

Abstract
The angiotensin II (AngII) type 1 receptor (AT1) plays a critical role in hypertrophy of vascular smooth muscle cells (VSMCs). Although it is well known that Gq is the major G protein activated by the AT1 receptor, the requirement of Gq for AngII-induced VSMC hypertrophy remains unclear. By using cultured VSMCs, this study examined the requirement of Gq for the epidermal growth factor receptor (EGFR) pathway, the Rho-kinase (ROCK) pathway, and subsequent hypertrophy. AngII-induced intracellular Ca2+ elevation was completely inhibited by a pharmacological Gq inhibitor as well as by adenovirus encoding a Gq inhibitory minigene. AngII (100nm)-induced EGFR transactivation was almost completely inhibited by these inhibitors, whereas these inhibitors only partially inhibited AngII (100nm)-induced phosphorylation of a ROCK substrate, myosin phosphatase target subunit-1. Stimulation of VSMCs with AngII resulted in an increase of cellular protein and cell volume but not in cell number. The Gq inhibitors completely blocked these hypertrophic responses, whereas a G protein-independent AT1 agonist did not stimulate these hypertrophic responses. In conclusion, Gq appears to play a major role in the EGFR pathway, leading to vascular hypertrophy induced by AngII. Vascular Gq seems to be a critical target of intervention against cardiovascular diseases associated with the enhanced renin-angiotensin system.
ANGIOTENSIN II (ANGII), the major bioactive peptide of the renin-angiotensin system, has been implicated in various cardiovascular diseases such as hypertension and atherosclerosis. Among the AngII receptor subtypes, the AngII type 1 receptor (AT1) is expressed in the cardiovascular system, such as in vascular smooth muscle cells (VSMCs). Through this receptor, AngII stimulates hypertrophic remodeling of VSMCs, which involves a variety of downstream signal transduction mechanisms (1,2,3,4). The AT1 receptor is traditionally described as mainly coupling to a heterotrimeric G protein, Gq (5). Activation of phospholipase C via Gq through the AT1 receptor leads to intracellular Ca2+ elevation and protein kinase C activation in VSMCs (5). Intracellular Ca2+ elevation and subsequent reactive oxygen species production via the AT1 receptor seem to be required for AngII-induced transactivation of the epidermal growth factor receptor (EGFR), a critical signal transduction event in mediating subsequent ERK activation and VSMC hypertrophy (6,7,8,9). Although coupling of the AT1 receptor to other heterotrimeric G proteins, including G12, G13, and Gi, have been demonstrated in VSMCs (10,11,12,13), participation of these G proteins in AngII functions of VSMCs remain unclear.
A small G protein, Rho, and its prototypical effector, Rho-kinase (ROCK), have also been implicated in various pathological responses of AngII (6) including the hypertrophy of VSMCs (14). In general, a G protein-coupled receptor activates Rho through a family of guanine nucleotide exchange factors (RhoGEFs) containing an regulator of G protein signaling domain that specifically interacts with G12 or G13 (15). However, Gq is also capable of similarly activating Rho (16), and a Gq-dependent signal may also participate in Rho and ROCK activation by the AT1 receptor in VSMCs (17). Furthermore, there is accumulating evidence suggesting the presence of heterotrimeric G protein-independent signal transduction originating via the AT1 receptors (18,19). In particular, G protein-independent mechanisms have been reported in activation of EGFR (20) as well as Rho (21) through the AT1 receptor, although the findings were mainly limited to artificial cell lines.
According to the above background information, we have used reagents specifically targeting Gq to test our hypothesis that Gq is the major G protein that participates in hypertrophic signal transduction in VSMCs stimulated by AngII. Here we demonstrate several lines of evidence supporting that Gq is indeed critical for the EGFR pathway and subsequent VSMC hypertrophy through the AT1 receptor.
Materials and Methods
Reagents
AngII was purchased from Sigma (St. Louis, MO). The main concentration of AngII used in the study (100 nm) was chosen according to our past study demonstrating its maximum ERK activation response in cultured rat VSMCs (22). Platelet-derived growth factor (PDGF)-BB was purchased from R&D Systems (Minneapolis, MN). [Sar1, Ile4, Ile8] AngII was purchased from Bachem (Torrance, CA). A23187 was purchased from Calbiochem (La Jolla, CA). YM-254890, a selective pharmacological inhibitor of Gq (23), was a gift from Astellas Pharma Inc. (Tokyo, Japan). The YM254890 concentration (1 or 10 μm) used in the study was chosen according to the original report of the inhibitor that established its Gq specificity (23). Rat intermedin-(8–47) was a kind gift from Dr. Nae Dun (Temple University). Phospho-specific antibody for Tyr1068-phosphorylated EGFR was purchased from Biosource International (Camarillo, CA). Phospho-specific antibody for Tyr204-phosphorylated ERK, and antibodies against EGFR and ERK2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-specific antibody for Thr853-phosphorylated myosin phosphatase target (MYPT) subunit 1 was purchased from Upstate (Lake Placid, NY), and antibody against MYPT subunit 1 was purchased from Covance (Vienna, VA). Antibody against glyceraldehyde-3-phosphate dehydrogenase was purchased from Chemicon (Temecula, CA).
Cell culture
Rat aortic VSMCs were prepared by the explant method and cultured in DMEM containing 10% fetal calf serum, penicillin, and streptomycin, as described in detail previously (24). Subcultured cells from passages 3–12 were used in the experiments and showed more than 99% positive immunostaining of smooth muscle α-actin antibody (22) and were negative for endothelial nitric oxide synthase expression (25). The dominant expression of AT1 receptors was confirmed on the basis of binding studies with a specific receptor antagonist (22). For each experiment, cells were cultured to 80–90% confluency and then made quiescent by incubating in serum-free media for 2–3 d (22).
Adenoviral infection
The adenoviral vectors encoding Gαq-(305–359), a specific inhibitor of Gq (GqI), was constructed as previously described (26). The adenovirus titer, plaque-forming unit, was determined by Adeno-X rapid titer kit (BD Biosciences, Franklin Lakes, NJ). VSMCs were infected with adenovirus at a multiplicity of infection (moi; plaque-forming unit per cell) of 20 for 2 d as previously described (27). Transfection efficiency was estimated to be greater than 90% as defined by infection of adenovirus encoding green fluorescent protein (25).
Immunoblotting
Immunoblotting was performed as previously described (22). The results were quantified by densitometry in the linear range of film exposure using CanoScan N670U (Canon, Lake Success, NY) and Un-Scan-It Gel 4.3 software (Silk Scientific, Salt Lake City, UT). Results were expressed as percent increase in which the maximum response to AngII is defined as 100% because the basal signals are more varied, depending on film exposure, than the stimulated signals (17).
Intracellular Ca2+ measurements
Intracellular Ca2+ was measured as described previously by using fura2 as an indicator (28).
Intracellular cAMP accumulation
After pretreatment with or without a GqI, VSMCs were incubated with 100 nm intermedin at 37 C for 20 min in the presence of 0.5 mm methylisobutylxanthine (29), and intracellular cAMP was determined by an enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI) following the manufacture’s instruction.
Protein assay
Subconfluent VSMCs on 12-well culture plates were incubated with serum-free DMEM for 3 d. For adenovirus infection, VSMCs were incubated with serum-free DMEM for 1 d and infected with adenovirus at 20 moi in serum-free DMEM for 2 d. The cells were further incubated with or without 100 nm AngII for 3 d. After aspiration of the medium, cells were washed twice with ice-cold Hanks’ balanced salt solution, and the total amount of cellular protein was measured as previously described (25).
Cell volume assay
After the pretreatments described in the protein assay, VSMCs were washed with Hanks’ balanced salt solution and trypsinized. The cells were then suspended in PBS, and the cell volume was measured by Z2 Coulter particle count and size analyzer as previously described (25).
Proliferation assay
After the pretreatments described in the protein assay, cell proliferation was measured using a CellTiter 96 Aqueous cell proliferation assay kit (Promega, Madison, WI) following the manufacturer’s protocol as previously described (25).
Statistics
Data were analyzed by using Student’s t test or one-way ANOVA. The mean ± sem was determined with a significance level of P < 0.05. Results are representative of at least three separate experiments.
Results
Requirement of Gq for the EGFR transactivation pathway induced by AngII
We have demonstrated that intracellular Ca2+ elevation participates in AngII-induced EGFR and ERK activation in VSMCs (7). Interestingly, it was reported that in addition to Gq, G12 (10) and Gβγ (11) might be involved in AngII-induced intracellular Ca2+ elevation in VSMCs. To identify the specific role of Gq in AngII signal transduction pathways in VSMCs, we first examined the effect of a selective pharmacological GqI (23) on intracellular Ca2+ elevation induced by AngII. The Ca2+ elevation induced by AngII was completely blocked by pretreatment with the GqI YM-254890 at 1 μm (Fig. 1A).
Figure 1.

The EGFR/ERK cascade induced by AngII was blocked by YM-254890, a pharmacological Gq inhibitor. A, VSMCs pretreated with vehicle (dimethylsulfoxide 0.1%) or YM-254890 (1 μm) for 10 min were stimulated with AngII (100 nm). Intracellular Ca2+ concentration was measured. B, VSMCs pretreated with or without YM-254890 (1 μm) for 10 min were stimulated with AngII (100 nm) or A23187 (10 μm) for 2 min. Cell lysates were immunoblotted with antibodies against Tyr1068-phosphorylated EGFR (EGFR-p) and total EGFR as indicated. Phosphorylation of EGFR was measured by densitometry. †, P < 0.05, compared with the stimulated control. C, VSMCs pretreated with or without various concentrations of YM-254890 for 10 min were stimulated with AngII (100 nm) for 10 min. Cell lysates were immunoblotted with antibodies against Tyr204-phosphorylated ERK and total ERK as indicated.
Our previous publication strongly suggests a critical role of Gq in mediating EGFR transactivation and subsequent ERK activation in VSMCs (7). However, there are reports suggesting a possibility that AngII activates EGFR and ERK through Gq-independent mechanisms involving β-arrestin (30) and the AT1 C-tail Tyr319 phosphorylation (20). Therefore, we tested the effects of YM-254890 on the EGFR transactivation pathway induced by AngII in VSMCs. Pretreatment with YM-254890 (1 μm) completely inhibited EGFR transactivation induced by AngII, but not by a Ca2+ ionophore, A23187, in VSMCs (Fig. 1B). ERK phosphorylation induced by AngII was also inhibited by pretreatment with 0.1–10 μm YM-254890 (Fig. 1C). YM-254890 at the concentrations less than 10 nm had no inhibitory effect on ERK phosphorylation (Fig. 1C) or EGFR transactivation (data not shown) induced by AngII.
Previously we and others have reported that AngII activates the Rho/ROCK pathway in VSMCs (14,17). This cascade appears to be in parallel with the EGFR/ERK cascade as judged by phosphorylation of MYPT, a selective ROCK substrate (17). The 100 nm AngII-induced maximum MYPT phosphorylation at 2 min was only partially inhibited by pretreatment with YM-254890 (1 μm) in VSMCs (Fig. 2A). In contrast, both MYPT and EGFR phosphorylations induced by 1 nm AngII were completely blocked by YM-254890 (1 μm) in VSMCs (Fig. 2A). These data suggest a distinct requirement of specific G protein to activate the Rho/ROCK cascade by AngII (Gq at low AngII concentration and G12/13 at high AngII concentration). In addition, to confirm a G protein selectivity of YM-254890, its effect on a Gs agonist-dependent cAMP production was examined. YM-254890 up to 10 μm did not interfere with an intracellular cAMP elevation in VSMCs induced by intermedin, a potent agonist for a family of Gs-coupled receptors (31) (Fig. 2B).
Figure 2.
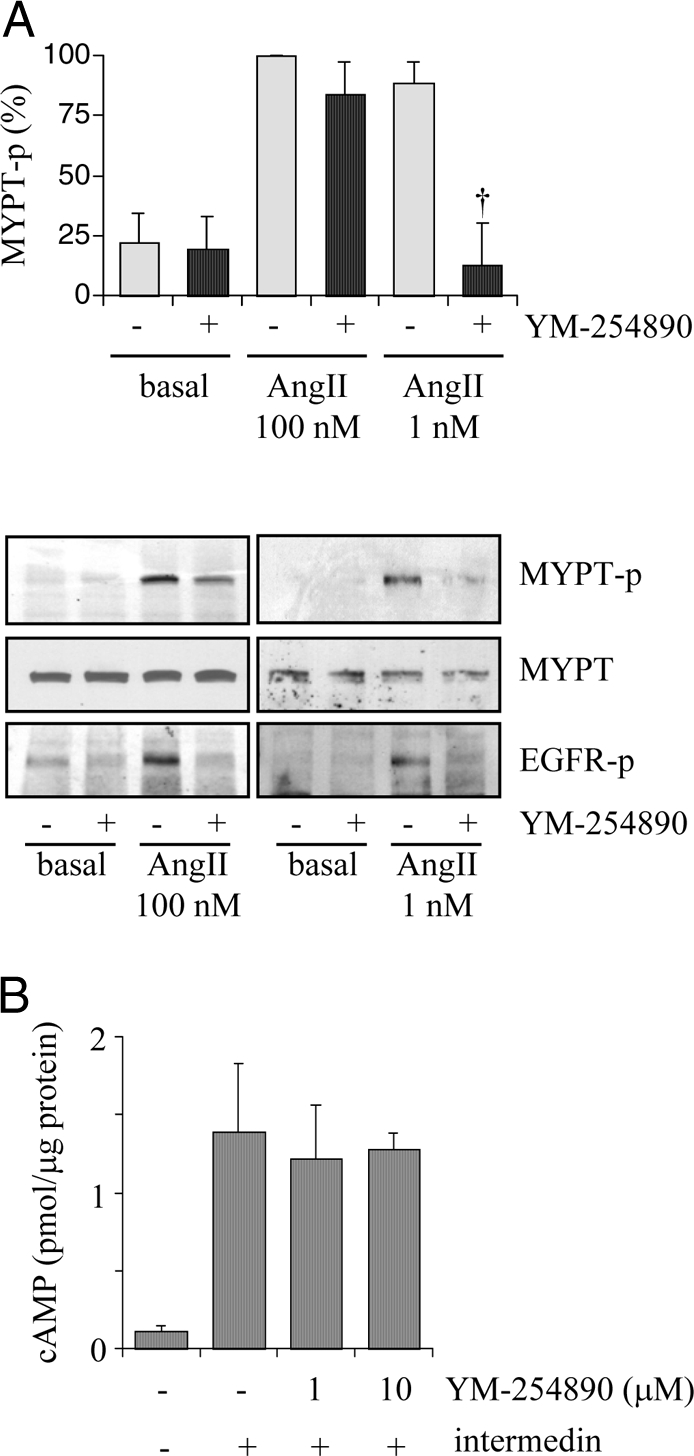
Effects of a Gq inhibitor on phosphorylation of a ROCK substrate, MYPT, by AngII or cAMP elevation by a Gs agonist, intermedin. A, VSMCs pretreated with or without YM-254890 (1 μm) for 10 min were stimulated with AngII (100 or 1 nm) for 2 min. Cell lysates were immunoblotted with antibodies against Thr853-phosphorylated MYPT (MYPT-p), total MYPT, and Tyr1068-phosphorylated EGFR (EGFR-p) as indicated. Phosphorylation of MYPT was measured by densitometry. †, P < 0.05, compared with the stimulated control. B, VSMCs pretreated with or without YM-254890 (1 and 10 μm) for 10 min were stimulated with intermedin (100 nm) for 20 min, and intracellular cAMP concentration was evaluated.
To compensate for any possible nonspecific effects of the pharmacological Gq inhibitor, we alternatively used an infection of adenovirus encoding a GqI minigene. The selectivity of this inhibitor has been well documented (32). Infection of GqI adenovirus inhibited the activation of EGFR and ERK induced by AngII (100 nm) almost completely. However, it only partially inhibited 100 nm AngII-induced MYPT phosphorylation (Fig. 3, A and B). GqI adenovirus also completely inhibited intracellular Ca2+ elevation induced by AngII in VSMCs (Fig. 3C). These data support the above findings obtained by the pharmacological Gq inhibitor.
Figure 3.

GqI selectively blocks the EGFR/ERK cascade induced by AngII. A and B, VSMCs infected with adenovirus encoding GqI minigene or control vector (20 moi) were stimulated with AngII (100 nm) for 2 min (A) or 10 min (B) and immunoblotted with antibodies against Tyr1068-phosphorylated EGFR (EGFR-p), total EGFR, and Thr853-phosphorylated MYPT (MYPT-p) (A) or Tyr204-phosphorylated ERK (ERK-p) and ERK2 (B). †, P < 0.05, compared with the stimulated control. C, VSMCs infected with adenovirus encoding GqI or control vector (20 moi) were stimulated with AngII (100 nm), and the intracellular Ca2+ level was measured.
It has been reported that [Sar1, Ile4, Ile8] AngII can activate ERK although a G protein-independent manner in cultured VSMCs (33). Also, G protein-independent Rho activation is known to occur in HEK cells overexpressing the AT1 receptor (21). However, [Sar1, Ile4, Ile8] AngII (10 μm) did not stimulate EGFR, ERK, or MYPT phosphorylation in VSMCs (Fig. 4A). To provide supporting evidence that the compound effectively interacted with the AT1 receptors in rat aortic VSMCs, we pretreated with [Sar1, Ile4, Ile8] AngII to see whether it inhibits the subsequent AngII response due to the receptor binding and subsequent internalization. Pretreatment of VSMCs with 10 μm [Sar1, Ile4, Ile8] AngII inhibited subsequent phosphorylations of EGFR and MYPT induced by AngII in VSMCs (Fig. 4B). Taken together, these data suggest that the G protein-independent cascade is insufficient to activate the EGFR/ERK or the Rho/ROCK cascade in VSMCs.
Figure 4.
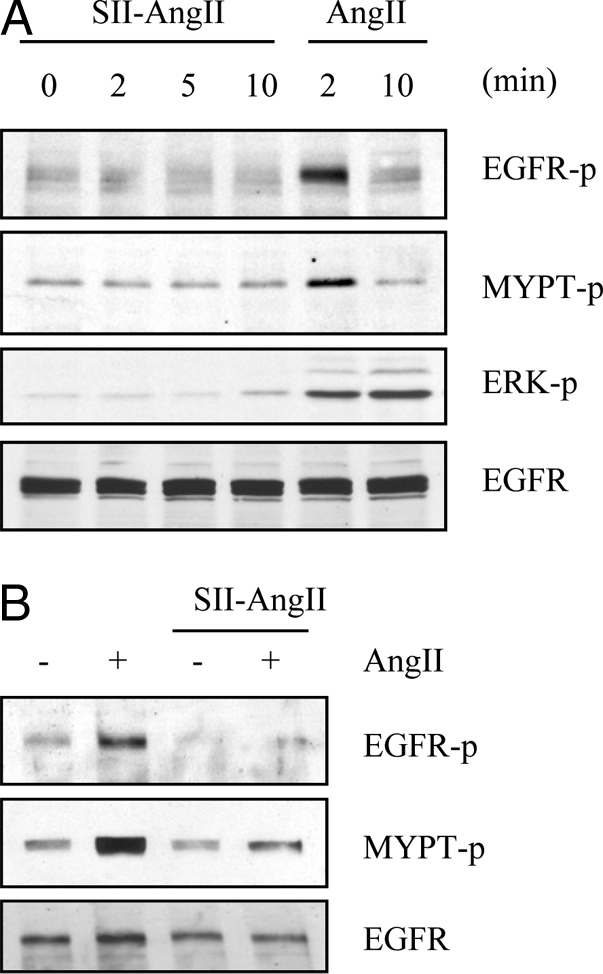
Effect of [Sar1, Ile4, Ile8] AngII on hypertrophic signal transduction in VSMCs. A, VSMCs were stimulated with 10 μm [Sar1, Ile4, Ile8] AngII (SII-AngII) or 100 nm AngII for the indicated time periods. B, VSMCs pretreated with or without 10 μm [Sar1, Ile4, Ile8] AngII (SII-AngII) for 30 min were stimulated with 100 nm AngII for 2 min. Cell lysates were immunoblotted with antibodies against Tyr1068-phosphorylated EGFR (EGFR-p), total EGFR, Thr853-phosphorylated MYPT (MYPT-p), and Tyr204-phosphorylated ERK as indicated.
Indispensable role of Gq for VSMC hypertrophy induced by AngII
Finally, we tested whether the inhibitors of Gq attenuate VSMC hypertrophy induced by AngII. We recently demonstrated that AngII-induced VSMC hypertrophy is quantitatively detectable by total cellular protein and cell volume measurements (25,34). YM-254890 (1 μm) inhibited the increase in cellular protein and cell volume induced by AngII in VSMCs. Neither AngII nor YM-254890 affected VSMC proliferation (Fig. 5, A–C). In contrast, YM-254890 had no inhibitory effect on the increase in cellular protein associated with PDGF-BB-induced VSMC growth (Fig. 5D). Infection with GqI adenovirus also inhibited protein accumulation induced by AngII in VSMCs (Fig. 6). However, 10 μm [Sar1, Ile4, Ile8] AngII did not increase total cellular protein (98 ± 10%, compared with 100% as basal, n = 3). These data suggest that activation of Gq is indispensable for VSMC hypertrophy induced by AngII.
Figure 5.

Effect of a Gq inhibitor, YM-254890, on protein synthesis, cell volume, and proliferation in VSMCs stimulated by AngII. A–C, After incubation with serum-free medium for 3 d, VSMCs were pretreated with YM-254890 (1 μm) for 10 min and then stimulated with 100 nm AngII for 3 d. Cell protein accumulation (A), cell volume (B), and cell proliferation (C) were measured. *, P < 0.05, compared with nonstimulated VSMCs. D, After incubation with serum-free medium for 3 d, VSMCs were pretreated with YM-254890 (1 μm) for 10 min and then stimulated with 50 ng/ml PDGF-BB for 3 d. Cell protein accumulation was measured.
Figure 6.
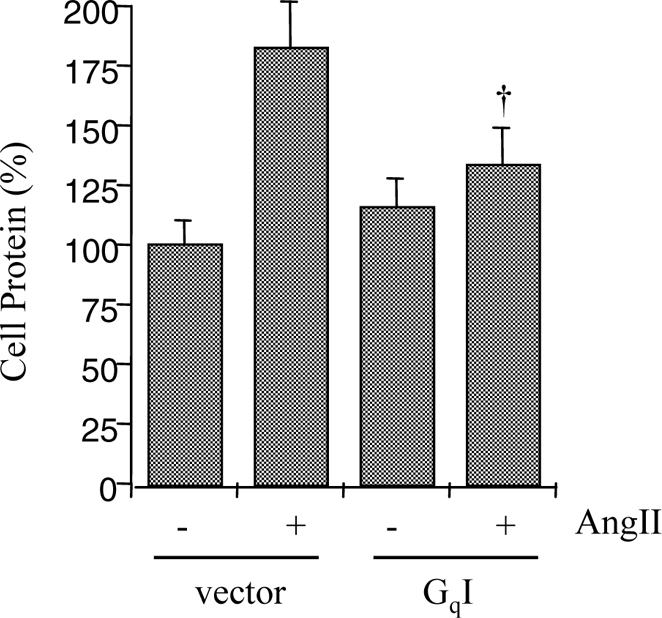
Effect of GqI on protein synthesis in VSMCs stimulated by AngII. After infection with adenovirus encoding GqI (20 moi) or the control vector (20 moi), VSMCs were stimulated by AngII (100 nm) for 3 d. Cell protein accumulation was measured. †, P < 0.05, compared with the stimulated control.
Discussion
In the present study, we have demonstrated a novel and significant finding that Gq coupling of the AT1 receptor is essential for the EGFR transactivation and subsequent VSMC hypertrophy induced by AngII. Various mutation studies of the AT1 receptor suggest that AngII could induce G protein-independent EGFR/ERK pathway activation (20,30,35). The mutations of AT1, such as D74F, Y292F, and N295S, diminished the receptor’s ability to activate phospholipase C, whereas these mutations did not have an effect on ERK activation by AngII in COS-1 cells (35). Also, an AT1 mutant, Y319F, does not induce EGFR transactivation (20), and a DRY/AAY AT1 mutant activates ERK through a β-arrestin-dependent mechanism (30). However, the role of Tyr319 in EGFR transactivation has been questioned (28,36), and most of the findings regarding G protein-independent functions of the AT1 receptor were restricted to artificial cell lines with forced overexpression of the AT1 receptor or its mutants. On the contrary, the EGFR transactivation induced by AngII through the endogenous AT1 receptor was completely blocked with two distinct but specific tools to inhibit Gq activity in our primary cultured VSMCs. In addition, an AngII analog, which has been shown to selectively induce G protein-independent signals via the AT1 receptors (21,33), failed to activate EGFR or ERK in the present study. These data therefore indicate that at least in a primary cultured model of vascular smooth muscle cells, the AT1 receptor-induced EGFR/ERK pathway activation is exclusively mediated through Gq.
It has been suggested that the AT1 receptor is coupled to G12/13 in VSMCs (10,11,13). In general, activation of G12/13 induces the Rho/ROCK signaling pathway through RhoGEFs such as PDZ-RhoGEF, p115 RhoGEF, and leukemia-associated RhoGEF (15) via interaction between G12/13 and the regulator of G protein signaling domain of these RhoGEFs. Although we have proposed a cytosolic tyrosine kinase, proline-rich tyrosine kinase (Pyk2), as a possible upstream mediator of the EGFR transactivation (37), we recently demonstrated clearly that the Rho/ROCK activation and not the EGFR transactivation by AngII requires Pyk2 activation in VSMCs (17). Moreover, AngII enhances association of Pyk2 with tyrosine-phosphorylated primary decidual zone-RhoGEF in VSMCs (17). In contrast to our earlier investigations suggesting that Pyk2 is activated via Gq by AngII in VSMCs (37,38), here YM-254890 as well as a GqI minigene only partially prevented MYPT phosphorylation induced by 100 nm AngII. This may be in line with a publication demonstrating association with and subsequent activation of Pyk2 by G13 (39). However, MYPT phosphorylation induced by 1 nm AngII was completely blocked by YM-254890. Therefore, these data indicate a distinct involvement of specific G proteins to activate the Rho/ROCK cascade by AngII. Gq may play a major role in activating ROCK at a low AngII concentration potentially through the Pyk2/RhoGEF axis, whereas at a high AngII concentration, G12/13 are sufficiently activated through the AT1 receptor to activate a RhoGEF without the Gq dependency.
It has been reported that not only the EGFR/ERK pathway (34,40,41) but also the Rho/ROCK pathway (14) induced by AngII contributes to VSMC hypertrophy. Thus, in vivo inhibition of ROCK by pharmacological ROCK inhibitors appears to suppress vascular and cardiac hypertrophy induced by AngII infusion in rats (42,43). In the present study with 100 nm AngII stimulation, specific inhibition of Gq was sufficient enough to block AngII-induced VSMC hypertrophy with minimum inhibitory effect on ROCK activation by AngII. In addition, recent genetic targeting of a ROCK isoform, ROCK1, rather suggests its specific role in mediating cardiovascular fibrosis (44,45). However, the in vivo requirement of the Rho/ROCK cascade for VSMC hypertrophy induced by AngII may depend on each vascular bed.
Regarding the experimental strategies used, we did not use a standard protein synthesis assay measuring a radiolabeled leucine incorporation in the present study. However, we believe that our two independent methods used here measures the hypertrophic effects of AngII just as sufficiently and perhaps more directly. Although the leucine incorporation may predict the subsequent cell hypertrophy, it can also be enhanced under cell proliferation. In addition, slight distinctions in control cell responses between Figs. 5 and 6 are likely due to different priming methods used before AngII stimulation.
Because the AT1 receptor is the major receptor expressed in our cultured VSMCs (22), we have not evaluated the possible confounding of these signal transductions by the AT2 receptor in the present study. To support this notion, we previously demonstrated that AngII-induced ERK activation was almost completely inhibited by an AT1 receptor antagonist, whereas it was unaffected by an AT2 antagonist in VSMCs. We have further shown that both EGFR transactivation and MYPT phosphorylation are mediated exclusively through the AT1 receptor in VSMCs (7,17). However, increasing evidence suggest the counterregulatory functions of the AT2 receptors toward the AT1 receptor-dependent functions including vascular hypertrophy in vivo (46,47). Also, present findings are limited in cultured rat aortic VSMCs. Therefore, it is important to examine a possible signal cross talk at the level of G protein between these subtype receptors in vivo and whether we can generalize the present findings in distinct vascular cell types/tissues.
Although the role of Gq in mediating cardiac hypertrophy has been well established (32), it has not been shown in VSMC hypertrophy induced by AngII. It has been reported that vascular specific overexpression of GqI resulted in decreased vascular wall thickness. This was associated with decreased arterial ERK activation, blood pressure elevation, and cardiac hypertrophy by G protein-coupled receptor agonists (48). Vascular Gq/11 are also critical for the development of salt-sensitive hypertension (49). Taken together, vascular Gq seems to be a critical target of intervention against cardiovascular diseases associated with the enhanced renin-angiotensin system.
Acknowledgments
We thank Kyoko Hinoki for her excellent technical assistance.
Footnotes
This work was supported by National Institutes of Health Grants HL076770 (to S.E.) and HL076575 (to G.D.F.); American Heart Association Established Investigator Award 0740042N (to S.E.); and W. W. Smith Charitable Trust Grant H0605 (to S.E.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online March 20, 2008
Abbreviations: AngII, Angiotensin II; AT1, AngII type 1 receptor; EGFR, epidermal growth factor receptor; GqI, inhibitor of Gq encoding Gαq-(305–359); moi, multiplicity of infection; MYPT, myosin phosphatase target; PDGF, platelet-derived growth factor; Pyk2, proline-rich tyrosine kinase; RhoGEF, Rho family of guanine nucleotide exchange factor; VSMC, vascular smooth muscle cell.
References
- Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S 2007 Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 112:417–428 [DOI] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL 2000 Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev 52:639–672 [PubMed] [Google Scholar]
- Yin G, Yan C, Berk BC 2003 Angiotensin II signaling pathways mediated by tyrosine kinases. Int J Biochem Cell Biol 35:780–783 [DOI] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK 2007 Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292:C82–C97 [DOI] [PubMed] [Google Scholar]
- Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW 1997 Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 29:366–373 [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Suzuki H, Nakashima H, Dhobale S, Frank GD, Motley ED, Eguchi S 2006 Angiotensin II signal transduction through small GTP-binding proteins: mechanism and significance in vascular smooth muscle cells. Hypertension 48:534–540 [DOI] [PubMed] [Google Scholar]
- Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, Motley ED, Kawakatsu H, Owada KM, Hirata Y, Marumo F, Inagami T 1998 Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J Biol Chem 273:8890–8896 [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW 2001 Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 21:489–495 [DOI] [PubMed] [Google Scholar]
- Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK 2002 Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 91:406–413 [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Griendling KK, Akers M, Lyons PR, Alexander RW 1998 Temporal dispersion of activation of phospholipase C-β1 and -γ isoforms by angiotensin II in vascular smooth muscle cells. Role of αq/11, α12, and βγ G protein subunits. J Biol Chem 273:19772–19777 [DOI] [PubMed] [Google Scholar]
- Macrez-Lepretre N, Kalkbrenner F, Morel JL, Schultz G, Mironneau J 1997 G protein heterotrimer Gα13β1γ3 couples the angiotensin AT1A receptor to increases in cytoplasmic Ca2+ in rat portal vein myocytes. J Biol Chem 272:10095–10102 [DOI] [PubMed] [Google Scholar]
- Okuda M, Kawahara Y, Yokoyama M 1996 Angiotensin II type 1 receptor-mediated activation of Ras in cultured rat vascular smooth muscle cells. Am J Physiol 271:H595–H601 [DOI] [PubMed] [Google Scholar]
- Gohla A, Schultz G, Offermanns S 2000 Role for G(12)/G(13) in agonist-induced vascular smooth muscle cell contraction. Circ Res 87:221–227 [DOI] [PubMed] [Google Scholar]
- Yamakawa T, Tanaka S, Numaguchi K, Yamakawa Y, Motley ED, Ichihara S, Inagami T 2000 Involvement of Rho-kinase in angiotensin II-induced hypertrophy of rat vascular smooth muscle cells. Hypertension 35:313–318 [DOI] [PubMed] [Google Scholar]
- Riobo NA, Manning DR 2005 Receptors coupled to heterotrimeric G proteins of the G12 family. Trends Pharmacol Sci 26:146–154 [DOI] [PubMed] [Google Scholar]
- Lutz S, Freichel-Blomquist A, Yang Y, Rumenapp U, Jakobs KH, Schmidt M, Wieland T 2005 The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem 280:11134–11139 [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Mifune M, Frank GD, Saito S, Inagami T, Kim-Mitsuyama S, Takuwa Y, Sasaki T, Rothstein JD, Suzuki H, Nakashima H, Woolfolk EA, Motley ED, Eguchi S 2005 Signal-crosstalk between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration of vascular smooth muscle cells stimulated by angiotensin II. Arterioscler Thromb Vasc Biol 25:1831–1836 [DOI] [PubMed] [Google Scholar]
- Hunyady L, Catt KJ 2006 Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol 20:953–970 [DOI] [PubMed] [Google Scholar]
- Smith NJ, Luttrell LM 2006 Signal switching, crosstalk, and arrestin scaffolds: novel G protein-coupled receptor signaling in cardiovascular disease. Hypertension 48:173–179 [DOI] [PubMed] [Google Scholar]
- Seta K, Sadoshima J 2003 Phosphorylation of tyrosine 319 of the angiotensin II type 1 receptor mediates angiotensin II-induced trans-activation of the epidermal growth factor receptor. J Biol Chem 278:9019–9026 [DOI] [PubMed] [Google Scholar]
- Barnes WG, Reiter E, Violin JD, Ren XR, Milligan G, Lefkowitz RJ 2005 β-Arrestin 1 and Gαq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem 280:8041–8050 [DOI] [PubMed] [Google Scholar]
- Eguchi S, Matsumoto T, Motley ED, Utsunomiya H, Inagami T 1996 Identification of an essential signaling cascade for mitogen-activated protein kinase activation by angiotensin II in cultured rat vascular smooth muscle cells. Possible requirement of Gq-mediated p21ras activation coupled to a Ca2+/calmodulin-sensitive tyrosine kinase. J Biol Chem 271:14169–14175 [DOI] [PubMed] [Google Scholar]
- Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, Kobori M 2004 A novel Gαq/11-selective inhibitor. J Biol Chem 279:47438–47445 [DOI] [PubMed] [Google Scholar]
- Eguchi S, Hirata Y, Imai T, Kanno K, Marumo F 1994 Phenotypic change of endothelin receptor subtype in cultured rat vascular smooth muscle cells. Endocrinology 134:222–228 [DOI] [PubMed] [Google Scholar]
- Suzuki H, Eguchi K, Ohtsu H, Higuchi S, Dhobale S, Frank GD, Motley ED, Eguchi S 2006 Activation of endothelial nitric oxide synthase by the angiotensin II type 1 receptor. Endocrinology 147:5914–5920 [DOI] [PubMed] [Google Scholar]
- Akhter SA, Skaer CA, Kypson AP, McDonald PH, Peppel KC, Glower DD, Lefkowitz RJ, Koch WJ 1997 Restoration of β-adrenergic signaling in failing cardiac ventricular myocytes via adenoviral-mediated gene transfer. Proc Natl Acad Sci USA 94:12100–12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi S, Iwasaki H, Ueno H, Frank GD, Motley ED, Eguchi K, Marumo F, Hirata Y, Inagami T 1999 Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J Biol Chem 274:36843–36851 [DOI] [PubMed] [Google Scholar]
- Mifune M, Ohtsu H, Suzuki H, Nakashima H, Brailoiu E, Dun NJ, Frank GD, Inagami T, Higashiyama S, Thomas WG, Eckhart AD, Dempsey PJ, Eguchi S 2005 G protein coupling and second messenger generation are indispensable for metalloprotease-dependent, heparin-binding epidermal growth factor shedding through angiotensin II type-1 receptor. J Biol Chem 280:26592–26599 [DOI] [PubMed] [Google Scholar]
- Eguchi S, Hirata Y, Imai T, Marumo F 1993 Endothelin receptor subtypes are coupled to adenylate cyclase via different guanyl nucleotide-binding proteins in vasculature. Endocrinology 132:524–529 [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ 2003 Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100:10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh J, Chang CL, Bhalla A, Klein C, Hsu SY 2004 Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem 279:7264–7274 [DOI] [PubMed] [Google Scholar]
- Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ 1998 Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280:574–577 [DOI] [PubMed] [Google Scholar]
- Miura S, Zhang J, Matsuo Y, Saku K, Karnik SS 2004 Activation of extracellular signal-activated kinase by angiotensin II-induced Gq-independent epidermal growth factor receptor transactivation. Hypertens Res 27:765–770 [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, Nakashima H, Eguchi K, Eguchi S 2006 ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol 26:e133–e137 [DOI] [PubMed] [Google Scholar]
- Hines J, Fluharty SJ, Yee DK 2003 Structural determinants for the activation mechanism of the angiotensin II type 1 receptor differ for phosphoinositide hydrolysis and mitogen-activated protein kinase pathways. Biochem Pharmacol 66:251–262 [DOI] [PubMed] [Google Scholar]
- Shah BH, Yesilkaya A, Olivares-Reyes JA, Chen HD, Hunyady L, Catt KJ 2004 Differential pathways of angiotensin II-induced extracellularly regulated kinase 1/2 phosphorylation in specific cell types: role of heparin-binding epidermal growth factor. Mol Endocrinol 18:2035–2048 [DOI] [PubMed] [Google Scholar]
- Eguchi S, Iwasaki H, Inagami T, Numaguchi K, Yamakawa T, Motley ED, Owada KM, Marumo F, Hirata Y 1999 Involvement of PYK2 in angiotensin II signaling of vascular smooth muscle cells. Hypertension 33:201–206 [DOI] [PubMed] [Google Scholar]
- Frank GD, Saito S, Motley ED, Sasaki T, Ohba M, Kuroki T, Inagami T, Eguchi S 2002 Requirement of Ca(2+) and PKCΔ for Janus kinase 2 activation by angiotensin II: involvement of PYK2. Mol Endocrinol 16:367–377 [DOI] [PubMed] [Google Scholar]
- Shi CS, Sinnarajah S, Cho H, Kozasa T, Kehrl JH 2000 G13α-mediated PYK2 activation. PYK2 is a mediator of G13α-induced serum response element-dependent transcription. J Biol Chem 275:24470–24476 [DOI] [PubMed] [Google Scholar]
- Eguchi S, Iwasaki H, Hirata Y, Frank GD, Motley ED, Yamakawa T, Numaguchi K, Inagami T 1999 Epidermal growth factor receptor is indispensable for c-Fos expression and protein synthesis by angiotensin II. Eur J Pharmacol 376:203–206 [DOI] [PubMed] [Google Scholar]
- Voisin L, Foisy S, Giasson E, Lambert C, Moreau P, Meloche S 2002 EGF receptor transactivation is obligatory for protein synthesis stimulation by G protein-coupled receptors. Am J Physiol Cell Physiol 283:C446–C455 [DOI] [PubMed] [Google Scholar]
- Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, Ichiki T, Takahashi S, Takeshita A 2003 Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ Res 93:767–775 [DOI] [PubMed] [Google Scholar]
- Kanda T, Hayashi K, Wakino S, Homma K, Yoshioka K, Hasegawa K, Sugano N, Tatematsu S, Takamatsu I, Mitsuhashi T, Saruta T 2005 Role of Rho-kinase and p27 in angiotensin II-induced vascular injury. Hypertension 45:724–729 [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK 2005 Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1 ± haploinsufficient mice. Circulation 112:2959–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YM, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, Michael LH, Schneider MD, Entman ML, Schwartz RJ, Wei L 2006 Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J 20:916–925 [DOI] [PubMed] [Google Scholar]
- Carey RM 2005 Cardiovascular and renal regulation by the angiotensin type 2 receptor: the AT2 receptor comes of age. Hypertension 45:840–844 [DOI] [PubMed] [Google Scholar]
- Johren O, Dendorfer A, Dominiak P 2004 Cardiovascular and renal function of angiotensin II type-2 receptors. Cardiovasc Res 62:460–467 [DOI] [PubMed] [Google Scholar]
- Keys JR, Greene EA, Koch WJ, Eckhart AD 2002 Gq-coupled receptor agonists mediate cardiac hypertrophy via the vasculature. Hypertension 40:660–666 [DOI] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S 2008 G12–G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14:64–68 [DOI] [PubMed] [Google Scholar]