

Abstract
TSH-secreting pituitary tumors (TSHomas) are pituitary tumors that constitutively secrete TSH. Molecular mechanisms underlying this abnormality are largely undefined. We recently created a knock-in mutant mouse harboring a mutation (denoted as PV) in the thyroid hormone receptor-β gene (TRβPV/PV mouse). As these mice age, they spontaneously develop TSHomas. Using this mouse model, we investigated the role of the phosphatidylinositol 3-kinase (PI3K)-AKT signaling pathway in the pathogenesis of TSHomas. Concurrent with aberrant growth of pituitaries, AKT and its downstream effectors, mammalian target rapamycin and p70S6K, were activated to contribute to increased cell proliferation and pituitary growth. In addition, activation of AKT led to decreased apoptosis by inhibiting proapoptotic activity of Bcl-2-associated death promoter, further contributing to the aberrant cell proliferation. These results suggest an activated PI3K-AKT pathway could underscore tumorigenesis, raising the possibility that this pathway could be a potential therapeutic target in TSHomas. Indeed, TRβPV/PV mice treated with a PI3K-specific inhibitor, LY294002, showed a significant decrease in pituitary growth. The progrowth signaling via AKT-mammalian target rapamycin-p70S6K and cyclin D1/cyclin-dependent kinase were inhibited, and proapoptotic activity of Bcl-2-associated death promoter was increased by LY294002 treatment. Thus, activation of the PI3K-AKT pathway mediates, at least in part, the aberrant pituitary growth, and the intervention of this signaling pathway presents a novel therapeutic opportunity for TSHomas.
TSH-SECRETING PITUITARY adenomas (TSHomas) are tumors that constitutively secrete TSH. They represent about 2% of all pituitary tumors in humans (1). TSHomas are usually large at diagnosis with significant headaches, visual field disturbances, and central hyperthyroidism (2). Pituitary surgery and irradiation are effective in improving the symptoms in two-thirds of patients (3). However, due to its local invasiveness, one of three patients may lack surgical opportunity and requires chemotherapy to control the tumor size and hyperthyroidism. Treatment with somatostatin analogs, such as octreotide or lanreotide, leads to the restoration of the euthyroid state with or without pituitary tumor shrinkage (4,5).
The molecular genetics underlying the development of TSHomas is not well understood. TSHomas could conceivably arise from activation or gain-of-function genetic mutations in the stimulatory pathways or from changes in the inhibitory pathways due to repression or loss-of-function mutations. Efforts to identify potential tumor promoter and suppressor genes involved in the pathogenesis of TSHomas have been hampered by a limitation of available pituitary tumor samples. However, the creation of a knock-in mouse that spontaneously develops TSHomas (TRβPV/PV mouse) has presented an unusual opportunity to elucidate the molecular basis leading to the development and progression of TSHomas (6).
The TRβPV/PV mouse harbors a knock-in mutation (denoted PV) in the thyroid hormone receptor (TR)-β gene. The PV mutation was identified in a patient with thyroid hormone resistance. It has a C insertion in codon 448 that leads to a frame-shift mutation in the C-terminal 14 amino acids of TRβ (7). As a result of this mutation, PV has completely lost thyroid hormone (T3) binding and transcription activity (8). TRβPV/PV mice exhibit severe dysregulation of the hypothalamic-pituitary-thyroid axis with 9- to 15-fold increased thyroid hormone associated with 400- to 500-fold elevated circulating serum TSH levels (9). As TRβPV/PV mice age, they spontaneously develop TSHomas with highly vascular and enlarged pituitaries, containing characteristic TSH-stained large thyroprival cells and adenomas (6). Using this mouse model of TSHomas, we identified cyclin D1 as one of the oncogenes that mediate the tumorigenesis of TSHomas (6). However, because tumorigenesis frequently results from multigenic alterations, we sought to identify other altered signaling pathways that could also contribute to the pathogenesis of TSHomas.
The phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway regulates a vast array of fundamental cellular responses and plays a critical role in controlling the balance between cell survival and apoptosis (10,11). AKT is the primary mediator of PI3K-initiated signaling by phosphorylation of a number of downstream substrates, such as mammalian target of rapamycin (mTOR), ribosomal protein S6 kinase (S6K), the forkhead family of transcription factors (FKHR/FOX), glycogen synthase kinase (GSK)-3, and proapoptotic Bcl-2-associated death promoter (BAD) protein (12). That overactivation of the PI3K/AKT signaling pathway has been found in numerous tumors (12,13,14) prompted us to investigate whether this pathway could be activated during the development of TSHomas in TRβPV/PV mice. In the present study, we found that indeed the PI3K/AKT signaling pathway was activated via increased phosphorylation of its downstream effectors that promote cell growth and decrease cell survival. Importantly, we demonstrated that the activation of this pathway was attenuated by treating TRβPV/PV mice with LY294002 (LY), a potent inhibitor of PI3K (15), leading to significant reduction in pituitary growth. These in vivo findings suggested that the PI3K/AKT pathway represents novel potential therapeutic targets for TSHomas.
Materials and Methods
Experimental animals and treatment
TRβPV/PV mice were prepared and genotyped as described previously (16). All animal experiments were performed according to protocols approved by the National Cancer Institute Animal Care and Use Committee. The reagent LY, a specific PI3K inhibitor, was a gift from Dr. J. Starling (Eli Lilly and Co., Indianapolis, IN). Beginning at the age of 2 months, TRβPV/PV mice and their wild-type siblings were given either 25 mg/kg LY or vehicle [dimethylsulfoxide (DMSO)] ip twice weekly for more than 10 months. Pituitaries were collected from euthanized mice and kept frozen at −80 C until use.
Western blot analysis
For Western blot analysis, pituitaries were dissected and frozen immediately until ready to be analyzed. Pituitary extracts were prepared in a similar manner as previously described (6). In short, frozen pituitary was homogenized in cell lysis buffer containing 20 mm Tris-HCl (pH 7.5), 125 mm NaCl, 2 mm EDTA, 0.5% Triton X-100, 0.2 μm okadaic acid, 2 mm Na3VO4, 100 mm NaF, and protease inhibitors (Complete mini-EDTA-free; Roche Diagnostics, Basel, Switzerland). The protein concentration was determined by the method of Bradford (Pierce Chemical Co., Rockford, IL). Approximately 40–50 μg of extract proteins were separated by SDS-PAGE, and the Western blot analysis was carried out as described by Furumoto et al. (6). Two sets of representative results from three to six independent experiments are shown in each figure. Primary antibodies for phosphorylated S473 AKT (no. 9271, 1:500 dilution), total AKT (no. 9272, 1:1000), phosphorylated S2448 mTOR (no. 2971, 1:500), total mTOR (no. 2972, 1:1000), phosphorylated Thr421/Ser424 p70S6K (no. 9204, 1:1000), total p70S6K (no. 9202, 1:500), phosphorylated Ser9/21 GSK3β/α (no. 9331, 1:1000), total GSK-3β (no. 9315, 1:500), phosphorylated Ser253 forkhead box (Fox)-O3a (no. 9466, 1:500), total FoxO3a (no. 9467, 1:500), phosphorylated Ser136 BAD (no. 9295, 1:500), and total BAD (no. 9292, 1:500) were from Cell Signaling Technology (Danvers, MA). Cyclin D1 (sc-450) and CDK4 (sc-260) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and used at a 1:200 dilution concentration. The Bcl-2 interacting mediator of cell death (Bim) antibody (OPAI-01021) from Affinity BioReagents (Golden, CO) was used at a 1:500 dilution concentration. Anti-dynamitin (dynactin 2.p50) antibody (AB5869P) from Chemicon (Temecula, CA) was used at 1:1000 dilution concentration.
Histopathological analysis of TSHomas in LY-treated TRβPV/PV mice
The percentage of anterior pituitary mass occupied by neoplastic cells was quantified using morphometry as follows. Multiple-step sections were prepared to include the entire mouse pituitary in five equally spaced separate sections for each mouse. Two mice were PV/PV animals untreated, and two were LY treated. Digital images of hematoxylin and eosin (H&E)-stained sections were captured using a ×4 objective. Each field had a total area of 4.3 × 106 m2. Neoplastic cells were easily identified as cells having foci of homogenous morphology with enlarged nuclei and increased cytoplasm that was often eosinophilic or clear. These foci occurred either as large solitary masses or small multifocal lesions. Normal areas of anterior pituitary showed the expected glandular arrangements of cells with small nuclei and variable tinctorial properties. The captured digital images were manually subdivided and selected using an overlay color based on either normal or neoplastic morphology. Middle lobe and posterior lobe were not included in the analysis. Using Photoshop 7.0 (Adobe, San Jose, CA), the selected areas were quantified by pixel area using the Histogram function. The normal and neoplastic areas were recorded for each section of each mouse, and the percentage of anterior pituitary occupied by neoplasia was calculated using the formula: percent neoplasia = number of pixels neoplastic/number of pixels neoplastic + number of pixels normal. The mean for each mouse was compiled and a sd determined among the five sections for each mouse.
Statistical analysis
All data are expressed as means ± sd. In statistical analysis, the Student’s t test was performed using GraphPad Prism (version 4.00 for Mac; GraphPad Software, San Diego, CA), and P < 0.05 was considered significant.
Results
Activation of AKT-mTOR-p70S6K pathway during development of TSHomas in TRβPV/PV mice
We have previously shown that as TRβPV/PV mice age, they spontaneously develop TSHomas with enlarged pituitaries (6). Because activation of the AKT-mTOR-p70S6K pathway leads to increased organ growth, we ascertained whether this pathway is affected during pituitary tumorigenesis of TRβPV/PV mice. Western blot analysis indicated that, compared with wild-type siblings, AKT was activated by increased phosphorylation in the pituitaries of TRβPV/PV mice (compare lanes 3 and 4 with lanes 1 and 2, Fig. 1a) without apparent effects on the respective total AKT protein levels (Fig. 1b). The downstream effectors, mTOR and p70S6K, were also activated by increased phosphorylation (compare lanes 3 and 4 with lanes 1 and 2, Fig. 1c; compare lanes 3 and 4 with lanes 1 and 2, Fig. 1e, respectively). No significant alterations in the protein levels of total mTOR and p70S6K were observed (Fig. 1, d and f, respectively). These data indicate that activation of the AKT-mTOR-p70S6K pathway could contribute to aberrant growth of pituitaries in TRβPV/PV mice.
Figure 1.

Activation of the AKT-mTOR-p70S6K pathway in pituitaries of TRβPV/PV mice. Protein levels of phosphorylated AKT (p-AKT) (a), total AKT (b), phosphorylated mTOR (p-mTOR) (c), total mTOR (d), phosphorylated p70S6K (p-p70S6K) (e), and total p70S6K (f) were determined in 40 μg of pituitary extract by Western blot analysis. Representative results from two wild-type (WT) and two TRβPV/PV mice are shown. Dynactin 2 (g) was used as a loading control.
LY294002 decreases the pituitary tumor weight in TRβPV/PV mice
That the AKT-mTOR-p70S6K pathway was activated provided us with an opportunity to assess whether this pathway could be a potential therapeutic target. We therefore treated TRβPV/PV mice with LY, a specific inhibitor of its immediate upstream regulator, PI3K, and determined its effects on the spontaneous development of TSHomas. Figure 2 shows that treatment of wild-type mice with LY had no significant effect on the growth of pituitaries (Fig. 2; compare bar 2 with bar 1). Consistent with our previous observations (6), the pituitaries of untreated TRβPV/PV mice were enlarged about 2.6-fold as compared with untreated wild-type siblings (Fig. 2, bar 3 vs. bar 1). This abnormal increase in pituitary growth in TRβPV/PV mice was blocked by LY treatment (Fig. 2, bar 3 vs. bar 4). These results indicate that inhibition of PI3K activity resulted in the inhibition of aberrant growth of pituitaries in TRβPV/PV mice.
Figure 2.
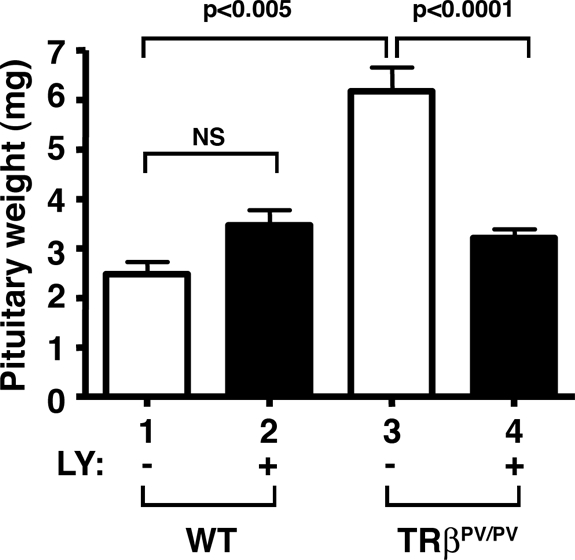
Reduced pituitary weight in TRβPV/PV mice treated with LY. Bars 1 and 2 represent pituitary weights of wild-type mice (WT) without or with LY treatment, respectively. Bars 3 and 4 represent the weight of the same tissue from TRβPV/PV mice without or with LY treatment, respectively. The data are expressed as means ± sd [n = 5 for DMSO treated wild-type mice (bar 1); n = 10 for LY treated wild-type mice (bar 2); n = 23 for DMSO treated TRβPV/PV mice (bar 3); and n = 24 for LY treated TRβPV/PV mice (bar 4)]. NS, Not significant.
To clearly demonstrate that the decreased pituitary tumor weight was due to reduced tumor growth by LY treatment, the area of pituitaries occupied by neoplastic cells was determined using morphometry analysis of five H&E-stained sequential step-sections to encompass each pituitary (see Materials and Methods). Figure 3A shows a representative H&E-stained micrograph of the untreated pituitary (Fig. 3A, a, c, and e) and treated pituitary (Fig. 3A, b, d, and f). On evaluation, the area of pituitaries occupied by neoplastic cells was marked in red and the area occupied by normal cells was marked in blue (Fig. 3A, c and d). The pathohistologic differences between neoplastic cells and normal cells are illustrated in the high magnification of the transition zone marked with as asterisk (Fig. 3A, e and f, for untreated and treated, respectively). The magnified area of the pituitary in the untreated animal shows homogeneous neoplastic cells that have enlarged eosinophilic or clear cytoplasm with a mild degree of nuclear enlargement and pleomorphism. Adjacent normal areas of anterior pituitary show cells in a glandular pattern with small nuclei and variable cytoplasmic tinctorial properties. In the treated animal, the areas of neoplastic morphology are small and heterogenous in distribution.
Figure 3.

LY treatment inhibited tumor cell growth (A). The area of pituitaries occupied by neoplastic cells was determined using morphometric analysis of five H&E-stained sequential step sections to encompass each pituitary as described in Materials and Methods. Representative examples from sections of untreated mice (panels a, c, and e) and LY-treated mice (panels b, d, and f) are shown. The neoplastic cells are marked in red (panels c and d) and normal cells are marked in blue. The abnormal and normal histologic characteristic details are shown in panels e and f. B, The areas occupied by the neoplastic cells and normal cells in the anterior lobe were quantified as described in Materials and Methods. The percentage of the pituitary mass occupied by neoplasia was dramatically reduced by LY treatment (n = 10).
The areas occupied by the neoplastic cells (e.g. marked in red, Fig. 3A, c and d) and the normal cells (marked in blue) in the anterior lobe were measured from H&E-stained sections, and the data are expressed as the percentage of anterior pituitary occupied by neoplasia. As shown in Fig. 3B, LY treatment led to a marked decrease in the area of the anterior lobe occupied by neoplastic cells. These data indicate that LY treatment effectively inhibited the tumor cell proliferation.
LY294002 inhibits the PI3K-AKT-mTOR-p70S6K signaling in the pituitary of TRβPV/PV mice
Activation of the PI3K downstream AKT-mTOR-p70S6K increases protein synthesis, cell growth, and proliferation, a process whereby cells accumulate mass and increase organ growth. The finding that LY treatment decreased pituitary growth of TRβPV/PV mice (Figs. 2 and 3) prompted us to examine whether this pathway was inhibited by LY treatment. Two representative sets of Western blot analyses from three to five independent experiments are shown in Fig. 4. Compared with untreated mice (Fig. 4a, lanes 1 and 3), TRβPV/PV mice treated with LY show decreased phosphorylated AKT (Fig. 4a, lanes 2 and 4). Compared with untreated mice (Fig. 4, c and e, lanes 1 and 3), concordant decreases in phosphorylated mTOR (Fig. 4c, lanes 2 and 4) and phosphorylated p70S6K (Fig. 4e, lanes 2 and 4) were detected in the treated mice. No significant changes in the total respective protein levels were observed in the pituitaries between treated and untreated mice (Fig. 4, b, d, and f). Dynactin-2, a subunit of the multiprotein complex dynactin, was used as a loading control in all Western blot analyses (Fig. 4g). These results indicate that LY treatment reduced the pituitary tumor growth of TRβPV/PV mice by attenuating AKT-mTOR-p70S6K signaling, mainly via phosphorylation cascades.
Figure 4.

LY attenuates activation of the AKT-mTOR-p70S6K pathway in pituitaries of TRβPV/PV mice. Pituitary extract (50 μg) from TRβPV/PV mice without or with LY treatment were used to determine phosphorylated and total protein levels of AKT (panels a and b), mTOR (panels c and d), and p70S6K (panels e and f) by Western blot analysis as described in Materials and Methods. Two sets (A and B) of representative results from three to six independent experiments are shown. Dynactin 2 (panel g) was used as a total protein loading control.
LY reduces cell proliferation by altering key cell cycle regulators in pituitaries of TRβPV/PV mice
To further understand molecular mechanisms by which LY treatment led to the reduction of pituitary tumors in TRβPV/PV mice, we evaluated several AKT-affected key cell cycle regulators by Western blot analysis (Fig. 5). GSK-3β is a downstream effector of the PI3K-AKT pathway (also see Fig. 7). Phosphorylation of GSK-3β by AKT inhibits its kinase activity to phosphorylate cyclin D1, thereby decreasing the proteasome degradation of cyclin D1 (17). Because cyclin D1 is a critical regulator of cell cycle (18), we evaluated the effect of LY treatment on the cellular abundance of phosphorylated GSK-3β and cyclin D1. As shown in Fig. 5 from two independent sets of experiments, LY treatment significantly reduced the phosphorylated GSK-3β (Fig. 5a, lane 2 vs. lane 1 and lane 4 vs. lane 3), and there was a concordant decrease in the cellular levels of cyclin D1 (Fig. 5c, lane 2 vs. lane 1 and lane 4 vs. lane 3). Furthermore, cellular levels of cyclin-dependent kinase 4 (CDK4) were also decreased (Fig. 5d, lane 2 vs. lane 1 and lane 4 vs. lane 3). The dynactin 2 loading controls are shown in Fig. 5e. These findings indicate that LY treatment reduced the cellular levels of cyclin D1 and CDK4 to decrease cell proliferation.
Figure 5.

LY reduces cell proliferation by altering key cell cycle regulators in TRβPV/PV mice. From 50 μg of pituitary extract, protein levels of phosphorylated GSK-3β (a), total GSK-3β (b), cyclin D1 (c), and CDK4 (d) were determined by Western blot analysis as described in Materials and Methods. Two sets (A and B) of representative results from three to six independent experiments are shown. Dynactin 2 (e) was used as a total protein loading control.
Figure 7.

Schematic representation of the activated PI3K-AKT pathway and its downstream effectors that contribute to the tumorigenesis of TSHomas in TRβPV/PV mice. Aberrant activation of the PI3K-AKT-mTOR-p70S6K pathway in pituitaries of TRβPV/PV mice increases protein synthesis and cell growth. AKT decreases cyclin D1 degradation by inhibiting GSK-3β activity through phosphorylation (17). AKT also inhibits cell apoptosis through phosphorylation of BAD and FoxO3a. Phosphorylated BAD is incapable of inhibiting the activity of antiapoptotic protein Bcl-2 (33,34). Similar to the phosphorylated BAD, phosphorylated FoxO3a is retained in cytoplasm and thus is incapable of activating the expression of the proapoptotic protein Bim (35). All these processes are reversed by LY294002 in vivo by deactivating the PI3K-AKT signaling pathway. By decreasing the subsequent phosphorylation events after the AKT activation, LY ultimately reduces tumor growth by inhibiting cell proliferation and promoting apoptosis in TSHomas of TRβPV/PV mice.
LY decreases cell survival by activating key proapoptotic regulators
Numerous studies indicate that activation of PI3K-AKT pathway promotes cell survival by directly phosphorylating key regulators of the apoptotic cascade. BAD, a member of the Bcl-2 family, is a direct phosphorylation target of AKT (see also Fig. 7). Its phosphorylation by AKT results in cytosolic accumulation, thereby preventing its interaction with and antagonizing the actions of prosurvival members of the family such as Bcl-2 at the mitochondrial membrane (19,20). We therefore examined the effect of LY on cellular protein levels of BAD and its phosphorylation status by Western blot analysis. Two representative sets of comparisons between LY-treated and untreated mice are shown in Fig. 6. Compared with pituitaries of untreated mice (Fig. 6a, lanes 1 and 3), pituitaries of treated mice show decreased phosphorylation of BAD (Fig. 6a, lanes 2 and 4) without apparent alterations of total cellular BAD protein levels in each set (Fig. 6b, lane 1 vs. lane 2 and lane 3 vs. lane 4). The reduction in the phosphorylation of BAD induced by LY treatment led to increased apoptosis (see Fig. 7).
Figure 6.

LY decreases key proapoptotic regulators in pituitaries of TRβPV/PV mice. Phosphorylated proapoptotic protein BAD (p-BAD; a), total BAD (b), p-Fox03a protein (c), total FoxO3a (d), and Bim (extra large isoform; e) protein levels were determined by Western blot analysis in 50 μg of pituitary extract as described in Materials and Methods. Two representative sets of results (A and B) are shown. Dynactin 2 (f) was used as the loading control.
Another means by which AKT promotes cell survival is by directly phosphorylating transcription factors that control the expression of pro- and antiapoptotic genes (14). The transcriptional factor FoxO family member such as FoxO3a is a direct phosphorylation target of AKT. When phosphorylated, it represses the expression of proapoptotic genes such as Bim (21). To understand whether LY treatment could lead to decreased cell survival via this mechanism, we also compared the phosphorylated Fox (p-Fox)-O3a in the pituitaries of untreated and treated TRβPV/PV mice (Fig. 6). Compared with pituitaries of untreated mice, pituitaries of TRβPV/PV mice treated with LY show reduced p-FoxO3a (Fig. 6c; compare lane 2 with lane 1 and lane 4 with lane 3). Total cellular FoxO3a protein levels were not significantly affected by LY treatment (Fig. 6d). The consequences of de-repression in the expression of its target genes mediated by the reduced p-FoxO3a were made evident by the increased levels of proapoptotic Bim in the pituitaries of treated mice (Fig. 6e; compare lane 2 with lane 1 and lane 4 with lane 3). Figure 6f shows the loading controls using dynactin 2. Taken together, these results indicate that LY treatment increased apoptosis by reduced phosphorylation of FoxO3a and increased proapoptotic regulator Bim (see Fig. 7).
Discussion
TSHomas are believed to derive from the monoclonal expansion of a single transformed thyrotrope that has lost growth regulation (21). The availability of a mouse model of TSHomas induced by radiothyroidectomy has helped understand the histopathological changes during the development of TSHomas (22). Studies of transplantable tumors have yielded valuable information of the secondary changes caused by the tumors and insights into effects of thyroid hormone in the development of tumors (23,24,25). However, studies to identify the gene alterations leading to TSHomas are limited. The discovery that the TRβPV/PV mouse spontaneously develops TSHomas has provided us with an opportunity to elucidate the molecular basis underlying the pathogenesis of TSHomas (6). Indeed, we found that overexpression of oncogenic cyclin D1 is one of the genetic changes that results in constitutive activation of the cyclin D1/cyclin-dependent kinase/retinoblastoma protein/E2F family of DNA-binding transcription factors (E2F) pathway to promote cell proliferation (6). The overexpression of cyclin D1 is mediated at the transcriptional level (6). In the present study, we found that AKT was activated and that phosphorylation of GSK-3β by AKT inhibited the kinase activity of GSK-3β to phosphorylate cyclin D1 (Fig. 7). Via the phosphorylation cascade, the degradation of cyclin D1 by the proteasome machinery is prevented (17). Thus, we identified an additional posttranslation mechanism by which cyclin D1 protein is stabilized to increase cell proliferation. It is important to point out that in addition to the activation of cyclin D1, the present study shows that activation of AKT-mTOR-p70S6K signaling also contributes to thyrotrope growth. Thus, the aberrant proliferation is mediated by at least two pathways: via cyclin D1 at cell cycle progression and via AKT-mTOR-p70S6K signaling to increase protein synthesis and cell growth and proliferation (see Fig. 7).
The molecular mechanisms by which PI3K-AKT is activated during the development of TSHomas are currently unknown. We have previously shown that in addition to TSHomas, TRβPV/PV mice also spontaneously develop follicular thyroid carcinoma similar to human thyroid cancer (26,27). One of the altered signaling pathways identified during thyroid carcinogenesis of TRβPV/PV mice is overactivation of the PI3K-AKT pathway, which was also reported in human thyroid cancer (28,29,30). The activation of PI3K is mediated via a novel nongenomic action by physical interaction of PV with the regulatory p85α subunit of PI3K (29). It is likely that PV could act in a similar fashion to activate PI3K-AKT in the pituitary. However, at present, the small size of the pituitary in TRβPV/PV mice has made extensive biochemical and molecular analysis very difficult to confirm this possibility. New methodologies are being developed to overcome this difficulty.
The currently available treatment options for TSHomas are surgery, radiotherapy, and somatostatin analog administration (2). Whereas each treatment or combinations of these treatments could be beneficial to patients, it would be valuable to explore other options that could improve treatment modalities. The finding that PI3K-AKT was activated in the pituitary of TRβPV/PV mice suggested that the PI3K-AKT pathway is a potential molecular target for TSHomas. Indeed, treatment of TRβPV/PV mice with a PI3K inhibitor, LY, led to a significant inhibition in the proliferation of neoplastic cell growth (Fig. 3), resulting in the reduction of pituitary size (Fig. 2). Biochemical analysis indicated that LY treatment attenuated PI3K-AKT-mTOR-p70S6K signaling (Fig. 4) and cyclin D1/CDK4 activity to reduce cell proliferation (Fig. 5). LY treatment also inhibited PI3K-AKT downstream signaling to decrease cell survival to further reduce cell proliferation (Fig. 6). The present finding that LY, a prototype of PI3K inhibitors, was effective in reducing pituitary tumor growth in vivo suggests that currently available second- or third-generation PI3K inhibitors such as ZSK474, IC187114, TG100–115, and others (31,32) could be tested as potential therapeutic approaches for TSHomas. The inhibitors that target specifically mTOR (e.g. CCI-779, RAD-001, and rapamycin) or AKT (e.g. perifosine and others) (32) could also be tested. The present study shows that the TRβPV/PV mouse is a preclinical mouse model of TSHomas. Therefore, the efficacy of these newer inhibitors of the PI3K-AKT-mTOR pathway could be tested in vivo using the TRβPV/PV mouse. These inhibitors hold promise as novel treatment modalities for TSHomas.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Disclosure Statement: The authors have nothing to disclose.
First Published Online March 20, 2008
Abbreviations: BAD, Bcl-2-associated death promoter; Bim, Bcl-2 interacting mediator of cell death; CDK4, cyclin-dependent kinase 4; DMSO, dimethylsulfoxide; Fox, forkhead box; GSK, glycogen synthase kinase; H&E, hematoxylin and eosin; LY, LY294002; mTOR, mammalian target of rapamycin; p-Fox, phosphorylated Fox; PI3K, phosphatidylinositol 3-kinase; PV, knock-in mutant mouse harboring a mutation; S6K, S6 kinase; TR, thyroid hormone receptor; TSHomas, TSH-secreting pituitary tumors.
References
- Mindermann T, Wilson CB 1993 Thyrotropin-producing pituitary adenomas. J Neurosurg 79:521–527 [DOI] [PubMed] [Google Scholar]
- McDermott MT, Ridgway EC 1998 Central hyperthyroidism. Endocrinol Metab Clin North Am 27:187–203 [DOI] [PubMed] [Google Scholar]
- Beck-Peccoz P, Brucker-Davis F, Persani L, Smallridge RC, Weintraub BD 1996 Thyrotropin-secreting pituitary tumors. Endocr Rev 17:610–638 [DOI] [PubMed] [Google Scholar]
- Comi RJ, Gesundheit N, Murray L, Gorden P, Weintraub BD 1987 Response of thyrotropin-secreting pituitary adenomas to a long-acting somatostatin analogue. N Engl J Med 317:12–17 [DOI] [PubMed] [Google Scholar]
- Chanson P, Weintraub BD, Harris AG 1993 Octreotide therapy for thyroid-stimulating hormone-secreting pituitary adenomas. A follow-up of 52 patients. Ann Intern Med 119:236–240 [DOI] [PubMed] [Google Scholar]
- Furumoto H, Ying H, Chandramouli GV, Zhao L, Walker RL, Meltzer PS, Willingham MC, Cheng SY 2005 An unliganded thyroid hormone β receptor activates the cyclin D1/cyclin-dependent kinase/retinoblastoma/E2F pathway and induces pituitary tumorigenesis. Mol Cell Biol 25:124–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrilla R, Mixson AJ, McPherson JA, McClaskey JH, Weintraub BD 1991 Characterization of seven novel mutations of the c-erbA β gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest 88:2123–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier CA, Parkison C, Chen A, Ashizawa K, Meier-Heusler SC, Muchmore P, Cheng SY, Weintraub BD 1993 Interaction of human β1 thyroid hormone receptor and its mutants with DNA and retinoid X receptor β. T3 response element-dependent dominant negative potency. J Clin Invest 92:1986–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S 2000 Mice with a targeted mutation in the thyroid hormone β receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA 97:13209–13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC 1997 PI3K: downstream AKTion blocks apoptosis. Cell 88:435–437 [DOI] [PubMed] [Google Scholar]
- Ullrich A, Schlessinger J 1990 Signal transduction by receptors with tyrosine kinase activity. Cell 61:203–212 [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC 2007 AKT/PKB signaling: navigating downstream. Cell 129:1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB 2005 Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 4:988–1004 [DOI] [PubMed] [Google Scholar]
- Nicholson KM, Anderson NG 2002 The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 14:381–395 [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF 1994 A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269:5241–5248 [PubMed] [Google Scholar]
- Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S 2000 Mice with a targeted mutation in the thyroid hormone β receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA 97:13209–13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ 1998 Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12:3499–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alao JP 2007 The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 6:24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G 1997 Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278:687–689 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME 1997 Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241 [DOI] [PubMed] [Google Scholar]
- Beck-Peccoz P, Persani L, Mantovani S, Cortelazzi D, Asteria C 1996 Thyrotropin-secreting pituitary adenomas. Metabolism 45:75–79 [DOI] [PubMed] [Google Scholar]
- Furth J 1954 Morphologic changes associated with thyrotrophin-secreting pituitary tumors. Am J Pathol 30:421–463 [PMC free article] [PubMed] [Google Scholar]
- Woodmansee WW, Kerr JM, Tucker EA, Mitchell JR, Haakinson DJ, Gordon DF, Ridgway EC, Wood WM 2006 The proliferative status of thyrotropes is dependent on modulation of specific cell cycle regulators by thyroid hormone. Endocrinology 147:272–282 [DOI] [PubMed] [Google Scholar]
- Kerr JM, Gordon DF, Woodmansee WW, Sarapura VD, Ridgway EC, Wood WM 2005 Growth arrest of thyrotropic tumors by thyroid hormone is correlated with novel changes in Wnt-10A. Mol Cell Endocrinol 238:57–67 [DOI] [PubMed] [Google Scholar]
- Wood WM, Sarapura VD, Dowding JM, Woodmansee WW, Haakinson DJ, Gordon DF, Ridgway EC 2002 Early gene expression changes preceding thyroid hormone-induced involution of a thyrotrope tumor. Endocrinology 143:347–359 [DOI] [PubMed] [Google Scholar]
- Suzuki H, Willingham MC, Cheng SY 2002 Mice with a mutation in the thyroid hormone receptor β gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid 12:963–969 [DOI] [PubMed] [Google Scholar]
- Ying H, Suzuki H, Furumoto H, Walker R, Meltzer P, Willingham MC, Cheng SY 2003 Alterations in genomic profiles during tumor progression in a mouse model of follicular thyroid carcinoma. Carcinogenesis 24:1467–1479 [DOI] [PubMed] [Google Scholar]
- Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, Ringel MD 2005 AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology 146:4456–4463 [DOI] [PubMed] [Google Scholar]
- Furuya F, Hanover JA, Cheng SY 2006 Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone β receptor. Proc Natl Acad Sci USA 103:1780–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko V, Saji M, Hardy E, Kruhlak M, Larin A, Savchenko V, Miyakawa M, Isozaki O, Murakami H, Tsushima T, Burman KD, De Micco C, Ringel MD 2004 Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. J Med Genet 41:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe T, Welham MJ, Ward SG 2007 The PI3K inhibitor arsenal: choose your weapon! Trends Biochem Sci 32:450–456 [DOI] [PubMed] [Google Scholar]
- Granville CA, Memmott RM, Gills JJ, Dennis PA 2006 Handicapping the race to develop inhibitors of the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin pathway. Clin Cancer Res 12:679–689 [DOI] [PubMed] [Google Scholar]
- Hsu SY, Kaipia A, Zhu L, Hsueh AJW 1997 Interference of BAD (Bcl-xL/Bcl-2-associated death promoter)-induced apoptosis in mammalian cells by 14–3-3 isoforms and P11. Mol Endocrinol 11:1858–1867 [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ 1995 Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80:285–291 [DOI] [PubMed] [Google Scholar]
- Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH 2002 The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol 168:5024–5031 [DOI] [PubMed] [Google Scholar]