more likely explanation is that the portion of thrombin that connects these two discontinuous sites, which spans residues 72–93, was not detected in our experiments. Indeed, as can be seen from Fig. 2B, this sequence corresponds to a disulfide-bonded region that was not detected in these experiments. It is also important that the binding affinity of TMEGF(4–5) for thrombin is >100 nM. Protein–ligand interactions in this affinity range are traditionally difficult to analyze, but the amide H/D exchange experiments simply required increasing the molar ratio of TMEGF(4–5) to thrombin to 2.5.
For these two very different proteins, rapid deuteration of primarily the surface accessible amides gave reliable information about the protein–protein interface. No unexplainable retention of deuteration was observed suggesting that rapid deuteration allows the measurement of primarily solvent accessibility changes without the added deuterium retention resulting from conformational changes. Further support for this contention is obtained by the good correlation (r = 0.81) that was obtained between the number of deuterons experiencing slowed exchange in each peptide and the change in solvent accessible surface area for that peptide. This result suggests that slowed off-exchange in the complex is indeed due to a decrease in solvent accessibility at the interface. Thus, measurement of decreases in H/D exchange rates of solvent-accessible amides can be used to monitor changes in solvent accessibility such as those occurring upon protein–protein complex formation.
Acknowledgments
We thank Susan Taylor for the gifts of PKA and PKI(5–24) as well as critical comments on the manuscript. We thank John Fenton for his generous gift of human α-thrombin. This work was supported by a National Institutes of Health Grant HL47463. Jeffrey Mandell acknowledges a National Institutes of Health/National Cancer Institute Grant T32 CA09523 and a fellowship from the La Jolla Interfaces in Science Program, funded by the Burroughs Wellcome Fund.
ABBREVIATIONS
- MALDI-TOF
Matrix-assisted laser desorption ionization-time-of-flight
- TM
thrombomodulin
- H/D
hydrogen/deuterium
- PKA
protein kinase A
References
- 1.Englander J J, Rogero J R, Englander S W. Anal Biochem. 1985;147:234–244. doi: 10.1016/0003-2697(85)90033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith D L, Deng Y, Zhang Z. J Mass Spectrom. 1997;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 3.Englander S W, Sosnick T R, Englander J J, Mayne L. Curr Opin Struct Biol. 1996;6:18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paterson Y, Englander S W, Roder H. Science. 1990;249:755–759. doi: 10.1126/science.1697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayne L, Paterson Y, Cerasoli D, Englander S W. Biochemistry. 1992;31:10678–10685. doi: 10.1021/bi00159a006. [DOI] [PubMed] [Google Scholar]
- 6.Benjamin D C, Williams D C, Smith-Gill S J, Rule G S. Biochemistry. 1992;31:9539–9545. doi: 10.1021/bi00155a005. [DOI] [PubMed] [Google Scholar]
- 7.Orban J, Alexander P, Bryan P. Biochemistry. 1994;33:5702–5710. doi: 10.1021/bi00185a006. [DOI] [PubMed] [Google Scholar]
- 8.Yi Q, Erman J E, Satterlee J D. Biochemistry. 1994;33:12032–12041. doi: 10.1021/bi00206a004. [DOI] [PubMed] [Google Scholar]
- 9.Williams D C, Jr, Benjamin D C, Poljak R J, Rule G S. J Mol Biol. 1996;257:866–876. doi: 10.1006/jmbi.1996.0207. [DOI] [PubMed] [Google Scholar]
- 10.Williams D C, Jr, Rule G S, Poljak R J, Benjamin D C. J Mol Biol. 1997;270:751–762. doi: 10.1006/jmbi.1997.1122. [DOI] [PubMed] [Google Scholar]
- 11.Werner M H, Wemmer D E. J Mol Biol. 1992;225:873–889. doi: 10.1016/0022-2836(92)90407-b. [DOI] [PubMed] [Google Scholar]
- 12.Dharmasiri K, Smith D L. Anal Chem. 1996;68:2340–2344. doi: 10.1021/ac9601526. [DOI] [PubMed] [Google Scholar]
- 13.Zheng J, Knighton D, Ten Eyck L, Karlsson R, Xuong N-H, Taylor S S, Sowadski J. Biochemistry. 1993;32:2154–2161. doi: 10.1021/bi00060a005. [DOI] [PubMed] [Google Scholar]
- 14.Mathews I I, Padmanabhan K P, Tulinsky A. Biochemistry. 1994;33:13547–13442. doi: 10.1021/bi00250a006. [DOI] [PubMed] [Google Scholar]
- 15.Mandell J G, Falick A M, Komives E A. Anal Chem. 1998;70:3987–3995. doi: 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- 16.White C E, Hunter M J, Meininger D P, White L R, Komives E A. Protein Eng. 1995;8:1177–1187. doi: 10.1093/protein/8.11.1177. [DOI] [PubMed] [Google Scholar]
- 17.Patterson D H, Tarr G E, Regnier F E, Martin S A. Anal Chem. 1995;67:3971–3978. doi: 10.1021/ac00117a024. [DOI] [PubMed] [Google Scholar]
- 18.Spengler B, Kirsch D, Kaufmann R. Rapid Commun Mass Spectrom. 1991;5:198–202. doi: 10.1002/(SICI)1097-0231(19960731)10:10<1199::AID-RCM643>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 19.Nicholls A, Sharp K, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 20.Narayana N, Cox S, Shaltiel S, Taylor S S, Xuong N-H. Biochemistry. 1997;36:4438–4448. doi: 10.1021/bi961947+. [DOI] [PubMed] [Google Scholar]
- 21.Narayana N, Cox S, Xuong N-H, ten Eyck L F. Structure (London) 1997;5:1–15. doi: 10.1016/s0969-2126(97)00246-3. [DOI] [PubMed] [Google Scholar]
- 22.Resing K A, Ahn N G. Biochemistry. 1998;37:463–475. doi: 10.1021/bi971750x. [DOI] [PubMed] [Google Scholar]
- 23.Wu Q Y, Sheehan J P, Tsiang M, Lentz S R, Birktoft J J, Sadler J E. Proc Natl Acad Sci USA. 1991;88:6775–6779. doi: 10.1073/pnas.88.15.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hrabal R, Komives E A, Ni F. Protein Sci. 1996;5:195–203. doi: 10.1002/pro.5560050202. [DOI] [PMC free article] [PubMed] [Google Scholar]