

Abstract
Although human and rodent telomeres have been studied extensively, very little is known about telomere dynamics in other vertebrates. Moreover, our current dependence on mice as a model for human tumorigenesis and aging poses a problem because human and mouse telomere biology is very different. To explore whether chickens might provide a more useful model, we have examined telomerase activity and telomere length in chicken tissues as well as in primary cell cultures. Although chicken telomeres resemble human telomeres in that they are 8–20 kb in length, the distribution of telomerase activity in chickens resembles what is found in mice. Active enzyme is present in germline tissue as well as in a wide range of somatic tissues. Because chicken cells exhibit extremely low rates of spontaneous immortalization, this finding indicates that constitutive telomerase expression does not necessarily lead to an increased immortalization frequency. Finally, we found that telomerase activity is greatly down-regulated when primary cultures are established from chicken embryos. Although this down-regulation explains the telomere loss and replicative senescence that we observed in fibroblast cultures, it raises questions concerning how relevant studies of senescence in primary cell cultures are to aging in whole animals.
Telomerase is the enzyme that maintains telomere length by adding new telomeric DNA onto the 3′ end of the chromosome (1). In humans, telomerase levels are highly regulated; whereas the enzyme is present in germline tissue, activity is undetectable in most somatic tissues and very low levels are present in cells with high proliferative potential (2, 3). A different situation exists in cancer cells where robust activity is present in the majority of tumors (4, 5). Up-regulation of telomerase appears to be a key step in cellular immortalization because it prevents replicative senescence (6–8).
The loss of telomeric DNA that occurs in primary cells at each round of replication appears to act as the mitotic clock that limits the number of times a population can divide before undergoing senescence (6–9). The link between telomere shortening and senescence is still unclear, but it is proposed that critically shortened telomeres trigger a cell cycle check point. Activation of the mitotic clock by telomere shortening appears to provide humans with an additional tumor suppressor mechanism by limiting the proliferative capacity of a cell. Thus, malignant transformation requires not only cumulative genetic changes to alter the growth characteristics of a cell, but also telomerase regulation.
Although mice provide a very valuable tool for studying many human diseases, they are less useful for exploring the role of telomeres in human tumorigenesis. Not only do mice express telomerase in most tissues (10, 11), but their telomeres are extremely long; telomeric restriction fragments are up to ≈150 kb in Mus musculus as compared with 5–15 kb in humans (12–14). Knockout mice, which lack the telomerase RNA gene, are viable, and cells from sixth generation animals can still be transformed and form tumors (10, 15). Thus, telomerase activity is not required for tumorigenesis in mice. This result may be because the cells from even sixth generation mice have sufficient telomeric DNA to undergo the many cell divisions required for tumor formation without reaching a limiting telomere length. Alternatively, the telomeres may be maintained by a nontelomerase based mechanism (5).
To date, mice and humans are the only vertebrates for which telomere dynamics has been systematically examined. To rectify this situation, we initiated studies in a different model system—the chicken. We chose to work with chickens because a number of observations suggested that these animals might be more useful than mice as a model for exploring the role telomeres play in human tumorigenesis and aging. First, chickens are much longer lived than mice [they live for up to 30 years (16)] and some of their telomeres were reported to be similar in length to humans telomeres (17). Moreover, primary cultures of chicken cells resemble those of human cells in that they undergo a clear-cut replicative senescence and exhibit very low rates of spontaneous immortalization (18, 19). These observations suggested that as in humans, chicken telomerase may be down-regulated in somatic tissues. However, we have found that telomerase regulation in chickens is in fact quite similar to what has been observed in mice, because enzyme activity is present in many somatic tissues. Unexpectedly, telomerase is down-regulated when primary cultures are established from embryos. Our data provide an explanation for the replicative senescence previously observed in cultures of chicken embryonic fibroblasts.
MATERIALS AND METHODS
Preparation of Extracts and TRAP Assays.
Chicken tissue (50–100 mg) was pulverized in liquid nitrogen; samples of tissue powder or pellets of tissue culture cells were then homogenized in 10 mM Tris⋅Cl (pH 7.5), 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 5 mM β-mercaptoethanol, 0.1 mM phenylmethylsulfonyl fluoride, 10 μM leupeptin, and 1 μM aprotinin and extracted for 30 min at 4°C. The extract was centrifuged at 16,000 × g and the supernatant divided into aliquots and frozen. The protein concentration was determined by Bradford assay.
The two-tube telomere repeat amplification protocol (TRAP) assay was performed essentially as described by Blasco et al. (20). The 40-μl telomerase extension reactions contained 50 mM Tris acetate (pH 8.0), 50 mM potassium acetate, 3 mM MgCl2, 1 mM spermidine, 1 mM EGTA, 5 mM β-mercaptoethanol, 0.5 μg of TS oligonucleotide primer, 2 mM dATP, dGTP, and dTTP, and 1–8 μl of telomerase extract. Reactions were incubated at 30°C for 60 min and then phenol/chloroform extracted. Control reactions were performed by using either water in the extension reaction instead of telomerase extract or extract pretreated with 10 μg RNase A for 25–30 min at 30°C. For the PCR reaction, 0.1 μg of CX or ACX primer were placed below a wax barrier, and the PCR mix containing 1–6 μl of telomerase extension reaction was placed above the barrier. The PCR reaction conditions were as described by Blasco et al. (20) for the CX primer and by Kim et al. (21) for the ACX primer. Control reactions contained both PCR primers but no telomerase extension products. After the PCR step, the reaction products were treated with proteinase K, phenol/chloroform extracted, ethanol precipitated, and separated on 10% polyacrylamide gels. To ensure that the TRAP assays were in the linear range, several different quantities of extract were tested in the telomerase extension reaction and a series of dilutions were used in the PCR step.
DNA Isolation and Measurement of Telomere Length.
Chicken tissue was pulverized in liquid nitrogen and the DNA isolated from the tissue powder by proteinase K digestion and phenol/chloroform extraction (22). Nuclei were extracted from chicken erythrocytes as described (23). DNA was then isolated from the erythrocyte nuclei and from embryonic fibroblasts by using either Pharmacia or Qiagen genomic DNA isolation kits.
To measure telomere length, genomic DNA was digested with HinfI and AluI, and the DNA fragments were separated by electrophoresis in nondenaturing agarose gels. The gels were then dried and hybridized with either 32P-labeled (T2AG3)4 or (C3TA2)4 oligonucleotides as described by Dione and Wellinger (24). Digestion of the G-strand overhang with mung bean nuclease, exonuclease I, or T4 DNA polymerase was as described by Wellinger et al. (25). Mean telomere length was calculated as described by Harley et al. (26). Restriction fragments containing telomeric DNA sequences were identified by in-gel Southern hybridization as previously described (27).
Culturing of Chicken Embryonic Fibroblasts.
Chicken embryonic fibroblasts were either obtained from BioWhittaker or isolated from 12-day-old embryos as described (28). They were cultured in Eagle’s modified essential medium supplemented with 5 or 10% fetal bovine serum and were passaged 1:3 when they reached 80–95% confluence. The population doublings were calculated as described (29) with minor modifications to account for plating efficiency (30). The equation used was as follows:
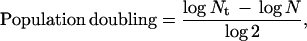 |
where Nt was the number of viable cells at the end of the growth period and N was the number of cells attached to the flasks after plating. N = Nif, where Ni is the number of cells seeded and f is the plating efficiency. Cultures were judged to be senescent when proliferation stopped and >90% of the cells showed β-galactosidase staining at pH 6.0 (31).
RESULTS
Expression of Telomerase in Chicken Tissues.
Given the striking difference between the levels of telomerase activity in human and mouse tissues, we set out to characterize the telomerase expression profile in an alternative vertebrate—the chicken. Tissues were isolated from 1-day-old chickens, 9-mo-old hens, and one 2-yr-old rooster. Extracts were made from the frozen tissue and assayed for telomerase activity by a modified two-tube TRAP assay. In the modified assay, the telomerase extension reaction is phenol/chloroform extracted to remove tissue-specific Taq polymerase inhibitors before the PCR step (32). As illustrated in Fig. 1, activity was readily detected in both germline (testis, lanes 5 and 19; ovary, lane 13) and a range of somatic tissues (e.g., kidney, spleen, liver, and lung, lanes 6–11). RNase A treatment abolished activity (lane 26) and no activity was observed in control reactions that lacked tissue extract (lanes 1–3). In general, robust telomerase activity could be detected in a wider range of tissues from the 1-day-old chickens than from the older birds (e.g., liver, compare lanes 8, 16, and 22, or lung, compare lanes 9 and 23). This may be because telomerase levels fall with age; however because additional older birds were unavailable, we could not test this hypothesis. Assays using several different extracts all failed to detect activity in heart and brain. The lack of enzyme activity in the brain samples and the low activity in some other tissues did not appear to be caused by telomerase or Taq polymerase inhibitors because when equal weights of chicken brain or liver were mixed with mouse liver before extract preparation, the level of the mouse telomerase activity was unaffected (data not shown). From these results, we conclude that the overall distribution of telomerase in chicken tissues is similar but not identical to that observed for M. musculus and M. spretus, where activity is generally absent from brain and muscular organs but present in a variety of other tissues (11, 33).
Figure 1.

Telomerase activity in chicken tissue. Lanes 1–4, PCR controls. Lane 1, PCR reaction with TS and CX primers but no input from the telomerase extension reaction; lanes 2 and 3, buffer rather than extract was used in the telomerase extension reaction. Lane 4, no TS primer was added to the telomerase extension reaction. Lanes 5–12, tissue samples from 1-day-old chicken. Lanes 13–18, tissue from a 9-mo-old hen. Lanes 19–24, tissues from a 24-mo-old rooster. T, testis; K, kidney; S, spleen; L, liver; U, lung; P, pancreas; Y, thymus; B, brain; O, ovary; H, heart. Lanes 25–26, extracts from the spleen of a 1-day-old chicken; −, no RNase A; +, pretreated with RNase A. Lane 27, extract from mouse liver. Lanes 28 and 29, extract from chicken DT40 cells, an avian leukosis virus-transformed B-cell line. +, Pretreated with RNase A; −, no RNase A. To remain in the linear range of the TRAP assay, each telomerase extension reaction contained 30 μg of extract, and 4 μl of the extension products were used in the PCR reactions.
Telomerase Activity During Replicative Senescence.
The presence of active telomerase in most tissues from 1-day-old chickens led us to ask whether active enzyme also was present in cultures of chicken embryonic fibroblasts. Like human primary cells, chicken embryonic fibroblasts have a finite life span in culture and undergo replicative senescence after a limited number of population doublings [usually 25–35 (18)]. However, the finite life span of human cell cultures appears to stem from the lack of telomerase in human somatic cells and the resulting telomere shortening that accompanies increased population doubling (6–8). If chicken embryonic fibroblasts have active telomerase, replicative senescence might have a completely different cause.
To explore this possibility, two separate cultures of chicken embryonic fibroblasts were passaged from population doubling 2 or 3 until the culture underwent replicative senescence. This occurred at population doubling 22 for culture 1 and population doubling 32 for culture 2 (Fig. 2A). Each time the cells were passaged, the number of population doublings were calculated and a fraction of the cells were frozen. Some of the frozen cells were used to make extracts for telomerase assays, whereas the remainder were used to isolate genomic DNA. The cultures were judged to have reached replicative senescence when the growth curve reached a plateau (Fig. 2A) and the majority of the cells (>90%) showed β-galactosidase staining at pH 6.0 (data not shown; 30). No survivor colonies grew out of the senescencent cultures; this fits with previous observations that chicken fibroblasts essentially never undergo spontaneous immortalization.
Figure 2.

Telomerase activity during the life span of a chicken embryonic fibroblast culture. (A) Growth curve for culture 2 (see text) showing the cumulative increase in population doubling with time in culture. (B) Telomerase activity at various population doublings for culture 2. Lane 1, control reactions in which buffer rather than extract was used in the telomerase extension reaction. Lanes 2, 3, and 15, RNase A-treated extracts from chicken embryonic fibroblasts at population doubling 6 (lane 2) or embryo tissue [(Emb) lanes 3 and 15]. Lanes 4 and 14, extract made from embryo tissue, no RNase A treatment; lanes 5–13, 16, and 17, extracts made from chicken embryonic fibroblasts throughout the lifespan of the culture, no RNase A treatment. The population doubling at the time of isolation is given at the top of each well. Thirty micrograms of protein was used in each telomerase extension reaction for lanes 2–13, and 80 μg was used in lanes 14–17.
As illustrated in Fig. 2B, the cultures of embryonic fibroblasts exhibited very low levels of telomerase activity throughout their life span (lanes 5–13). However, we observed robust activity when extracts were made from the tissue of a 12-day-old embryo (lanes 4 and 14). Because the embryonic fibroblast cultures were generated from 10- to 11-day-old embryos, this finding suggested that telomerase might be down-regulated when cell cultures were established from embryonic tissue. To further investigate this possibility, we isolated four 12-day-old embryos from fertilized eggs and used one-half of each embryo to establish a fibroblast culture. These cultures were passaged five times and telomerase extracts were made from the cells at passage 2 and 4. The remaining embryo tissue was frozen and used later to make telomerase extracts. For each of the embryos, the extracts made from frozen tissue exhibited high levels of telomerase activity, but the amount of activity was greatly decreased in the corresponding cell culture. The data for embryos 1 and 2 are shown in Fig. 3. In some cases, low levels of activity still remained (see over exposure of lane 6), whereas in other cases no activity could be detected. To ensure that we were not merely isolating and culturing a telomerase negative population of cells from the embryos, we next assayed the cells used to establish the fibroblast cultures soon after they were isolated from the embryo. Four embryos were digested with trypsin, and the resulting cell suspensions were allowed to adhere to tissue culture dishes for 4 hr. We then harvested both the adherent cells that give rise to the long term fibroblast cultures and the nonadherent cells that are usually discarded with the supernatant. For each embryo, the adherent cells showed robust telomerase activity. The data for one embryo is shown in Fig. 3B.
Figure 3.

Telomerase activity in 12-day-old embryos and fibroblast cultures established from the embryos. (A) The activity present in fibroblast cultures after two or four passages. Lanes 1 and 2 controls; lane 1, PCR reaction with TS and ACX primers but no input from the telomerase extension reaction; lane 2, buffer rather than extract was used in the telomerase extension reaction; lanes 3–7, tissue and cell cultures samples from embryo 1; lanes 8–12, tissue and cell cultures samples from embryo 2; lanes 13–17, over exposure of lanes 3–7. Lanes 3, 5, 8,10, 13, and 15, extracts were made directly from embryo tissue. Lanes 4, 6, 9, 11, 14, and 16, extracts were made from embryonic fibroblast cultures after two passages. Lanes 7, 12, and 17, extracts were made from embryonic fibroblasts after four passages. +, Pretreatment with RNase A; −, no RNase A treatment. (B) The activity present in embryo cells 4 hr after isolation. Lane 1, extract was made directly from embryo tissue. Lane 2, extract was made from the population of cells that had not adhered to the tissue culture dishes after a 4-hr incubation. Lane 3, extract was made from the population of cells that had attached to the tissue culture dish after 4 hr.
Telomere Length During Replicative Senescence.
Our finding that telomerase is down-regulated in cultures of chicken embryonic fibroblasts suggested that replicative senescence might well be the result of progressive telomere shortening with increased population doubling. To look for such telomere shortening, we attempted to examine telomere length by Southern hybridization using restriction-digested genomic DNA and a (T2AG3)4 probe. However, we found that the characteristic smear of telomeric restriction fragments was obscured by a series of sharp bands that were mostly Bal 31 resistant (Fig. 4A, lanes 15). The Bal 31 resistance indicated that these bands arise from the abundant interstitial telomeric sequence that can be seen by fluorescense in situ hybridization on many chicken chromosomes (ref. 34 and C. Price, unpublished observations). Digestion of genomic DNA with enzymes exhibiting different sequence biases (e.g., HinfI + MspI versus HinfI + AluI) caused a slight alteration in the pattern of the bands but did not prevent them from obscuring the signal from the telomeric restriction fragments (Fig. 4A, lanes 6–7). Pulsed field gels revealed that the largest fragments generated by HinfI + AluI digestion were <50 kb and most were under 25 kb (data not shown).
Figure 4.

Measuring telomere length. (A) Southern blot showing telomeric restriction fragments and interstitial telomeric sequences. Genomic DNA from chicken erythrocytes was digested with HinfI + AluI (lanes 1–5 and 7) or HinfI + MspI (lane 6), and telomeric sequences were identified by in-gel Southern hybridization using a (T2AG3)4 probe and a standard denaturation step. In lanes 1–5, the DNA was digested with Bal 31 before restriction digestion. The duration of the Bal 31 digestion is shown above each lane. (B) Identification of telomeric restriction fragments by hybridization to the G-strand overhang. Genomic DNA from chicken erythrocytes was digested with HinfI and AluI, the DNA was separated in an agarose gel, and the gel was incubated with 32P-labeled (C3AT2)4 under nondenaturing conditions. Lanes 3, 5, and 7; the DNA was treated with mung bean nuclease, Exonuclease 1, or T4 DNA polymerase before restriction digestion. Size markers are shown in lane 1. Sizes in kilobases indicated to the left of each gel.
Because standard Southern hybridization was uninformative, we instead made use of the telomeric G-strand overhang to analyze telomere length (24, 35). In this assay, genomic DNA was digested with restriction enzymes, the DNA separated in a nondenaturing gel, and an end-labeled (C3TA2)4 probe hybridized under nondenaturing conditions to the G-strand overhang (24). The probe did not hybridize to interstitial telomeric sequences because they are double-stranded. As shown in Fig. 4B, the telomeric restriction fragments identified by the G-overhang assay were in the 8–20 kb range for DNA isolated from chicken red blood cells (lanes 2, 4, and 6). No fragments in this size range were identified by the complementary (T2AG3)4 probe (data not shown). As expected, removal of the G-strand overhang by digesting the genomic DNA with Mung Bean nuclease or the 3′ → 5′ nucleases Exonuclease 1 or T4 DNA polymerase before restriction digestion abolished hybridization to the (C3TA3)4 probe.
When we used the G-overhang assay to analyze telomere length throughout the life span of chicken embryonic fibroblast cultures, we found that there was an overall decline in telomere length with increased population doubling (Fig. 5). Initially the majority of the telomeres were in the 9–20 kb range. However, over time, the telomeres became much more heterogeneous and the mean length decreased by ≈60 bp per population doubling.
Figure 5.

Changes in telomere length as a function of population doubling. DNA was isolated from chicken embryonic fibroblasts at points throughout the lifespan of the culture. The DNA was digested with HinfI + AluI, and telomere length was measured by hybridization to the G-strand overhang. The population doubling at the time of sample isolation is shown above each lane. Size markers are shown in lanes 1 and 2. Sizes in kilobases are indicated on the left. (B) Mean telomere length was calculated from the data shown in A and plotted as a function of population doubling.
DISCUSSION
Although humans and rodent telomeres have been studied extensively, this study using chicken cells provides the first comprehensive analysis of telomere dynamics in a vertebrate that is not a mammal. Our finding that telomerase is present in a wide variety of somatic tissues indicates that telomerase regulation in chickens more closely resembles the situation observed in mice as opposed to humans. This finding was unexpected given that immortalization frequency, telomere length and animal lifespan are more similar for chickens and humans than for chickens and mice. Thus, contrary to previous suggestions (33), constitutive telomerase expression does not necessarily result in increased ease of immortalization. Moreover, as previously observed for M. spretus, abundant telomerase activity does not result in long telomeres (11).
Our studies of telomere length clearly demonstrate that telomeres from several different chicken tissues and transformed cell lines (Fig. 4 and data not shown) are in the 8- to 20-kb range. We find no evidence of a bimodal distribution with some telomeres of ≈10 kb and the remainder in to ≈100-kb range as was reported previously (17). Because the previous study used Southern hybridization to detect telomeric DNA, the 100-kb fragments might have been a consequence of limited digestion of interstitial telomeric DNA. The short length of chicken as compared with mouse telomeres means that chicken cells will provide a useful system for examining how telomere length is regulated by telomere-binding proteins and other telomerase-modifying activities.
The observation that telomerase is greatly down-regulated when cultures are established from chicken embryos has important implications for studies that depend on tissue culture cells to study senescence. Although chicken embryonic fibroblasts have been used extensively as a model to study aging (18, 19, 36), the relevance of studies of senescence in tissue culture cells to aging in whole animals has been questioned. Some concerns were alleviated by the demonstration that there is an age-dependant increase in senescent cells (as revealed by β-galactosidase staining) in a variety of tissues (31). However, our findings suggest that the cause of senescence may be quite different in cell cultures versus whole animals. Whereas senescence is most likely the result of telomerase down-regulation and telomere shortening in chicken fibroblast cultures, the presence of telomerase in many normal somatic tissues suggests that senescence in whole animals may be caused by other types of genotoxic damage.
Acknowledgments
We are grateful to Sheila Sheidler and Lyle Robson for providing us with chicken blood and chicken tissues and Clinton Jones for his gifts of chicken embryonic fibroblasts. We thank Titia de Lange and Bibo Lee for helpful suggestions. This work was supported by a seed grant from the University of Nebraska Center for Biotechnology and Grant 98–37 from the Nebraska Department of Health (to C.P.).
ABBREVIATION
- TRAP
telomere repeat amplification protocol
References
- 1.Greider C W. In: Telomerase Biochemistry and Regulation. Blackburn E H, Greider C W, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. pp. 35–68. [Google Scholar]
- 2.Greider C W. Proc Natl Acad Sci USA. 1998;95:90–92. doi: 10.1073/pnas.95.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolquist K A, Ellisen L W, Counter C M, Meyerson M, Tan L K, Weinberg R A, Haber D A, Gerald W L. Nat Genet. 1998;19:182–186. doi: 10.1038/554. [DOI] [PubMed] [Google Scholar]
- 4.Shay J W, Bacchetti S. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 5.Bryan T M, Englezou A, Dunham M A, Reddel R R. Exp Cell Res. 1998;239:370–378. doi: 10.1006/excr.1997.3907. [DOI] [PubMed] [Google Scholar]
- 6.Wen J, Cong Y S, Bacchetti S. Hum Mol Genet. 1998;7:1137–1141. doi: 10.1093/hmg/7.7.1137. [DOI] [PubMed] [Google Scholar]
- 7.Vaziri H, Benchimol S. Curr Biol. 1998;8:279–282. doi: 10.1016/s0960-9822(98)70109-5. [DOI] [PubMed] [Google Scholar]
- 8.Bodnar A G, Ouellette M, Frolkis M, Holt S E, Chiu C P, Morin G B, Harley C B, Shay J W, Lichtsteiner S, Wright W E. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 9.Harley C B, Vaziri H, Counter C M, Allsopp R C. Exp Gerontol. 1992;27:375–382. doi: 10.1016/0531-5565(92)90068-b. [DOI] [PubMed] [Google Scholar]
- 10.Blasco M A, Lee H W, Hande M P, Samper E, Lansdorp P M, DePinho R A, Greider C W. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 11.Chadeneau C, Siegel P, Harley C B, Muller W J, Bacchetti S. Oncogene. 1995;11:893–898. [PubMed] [Google Scholar]
- 12.Kipling D, Cooke H J. Nature (London) 1990;347:400–402. doi: 10.1038/347400a0. [DOI] [PubMed] [Google Scholar]
- 13.Zijlmans J M, Martens U M, Poon S S S, Raap A W, Tanke H J, Ward R K, Lansdorp P M. Proc Natl Acad Sci USA. 1997;94:7423–7428. doi: 10.1073/pnas.94.14.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Lange T, Shiue L, Myers R M, Cox D R, Naylor S L, Killery A M, Varmus H E. Mol Cell Biol. 1990;10:518–527. doi: 10.1128/mcb.10.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee H W, Blasco M A, Gottlieb G J, Horner J W, II, Greider C W, DePinho R A. Nature (London) 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 16.Strehler B L. Time, Cells and Aging. 2nd Ed. New York: Academic; 1977. pp. 37–41. [Google Scholar]
- 17.Lejnine S, Makarov V L, Langmore J P. Proc Natl Acad Sci USA. 1995;92:2393–2397. doi: 10.1073/pnas.92.6.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hay R. In: Aging in Cell and Tissue Culture. Holeckova E, Cristofalo V J, editors. New York: Plenum; 1970. [Google Scholar]
- 19.Williams J R, Dearfield K L. In: Handbook of Cell Biology of Aging. Cristofalo V J, editor. Boca Raton, FL: CRC; 1985. pp. 433–450. [Google Scholar]
- 20.Blasco M A, Rizen M, Greider C W, Hanahan D. Nat Genet. 1996;12:200–204. doi: 10.1038/ng0296-200. [DOI] [PubMed] [Google Scholar]
- 21.Kim N W, Wu F. Nucleic Acids Res. 1997;25:2595–2597. doi: 10.1093/nar/25.13.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strauss W M. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J, G, Smity J A, Struhl K, editors. Vol. 1. New York: Wiley; 1994. p. 2.22. [Google Scholar]
- 23.Salvz H P, Host J P. A Laboratory Guide for in Vivo Studies of DNA Methylation and Protein–DNA Interactions. Basel: Birkhauser; 1990. pp. 22–25. [Google Scholar]
- 24.Dionne I, Wellinger R J. Proc Natl Acad Sci USA. 1996;93:13902–13907. doi: 10.1073/pnas.93.24.13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wellinger R J, Wolf A J, Zakian V A. Cell. 1993;72:51–60. doi: 10.1016/0092-8674(93)90049-v. [DOI] [PubMed] [Google Scholar]
- 26.Harley C B, Futcher A B, Greider C W. Nature (London) 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 27.Coviello-McLaughlin G M, Prowse K R. Nucleic Acids Res. 1997;25:3051–3058. doi: 10.1093/nar/25.15.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eric H. In: Methods in Enzymology. Jakoby W B, Pastan I H, editors. Vol. 58. New York: Academic; 1979. pp. 381–393. [Google Scholar]
- 29.Patterson M K. In: Methods in Enzymology. Jakoby W B, Pastan I H, editors. Vol. 58. New York: Academic; 1979. pp. 150–151. [Google Scholar]
- 30.Reznikoff C A, Loretz L J, Pesciotta D M, Oberely T D, Ignjatovic M M. J Cell Physiol. 1987;131:258–301. doi: 10.1002/jcp.1041310302. [DOI] [PubMed] [Google Scholar]
- 31.Dimri G P, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano E E, Linskins M, Rubeli I, Pereira-Smith O, et al. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiyama E, Hiyama K, Tatsumoto N, Kodama T, Shay J W, Yokoyama T. Int J Oncol. 1996;9:453–458. doi: 10.3892/ijo.9.3.453. [DOI] [PubMed] [Google Scholar]
- 33.Prowse K R, Greider C W. Proc Natl Acad Sci USA. 1995;92:4818–4822. doi: 10.1073/pnas.92.11.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nanda I, Schmid M. Cytogenet Cell Genet. 1994;65:190–193. doi: 10.1159/000133630. [DOI] [PubMed] [Google Scholar]
- 35.van Steensel B, Smogorzewska A, de Lange T. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 36.Nielsen P J, Ryan J M. J Cell Physiol. 1981;107:371–378. doi: 10.1002/jcp.1041070308. [DOI] [PubMed] [Google Scholar]