

Abstract
Rapid, sensitive, and sequence-specific DNA detection can be achieved in one step using an engineered intrasterically regulated enzyme. The semi-synthetic inhibitor-DNA-enzyme (IDE) construct (left) rests in the inactive state, but upon exposure to a complementary DNA sequence undergoes a DNA hybridization-triggered allosteric enzyme activation (right). The ensuing rapid substrate turnover provides the built-in signal amplification mechanism for detecting approximately 10 femtomoles of DNA in less than three minutes under physiological conditions.
We describe the design, synthesis, and functional characterization of an intrasterically regulated semi-synthetic enzyme and its application in sequence specific DNA detection.1–2 The system is composed of covalently associated inhibitor-DNA-enzyme (IDE) modules and functions via DNA hybridization-triggered allosteric enzyme activation and signal amplification through substrate turnover.3 Its functional capacity is highlighted by the sequence specific detection of approximately 10 femtomoles of DNA in less than three minutes under physiological conditions. These studies suggest that rationally designed intrasterically regulated enzymes may constitute a promising new class of reagents for highly sensitive, rapid, and PCR-independent one-step detection of label-free DNA sequences.
The principles that govern intrasteric regulation in natural enzymes4 are firmly rooted in the physicochemical advantages of intramolecular binding. These enzymes often use an appending N- or C-terminal polypeptide pseudosubstrate to block access to their active sites. Enzyme regulation (activation) occurs at an allosteric site, typically the site between the enzyme and pseudosubstrate, where a conformational change or cleavage event removes the pseudosubstrate from the active site. We sought to mimic the intrasteric regulatory features of natural enzymes by designing novel semi-synthetic constructs that can be activated in the presence of complementary DNA sequences. The IDE complex described employs a single stranded DNA probe to covalently tether Cereus neutral protease5 (CNP) to its small molecule phosphoramidite inhibitor.6 We anticipated that the conformational flexibility of the ss-DNA tether in an appropriately functionalized system would allow facile intramolecular binding of the inhibitor to the enzyme active site to furnish the inactive state of the IDE (Figure 1). Likewise, we postulated that IDE should rapidly convert to an active state in the presence of its complementary DNA sequence because formation of a high-affinity DNA duplex structure was expected to drastically alter the conformation of the tether and favor the liberation of the inhibitor from the enzyme active site. Once activated, the enzyme would act upon its fluorogenic substrate, present in situ, producing an optical output signal. Moreover, because the enzyme turns over many copies of the substrate, in principle each probe hybridization event can be chemically amplified to furnish high detection sensitivity.
Figure 1.
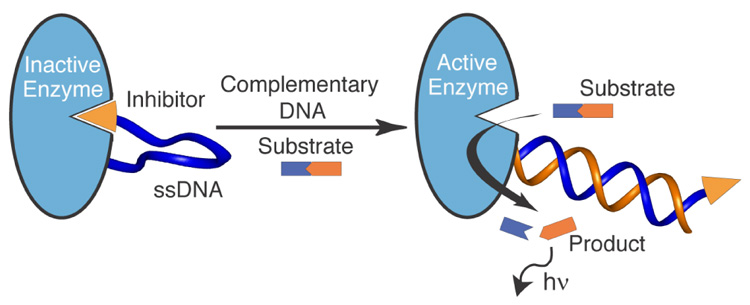
Schematic representation of an intrasterically inactivated inhibitor-DNA-enzyme (IDE) construct (left) and the DNA hybridization-triggered enzyme activation (right). IDE can be used to sense low concentrations of complementary DNA because of its built-in capacity for signal amplification via rapid substrate turnover.
The foremost variable in the design of an IDE complex is the enzyme module, which must be compatible with the allosteric DNA tether and possess high intrinsic catalytic activity to afford rapid rates of signal evolution. The choice of the enzyme in turn dictates the type of inhibitor and substrate to be used. In the present design we have employed CNP5, an endolytic extracellular zinc metalloprotease from Bacillus cereus, as the enzyme constituent of the IDE. Its high structural homology to thermolysin7 was invaluable in selecting the inhibitor module6 and the inhibitor-DNA and DNA-enzyme attachment sites used in our design (vide infra). Molecular modeling was used to determine the attachment sites that seemed to minimize DNA strain and unfavorable steric interactions in the intramolecular binding of the inhibitor to the enzyme active site. Based on the modeling analyses, the surface exposed glutamic acid 151 of CNP was chosen as the DNA-enzyme ligation site. Furthermore, because the wild type CNP lacks any cysteine residues, the CNPE151C mutant was chosen as our target enzyme to afford a chemoselective handle for DNA attachment. The CNP gene8 was amplified by PCR using genomic DNA from B. cereus (strain DSM3101) as the template. The resulting gene, which coded for the proenzyme of cereus neutral protease, was then mutated (E151C) using overlap extension-PCR method9 and inserted into pET19b (Novagen) to afford the expression plasmid pCNP151C. CNPE151C was overexpressed in E. coli BL21(DE3) (Novagen), isolated as inclusion bodies, refolded, and purified by D-phenylalanine affinity chromatography.
The enzyme substrate, DABCYL-βAla-Ala-Gly-Leu-Ala-βAla-EDANS, was designed based on CNP’s substrate selectivity and prepared in two steps by the standard solid- and solution-phase methods.10 Protease activity was measured as a function of increasing EDANS fluorescence in time as the result of the endolytic cleavage of the peptide substrate separating the EDANS fluorophore from DABCYL quencher. The measured kcat and Km values of 165 s−1 and 15 µM, respectively (20 mM Tris, pH 7.0) are in good agreement with literature values for the wild type protease.
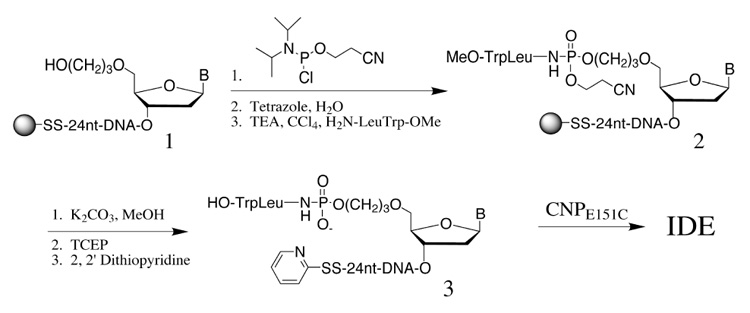
Naturally occurring inhibitors of this class of proteases often utilize the phosphoramidite as the transition state mimic.6 By adopting this class of inhibitors to our IDE design, we were also able to develop a convenient solid-phase synthetic process for the production of the desired inhibitor-DNA conjugate (Scheme 1). The support-bound 24-mer oligonucleotide11 1 having a 3’-thiol-modifier C3 S-S CPG and a 5’-C3 spacer was synthesized according to the reagents and procedures recommended for ultramild DNA synthesis (Glen Research). Subsequent reaction with 2-cyanoethyl diisopropyl chlorophophoramidite followed by oxidation afforded the corresponding 5’-H-phosphonate diester intermediate. The H-phosphonate was then reacted with H2N-LeuTrp-OMe in the presence of triethylamine and carbon tetrachloride12 to give the protected phosphoramidite-containing DNA-phosphoryl-LeuTrp-OMe 2. The DNA construct was then deprotected in aqueous MeOH/K2CO3 and cleaved from the solid support by disulfide reduction in the presence of tris(2-carboxyethyl) phosphine (TCEP). The free 3’-thiol moiety was then activated as a pyridyl disulfide and the resulting oligonucleotide-inhibitor complex 3 was purified by polyacrylamide gel electrophoresis and RP-HPLC and characterized by MALDI-MS. The coupling of inhibitor-DNA conjugate 3 to CNPE151C took place smoothly and in high yields under slightly basic conditions. The desired IDE complex was purified to homogeneity by anion exchange chromatography (FPLC) and further characterized by gel electrophoresis.
scheme 1.
IDE was found to be remarkably sensitive in detecting low concentrations of complementary DNA. The correlation between IDE activity and complementary DNA concentration (10 µM to 10 pM) was examined by equilibrating complementary DNA with IDE (1.5 nM) for 5 minutes prior to addition of 40 µM substrate (Figure 2a). Even at 100 pM complementary DNA concentrations (10 femtomoles), a five fold increase in initial rate over IDE alone was observed giving rise to a detectable signal in less than 3 minutes. Furthermore, 10 pM DNA concentrations could be distinguished over the background signal if the assay was extended to 80 minutes (Figure 2b). Conversely, IDE could not be activated in the presence of noncomplementary DNA11 even at the highest tested concentration (10 µM). Moreover, noncovalently associated ID-enzyme complexes did not display increased protease activity in the presence of complementary DNA further supporting the conformational and the covalent anchoring requirements of the intrasterically regulated IDE.
Figure 2.
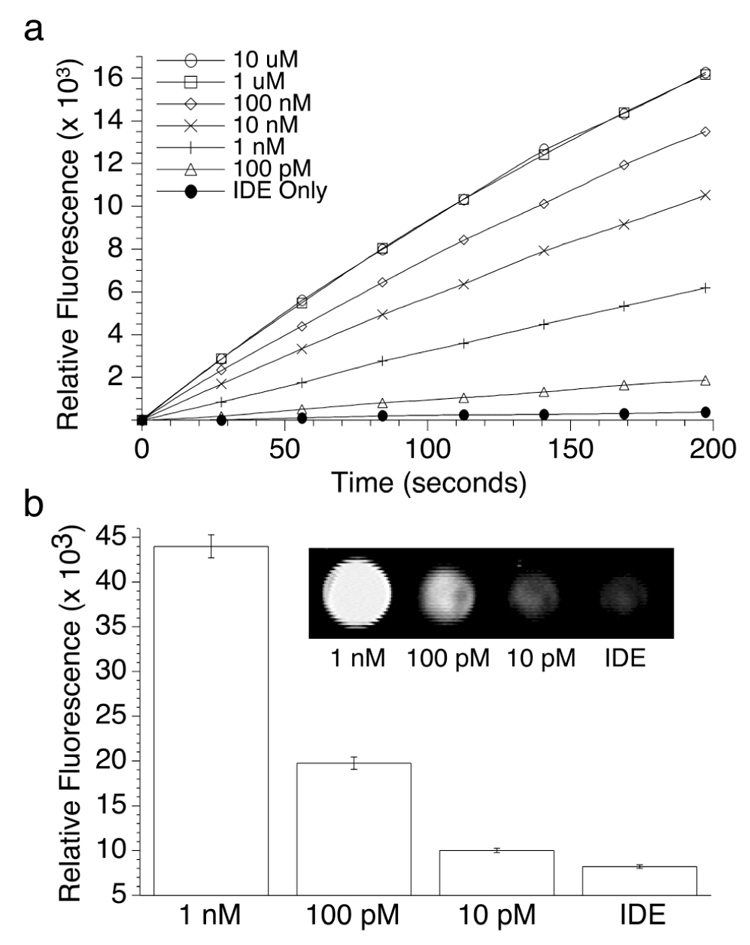
Effect of complementary DNA concentration on IDE activity. (a) Activity was measured through endolytic cleavage of substrate (40 mM) in the presence of 1.5 nM IDE (1M NaCl, 20 mM TRIS, pH 8) using a fluorescent plate reader (λex=360, λem=465). (b) Fluorescence measurements 80 minutes after addition of substrate showed a lower detection limit of 10 pM complementary DNA. Additionally, IDE activity could be visualized using a broad band UV lamp and CCD detector (inset), which has important practical applications for high-throughput screening.
The above experiments highlight the potential of IDE in practical applications where sensitivity and simple detection methods are a requirement.
Supplementary Material
Supporting Information. Cloning and expression of CNPE151C, synthesis and purification of IDE and substrate (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We thank ONR for financial support (N00014-98-1-0622), NIH for a postdoctoral fellowship (KMG), and NSF for Predoctoral Fellowship (DAT). A.S. is the recipient of the Norton B. Gilula Graduate Student Fellowship.
Contributor Information
Alan Saghatelian, Contribution from the Departments of Chemistry and Molecular Biology and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, California 92037..
Kevin M. Guckian, Contribution from the Departments of Chemistry and Molecular Biology and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, California 92037.
Desiree A. Thayer, Contribution from the Departments of Chemistry and Molecular Biology and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, California 92037.
M. Reza Ghadiri, Contribution from the Departments of Chemistry and Molecular Biology and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, California 92037. ghadiri@scripps.edu.
References
- 1.PCR-based techniquesHacia JG, Fan J-B, Ryder O, Jin L, Edgemon K, Ghandour G, Mayer RA, Sun B, Hsie L, Robbins CM, Brody LC, Wang D, Lander ES, Lipshutz R, Fodor SPA, Collins FS. Nat. Genet. 1999;22:164–167. doi: 10.1038/9674.Kostrikis LG, Tyagi S, Mhlanga MM, Ho DD, Kramer FR. Science. 1998;279:1228–1229. doi: 10.1126/science.279.5354.1228.Nuovo GJ. Rapid Detect. Infect. Agents. 1998:191–206.Nuovo GJ. Methods Mol. Biol. 2000;123:217–238. doi: 10.1385/1-59259-677-0:217.Post JC, Ehrlich GD. JAMA. 2000;283:1544–1546. doi: 10.1001/jama.283.12.1544.
- 2.PCR-independent techniquesElghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Science. 1997;277:1078–1081. doi: 10.1126/science.277.5329.1078.Taton TA, Mirkin CA, Letsinger RL. Science. 2000;289:1757–1760. doi: 10.1126/science.289.5485.1757.Player AN, Shen LP, Kenny D, Antao VP, Kolberg JA. J. Histochem. Cytochem. 2001;49:603–611. doi: 10.1177/002215540104900507.Stojanovic MN, De Prada P, Landry DW. ChemBioChem. 2001;2:411–415. doi: 10.1002/1439-7633(20010601)2:6<411::AID-CBIC411>3.0.CO;2-I.Umek RM, Lin SW, Vielmetter J, Terbrueggen RH, Irvine B, Yu CJ, Kayyem JF, Yowanto H, Blackburn GF, Farkas DH, Chen Y-P. J. Mol. Diagn. 2001;3:74–84. doi: 10.1016/S1525-1578(10)60655-1.
- 3.For a recent review and comparison of techniques combining nucleic acid amplification and detection seeSchweitzer B, Kingsmore S. Current Opinion in Biotechnology. 2001;12:21–27. doi: 10.1016/s0958-1669(00)00172-5.
- 4.Kobe B, Kemp BE. Nature. 1999;402:373–376. doi: 10.1038/46478. [DOI] [PubMed] [Google Scholar]
- 5.Feder J, Keay L, Garrett LR, Cirulis N, Moseley MH, Wildi BS. Biochim Biophys Acta. 1971;251:74–78. doi: 10.1016/0005-2795(71)90061-4. [DOI] [PubMed] [Google Scholar]
- 6.(a) Matthews BW. Acc. Chem. Res. 1988;21:333–340. [Google Scholar]; (b) Komiyama T, Suda H, Aoyagi T, Takeuchi T, Umezawa H. Arch. Biochem. Biophys. 1975;171:727–731. doi: 10.1016/0003-9861(75)90085-5. [DOI] [PubMed] [Google Scholar]
- 7.Stark W, Pauptit RA, Wilson KS, Jansonius JN. Eur J Biochem. 1992;207:781–791. doi: 10.1111/j.1432-1033.1992.tb17109.x. [DOI] [PubMed] [Google Scholar]
- 8.Wetmore DR, Wong SL, Roche RS. Mol Microbiol. 1992;6:1593–1604. doi: 10.1111/j.1365-2958.1992.tb00884.x. [DOI] [PubMed] [Google Scholar]
- 9.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 10.Matayoshi ED, Wang GT, Krafft GA, Erickson J. Science. 1990;247:954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 11.The sequence of the IDE tether = AGTTGATTTGTGCTTCAGTGTGCT and the complement = AGCACACTGAAGCACAAATCAACT and nonspecific = AGTGGAGATGACAGCTACTCGTGC
- 12.Gryaznov S, Chen JK. J. Am. Chem. Soc. 1994;116:3143–3144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information. Cloning and expression of CNPE151C, synthesis and purification of IDE and substrate (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.