

Abstract
Cell migration is a fundamental process occurring during embryonic development and tissue morphogenesis. In the nematode Caenorhabditis elegans, morphogenesis of the body-wall musculature involves short-range migrations of 81 embryonic muscle cells from the lateral surface of the embryo towards the dorsal and ventral midlines. This study shows that mutations in ina-1 (α-integrin), as well as vab-1 (Eph receptor), and vab-2 (ephrin), display defects in embryonic muscle cell migration. Furthermore, an RNAi-based enhancer screen in an ina-1 weak loss-of-function background identified mnp-1 (matrix non-peptidase homologue-1) as a previously uncharacterized gene required for promoting proper migration of the embryonic muscle cells. mnp-1 encodes a membrane associated metalloproteinase homologue that is predicted to be catalytically inactive. Our data suggest that MNP-1 is expressed in migrating muscle cells and localizes to the plasma membrane with the non-peptidase domain exposed to the extra-cellular environment. Double-mutant analysis between mnp-1(RNAi), ina-1, and vab-1 mutations; as well as tissue specific rescue experiments; indicated that each of these gene products function predominantly independent of each other and from different cell types to affect muscle cell migration. Together these results suggest complex interactions between the adjacent epidermal, neuronal, and muscle cells are required to promote proper muscle cell migration during embryogenesis.
Introduction
Cell migrations are essential for many biological processes, including embryonic development, immune response, wound healing, and cell metastasis. For cells to properly navigate through complex environments, such as those encountered during these processes, complicated interactions between the migrating cell, the extra-cellular matrix (ECM), and the surrounding tissues are essential. Not only does the migrating cell need to adhere and move across the surrounding ECM, it also must identify and respond to a variety of guidance cues, actively degrade confining components of its pericellular environment, and navigate an intricate three-dimensional landscape. We are only just beginning to understand how cells find their way through these complex surroundings and the influence adjacent tissues may exert during cell migrations in vivo.
In an effort to further our understanding of how cells migrate during development, we turned our attention to the movements of the 81 embryonic body-wall muscle cells of C. elegans. The muscle cells are born on the lateral sides of the embryo and subsequently migrate to take up positions flanking the dorsal and ventral midlines (fig. 1) (Sulston et al., 1983; Hresko et al., 1994; Moerman et al., 1996). Migration of these cells occurs during embryogenesis, between roughly 290 and 350 minutes after the first cell division, concurrent with the onset of embryonic elongation (Hresko et al., 1994). The net result of these migration events is the formation of four distinct muscle quadrants running the entire length of the worm that make possible the locomotion of the organism. Our investigation identified four genes required for proper embryonic muscle cell migration, including an α-integrin subunit (ina-1) (Baum and Garriga, 1997), an Eph receptor (vab-1) (George et al., 1998), an ephrin ligand (vab-2) (Chin-Sang et al., 1999), and a previously uncharacterized, inactive extracellular peptidase (mnp-1).
Figure 1. Migration of the embryonic body-wall muscle cells in C. elegans.
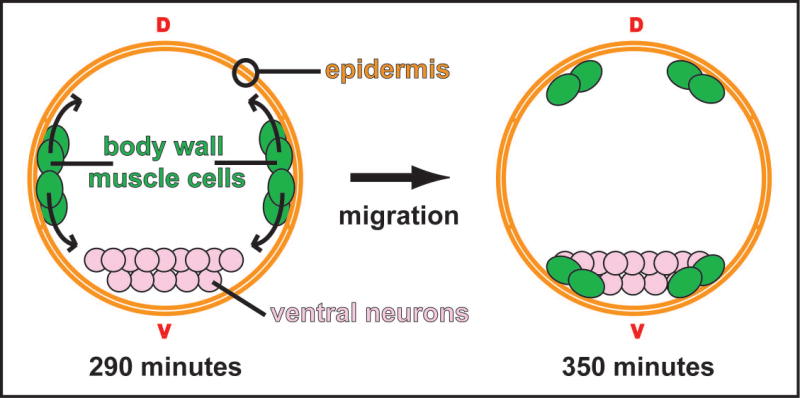
A cross-sectional schematic depicting the migrations of the body-wall muscle cells during embryogenesis. The muscle cells are born as two clusters on either side of the embryo. At 290 minutes (25°C; approximate bean stage) the cell clusters separate at the lateral axis and migrate towards both the dorsal (d) and ventral (v) midlines. The migrating cells pass between the overlying epidermal sheet and the underling cells, which include neuronal precursors. By 350 minutes (25°C; approximate late comma stage) the cells have taken up positions flanking what will become the dorsal and ventral nerve cords (Hresko et al., 1994).
Integrins are heterodimeric cell surface proteins comprised of α- and β-subunits that can recognize both soluble and surface-bound ligands at the cell surface. The primary role of integrins is to provide a direct physical connection between the cytoskeleton and the ECM. In this capacity integrins are able to perform a multitude of functions at the cell surface (Bökel et al., 2002; Arnaout et al., 2005). First, integrins are able to promote tissue integrity by providing a strong mechanical link to the ECM. Second, integrins function as signaling receptors, relaying information across the cell membrane in either direction. Third, integrins directly affect cell migration and cell shape by their ability to influence both the contractile apparatus within the cell and the binding of the cell to rigid extracellular surfaces. Together, these influences can result in the generation of force and subsequent cell motility. Finally, integrins are also able to affect the assembly and remodeling of the ECM by binding soluble secreted matrix proteins and subsequently affecting the assembly of fiber networks. Cell culture experiments have provided considerable insight into the direct role integrins play during cell migration. However, analysis of integrin function from genetic models, including C. elegans, has also highlighted the complexity of roles these receptors play during cell movements within the context of a developing organism (Bökel et al., 2002; Meighan and Schwarzbauer, 2007).
The Eph receptors are transmembrane receptor tyrosine kinases that play a leading role in controlling both developmental cell movements and stabilizing the patterns of cell organization (Poliakov et al., 2004; Sela-Donenfeld and Wilkinson, 2005). Activity of these receptors is modulated in part via the interaction with the ephrin family of ligands. When present on opposing cell surfaces, the Eph receptor/ ephrin ligand complex can mediate cell contact-dependent signaling events. It is through their ability to communicate cell contact-dependent information that the Eph receptors/ ephrin ligands are able to modulate both repulsive and attractive responses between adjacent cells. These responses are achieved via the combined ability of the Eph pathway to influence actin cytoskeletal dynamics and modify the adhesive properties of cells. In this way the Eph receptors and ephrin ligands are thought to influence cell migrations during development (Poliakov et al., 2004; Sela-Donenfeld and Wilkinson, 2005).
Extracellular peptidases have also been shown to play a critical role in promoting cell motility and invasion during development and cancer metastasis (Mott and Werb, 2004). This task is accomplished via their ability to directly modify the extra-cellular environment. The extracellular milieu is a complex mixture of both soluble molecules and the insoluble ECM. The ECM influences cellular behavior by not only providing attachment sites and physical support for cells, but also harboring embedded information in the form of bioactive molecules, cytokines, and growth factors. Extracellular peptidases can cleave components of the ECM as well as non-ECM molecules, including cell surface molecules and other non-matrix proteins. In this capacity peptidases are able to directly modulate cell function in a number of ways. First, peptidases are required to maintain and/or promote changes in the ECM architecture. Second, peptidases are also able to modulate cellular adhesion and influence the linkage between the cell surface and the ECM. Finally, peptidases can also release sequestered active peptides, cytokines and growth factors from ECM macromolecules and subsequently elicit a host of cellular responses. Through their ability to regulate the extracellular environment, peptidase are intimately involved in the regulation of cell shape, cell migration, tissue morphogenesis, and tumor progression.
Materials and Methods
Strains
All strains were maintained according to standard protocol (Brenner, 1974). Strains used: rrf-3(ok629)II, vab-1(dx31)II, mnp-1(ok2434)III, ina-1(gm86)III, ina-1(gm144)III, vab-2/efn-1(e96)IV, and sid-1(qt2)V. Integrated arrays: hlh-1::GFP (strain PD7962, a gift from A. Fire) and myo-3::GFP (ccIs4251) (Fire et al., 1998).
Expression constructs
The mnp-1p::GFP reporter construct was generated by inserting 3kb of upstream regulatory sequence into the pPD122.56 vector (a gift from A. Fire), which contains four copies of a nuclear localization signal (NLS), the GFP coding sequence (Chalfie et al., 1994) and the 3′ untranslated region of unc-54 (Fire et al., 1990) at the 3′ end. This construct was injected into N2 worms at 10ng/ml along with a dominant Roller marker, pRF4 containing rol-6 (su1006), at 100ng/ml to generate kuEx166. The mnp-1p::mnp-1tm::GFP reporter construct was generated by inserting 123bp of the mnp-1 coding sequence into the kuEx166 vector, replacing the NLS sequences. This construct was injected into N2 worms at 10ng/ml along with a dominant Roller marker, pRF4 containing rol-6 (su1006), at 100ng/ml to generate kuEx167. The mnp-1p::mnp-1tm::β-gal reporter construct was generated by replacing the NLS-GFP sequence of the kuEx167 vector with the β-gal coding sequence from the vector pPD21.28 (a gift form A. Fire). This construct was injected into N2 worms at 10ng/ml along with a dominant Roller marker, pRF4 containing rol-6 (su1006), at 100ng/ml to generate kuEx168. The mnp-1p::mnp-1tm(2x)::β-gal reporter construct was generated by replacing the GFP sequence of the kuEx168 vector with the β-gal and synthetic trans-membranes coding sequences from the vector pPD34.110 (a gift form A. Fire). This construct was injected into N2 worms at 10ng/ml along with a dominant Roller marker, pRF4 containing rol-6 (su1006), at 100ng/ml to generate kuEx169. The ajm-1p::ina-1, F25B3.3p::ina-1, and hlh-1p::ina-1 rescuing constructs were generated by inserting 1690bp of the ajm-1 promoter sequence, 2582bp of the F25B3.3-1 promoter sequence, or 3071bp of the hlh-1 promoter sequence upstream of the full-length ina-1 open reading frame in the pBS SK(-) vector. This construct was injected into N2 worms at 1ng/ml along with a sur-5::GFP marker (pTG96.2) (Yochem et al., 1998) at 50ng/ml to generate kuEx170, kuEx171, and kuEx172 respectively. The hlh-1p::mnp-1hp RNAi consruct was generated by replacing the ina-1 sequence of the kuEx172 vector with an mnp-1 hairpin loop consisting of 833bp of the mnp-1 open reading frame (ctggagtcatcacattcctga to ctgctcgacacgtttttcca) joined with 630bp of the mnp-1 open reading frame (ctggagtcatcacattcctga to cgaaaccatctgcgtgtccc) in reverse orientation, forming a 630bp stem separated by a 203bp loop sequence. This construct was injected into N2 worms at 1ng/ml along with a dominant Roller marker, pRF4 containing rol-6 (su1006), at 100ng/ml to generate kuEx174.
RNAi Analysis
All RNAi experiments were done in strains containing the rrf-3(ok629) allele (Simmer et al., 2002). Double-stranded (ds)RNA was applied to worms by feeding (Kamath et al., 2001; Timmons et al., 2001). Briefly, feeding RNAi bacterial strains were seeded on NGM agar plates containing IPTG and ampicillin. Adult worms were added to the plates on the following day and cultured at 20°C. Control animals were fed with bacteria carrying an empty vector. Phenotypes were observed in the F3 progeny of worms that were fed with the respective dsRNA.
β-gal assays and Immunofluorescence
Embryos for β-gal assays were fixed using the “freeze-crack” method and assayed for β-gal activity as described previously (Epstein and Shakes, 1995). All images were collected using an Axioplan2 microscope (Carl Zeiss, Thorton, NY) and a Hamamatsu C4742-95 CCD camera (Hamamatsu Photonics KK, Bridgewater, NJ). Images were analyzed using Openlab 3.1.7 (Improvision, Lexington, MA) software, and figures were compiled using Photoshop 8.0 and Illustrator 11.0 (Adobe, San Jose, CA).
Results
Mutations in ina-1, vab-1 and vab-2 have defects in muscle cell migration
In an effort to identify genes involved in the embryonic muscle cell migrations we first implemented a candidate approach. Muscle cell position was assayed in mutant strains with known functions in cell migration that also displayed body morphology defects that might indicate deformations of the body-wall muscle cell quadrants. Mutations in ina-1, vab-1, or vab-2 each result in a distinct anterior morphological phenotype called a ‘notched head’ (fig. 2) (Baum and Garriga, 1997; George et al., 1998; Chin-Sang et al., 1999). We determined if muscle cell nuclei were misplaced in newly hatched L1 larva displaying a notched head phenotype. In order to visualize muscle cell positions, these strains carried a transgene that expressed GFP from the myo-3 promoter (Fire et al., 1998).
Figure 2. Muscle cells are mispositioned in notched head worms.
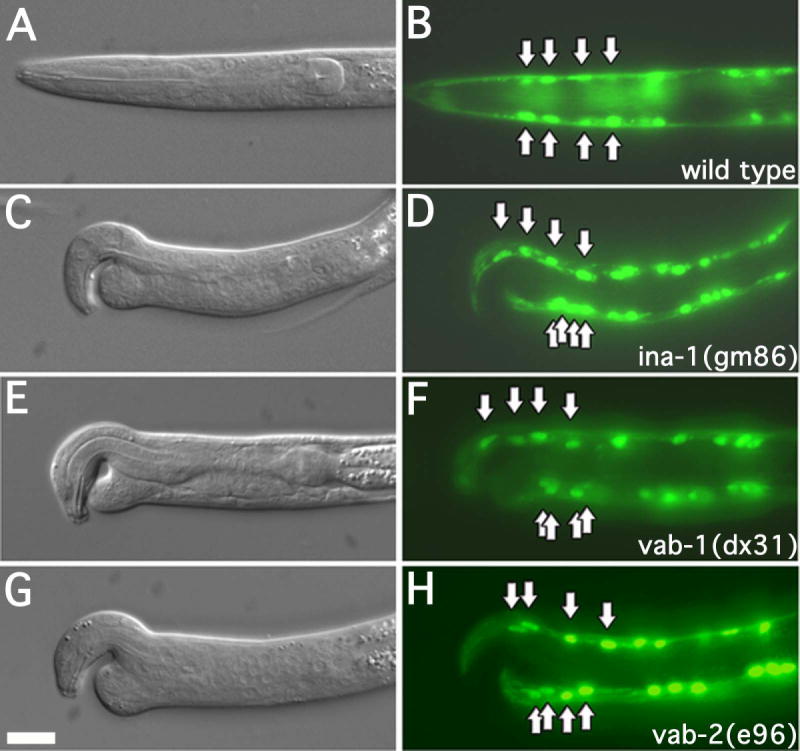
DIC and fluorescence images of L1 larva expressing myo-3::GFP. Arrows indicate position of the anterior most muscle cell nuclei. The genotypes of the strains are as indicated. Every case in which larva displayed an apparent notched head phenotype, the anterior most ventral muscle cell nuclei were mispositioned. More than 50 L1 larva were examined for each strain. In this and all subsequent figures dorsal is to the top, anterior is to the left. Scale bar represent 10μm.
We observed that ina-1(gm86), vab-1(dx31), or vab-2(e96) mutant L1 larva displaying a notched head phenotype exhibit misplaced muscle cell nuclei (n >50 for each strain). Specifically the dorsal muscle cells appeared properly positioned, with a similar spacing between nuclei identical to that of wild type (fig. 2). However, the anterior-most ventral muscle cells inevitably clustered just posterior of the notched head defect (fig. 2).
To determine if the misplaced muscle cells were a result of failed or improper migration of the embryonic body-wall muscle cells, we visualized these cells prior to and after their migrations. We examined embryos from ina-1(gm86), vab-1(dx31), or vab-2(e96) that contained a transgene expressing hlh-1::GFP (Chen et al., 1994).
In all embryos (n >30 for each strain), the embryonic muscle cells were born at the proper time and properly positioned (not shown). By the stage at which most of the embryonic muscle cells had finished their migrations in wild type (2 of 30, 7% embryos with misplaced muscle cells); the ina-1(gm86), vab-1(dx31), and vab-2(e96) mutants displayed a greater than eight-fold increase in misplaced muscle cells (21 of 30, 70%; 23 of 30, 77%; and 18 of 30, 60% respectively) (fig. 3). Specifically, the anterior dorsal quadrant was typically devoid of GFP expression (brackets, fig. 3) and the corresponding cells were inappropriately positioned to the lateral sides or the posterior (arrows, fig. 3). The proper expression of GFP from the muscle specific promoters, myo-3 and hlh-1 (fig. 2 and 3), suggested the muscle cell fate was being properly executed in these strains. These observations are consistent with a defect in muscle cell migration. It is worth noting that the embryonic region that lacked muscle cells in these mutants is consequently where the notched head defect appears in L1 larva.
Figure 3. Muscle cell migration defects in ina-1, vab-1 and vab-2 mutant worms.
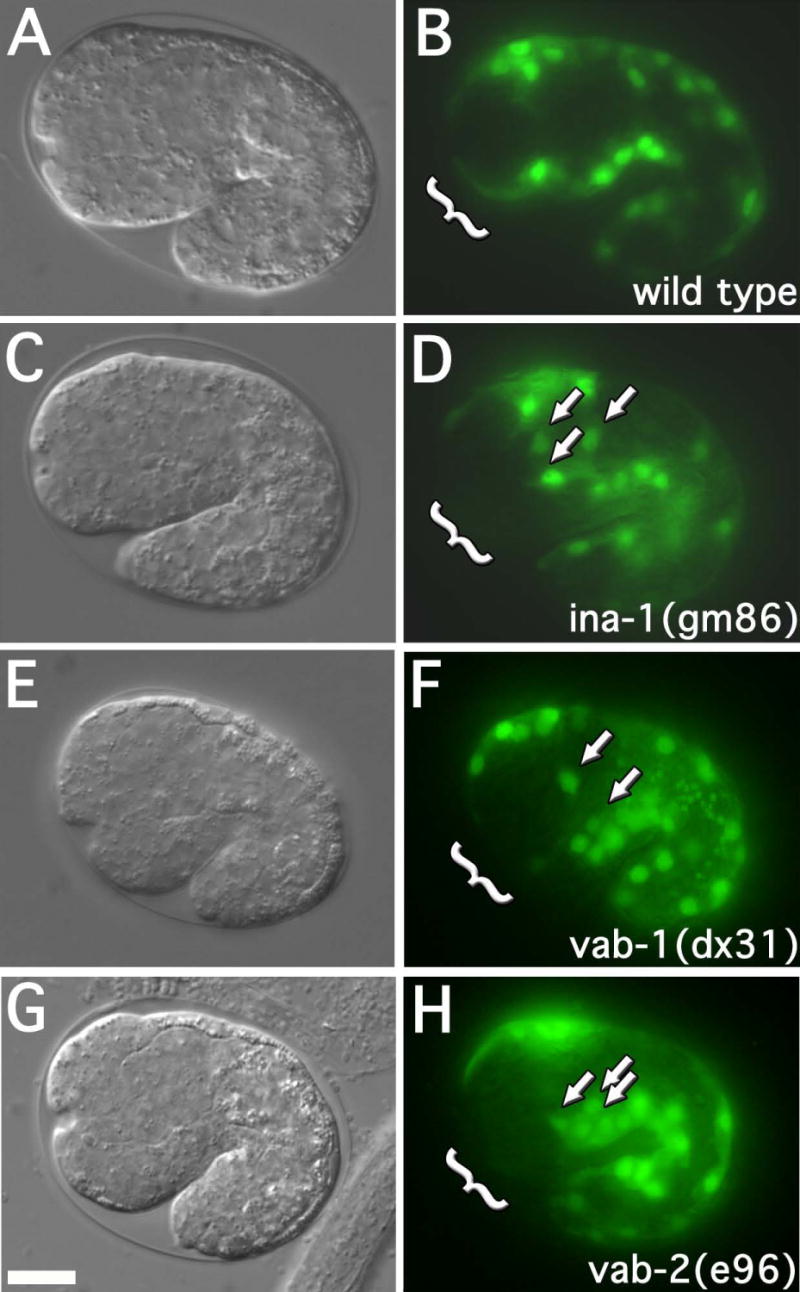
DIC and fluorescence images of approximate 1½ fold embryos (25°C; approximately 380 minutes) expressing hlh-1::GFP. The genotypes of strains are as indicated. Arrows indicate mispositioned muscle cell nuclei. Brackets indicate the anterior-ventral quadrant. Embryos at this stage were assayed for anterior muscle cell nuclei misplaced towards the posterior or lateral surface. 30 embryos were examined for each strain. The frequency of misplaced muscle cell nuclei were as follows: wild type (2 of 30, 7%), ina-1(gm86) (21 of 30, 70%, p<0.001), vab-1(dx31) (23 of 30, 77%, p<0.001), and vab-2/efn-1(e96) (18 of 30, 60%, p<0.001). p-values represent comparison to wild type. Scale bar represent 10μm.
MNP-1 is required for muscle cell migration
To identify new genes involved in muscle cell migration we took advantage of the close association between the notched head phenotype and defects in embryonic muscle cell migration. We found that ina-1(gm144);rrf-3(ok629), a weak loss of function mutant that does not display a notched head phenotype (0 of 200, 0% notched head), when treated with vab-1(RNAi) or vab-2(RNAi) showed a greater than 3.5 fold increase in the penetrance of the notched head phenotype (78 of 200, 39% and 66 of 200, 33%, respectively) when compared to a rrf-3(ok629) strain treated with the same RNAi (22 of 200, 11% and 8 of 200, 4%, respectively) (fig. 4a). We hypothesized that the ina-1(gm144) allele would function as a sensitive background in which to detect the role of other genes influencing the notched head phenotype. We performed an RNAi based screen, encompassing a majority of the genes found on chromosome III, using an ina-1(gm144);rrf-3(ok629) strain background and identified B0285.7 (fig. 4a) as a notched head enhancer (60 of 200, 30%). Also, RNAi treatment of B0285.7 in an rrf-3(ok629) strain alone, resulted in a low penetrance (10 of 200, 5%) notched head phenotype (fig. 4a-c). We named the gene corresponding to B0285.7, mnp-1 (matrix non-peptidase homologue 1). A recently obtained genomic deletion (ok2434), encompassing a portion of the mnp-1 open reading frame, also displays a notched head phenotype (not shown). This observation supports our assertion that mnp-1(RNAi) indeed results in a knockdown of B0285.7 function.
Figure 4. Notched head and muscle cell migration defects in mnp-1(RNAi) worms.
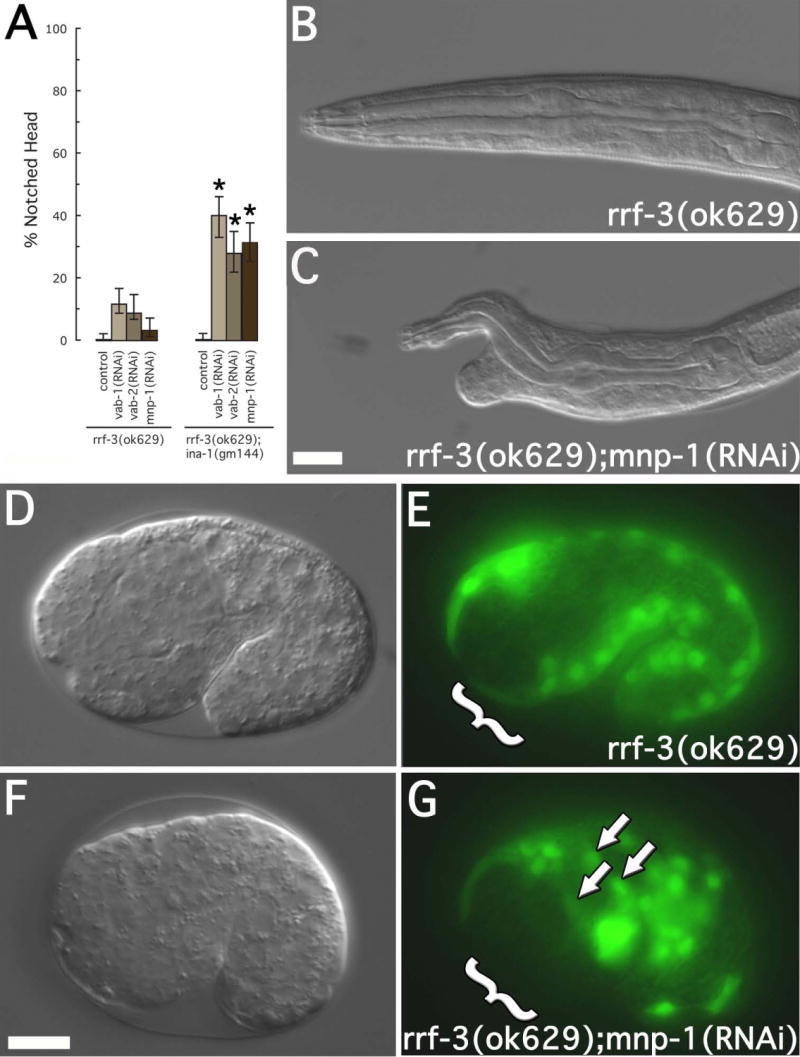
(A) Notched head RNAi phenotype. Strains include rrf-3(ok629) and ina-1(gm144);rrf-3(ok629). RNAi treatment included vab-1(RNAi), vab-2(RNAi), and mnp-1(RNAi). For each treatment, 200 L1 larval were examined. Asterisks indicate a significant difference (p<0.0002) between rrf-3(ok629) and ina-1(gm144);rrf-3(ok629) for a given RNAi treatment. Error bars represent 95% confidence intervals. (B-C) DIC images of L1 larvae showing the mnp-1(RNAi) notched head phenotype. Genotypes and RNAi treatment are as indicated. Scale bar represent 10μm. (D-G) DIC and fluorescence images of approximate 1½ fold embryos expressing hlh-1::GFP. Arrows indicate mispositioned muscle cell nuclei. Brackets indicate the anterior-ventral quadrant. Embryos at this stage were assayed for anterior muscle cell nuclei misplaced towards the posterior or lateral surface. The frequency of misplaced muscle cell nuclei were as follows: rrf-3(ok629) (7 of 129, 5.4%) and rrf-3(ok629)::mnp-1(RNAi) (39 of 339, 11.5%, p<0.05). Scale bars represent 10μm.
In an effort to assay the role of MNP-1 in the process of embryonic muscle cell migration, we monitored muscle-cell placement after treatment of rrf-3(ok629) with mnp-1(RNAi). We observed that in all embryos (n >30), the muscle cells were born at the proper time and at the proper position (not shown). However, at the stage during which all muscle cells have finished migrations in rrf-3(ok629) treated with control(RNAi) (7 of 129, 5.4% embryos with misplaced muscle cells), the mnp-1(RNAi);rrf-3(ok629) strain displayed misplaced muscle cells at a low but significant frequency (39 of 339, 11.5%; p<0.05). More specifically, the anterior dorsal quadrant was typically devoid of muscle cells (brackets, fig. 4d-g), in those embryos displaying defects, and these were inappropriately positioned to the lateral sides or to the posterior (arrows, fig. 4d-g). The proper expression of GFP from the muscle specific promoters, myo-3 and hlh-1 (fig. 6e and 4g), suggested the muscle cell fate was being properly executed in the mnp-1(RNAi);rrf-3(ok629) strains.
Figure 6. Double mutant analysis and tissue specificity of notched head genes.
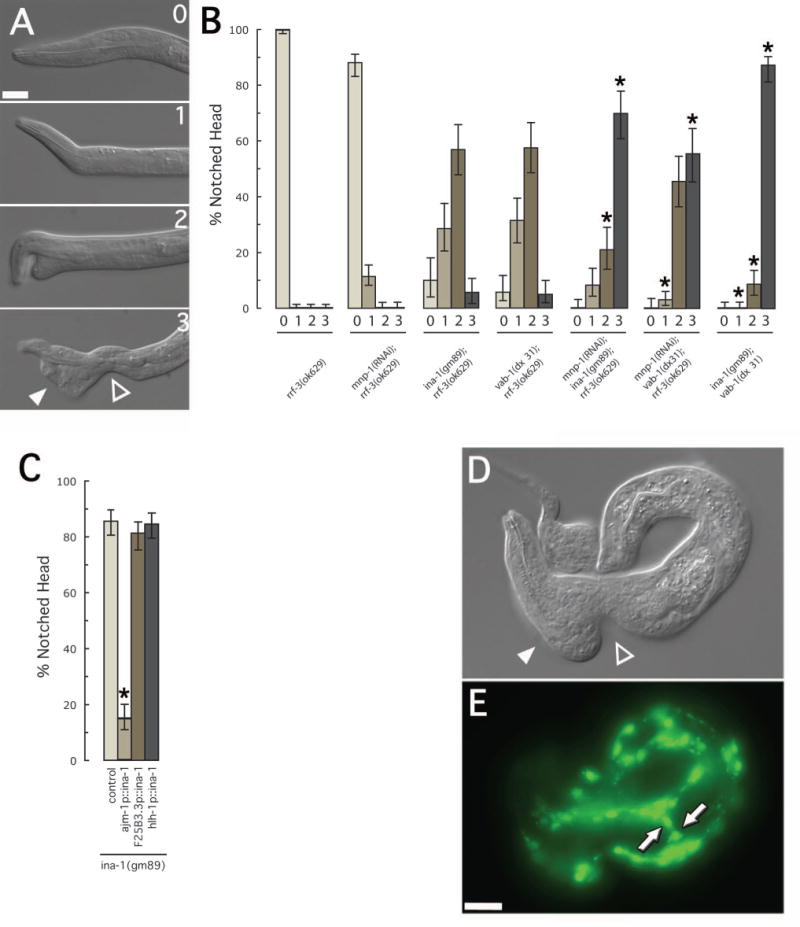
(A) DIC images of L1 mutant larva showing the range of defects associated with the notched head phenotype. The most severe phenotype (3) is characterized by a thinning of tissue relative to the isthmus of the pharynx (open triangle) and an accumulation of cells towards the anterior (closed triangle). (B) Severity of notched head phenotype in single and double mutant combinations. The genotypes of strains assayed are as indicated. For each strain, more than 100 L1 larval were examined. Asterisks indicate a significant change (p<0.0002) from that of either of the individual alleles alone. Error bars represent 95% confidence intervals. (C) Tissue specific rescue of the notched head RNAi phenotype in ina-1(gm89). The ajm-1 promoter drives expression in epidermal tissue (Koppen et al., 2001)), the F25B3.5 promoter drives expression in neuronal tissue (Altun-Gultekin et al., 2001), and the hlh-1 promoter drives expression in embryonic muscle cells (Chen et al., 1994)). For each strain, more than 200 L1 larval were examined. Asterisks indicate a significant difference (p<0.0002) from that of the control strain alone. Error bars represent 95% confidence intervals. (D) DIC and (E) fluorescence images of an rrf-3(ok629) L1 larva, containing the hlh-1p::mnp-1hp, expressing myo-3::GFP. The body morphology seen in these larva were often characterized by a thinning of tissue relative to the isthmus of the pharynx (open triangle) and an accumulation of cells towards the anterior (closed triangle). This is similar to the most severe phenotype associated with notched head mutations. Compare to figure 5a.3 above. Muscle cells were often seen located to the lateral surfaces of the larva (arrows). Scale bars represent 10μm.
MNP-1 encodes a membrane associated, M1 family non-peptidase homologue
The mnp-1 gene encodes a 781 amino acid protein with a putative transmembrane domain proximal to the NH2-terminus. A BLAST search indicated the remaining bulk of MNP-1 had a high similarity to the M1 family of peptidases that by definition contain the HENNH+E sequence motif required for coordination of the essential zinc ion within the catalytic pocket (Nigel and Hooper, 1994)). The canonical member of the M1 family of peptidases is aminopeptidase A of Homo sapiens. ClustalW alignment of the M1 peptidase domain of MNP-1 with a similar sequence from aminopeptidase A indicated a significant similarity between these two protein domains (17.2% identity and 33.9% similarity). Importantly, these alignments indicated three of the four absolutely conserved residues of the HENNH+E motif are not retained in MNP-1 (fig. 5a). This observation suggests strongly that MNP-1, though closely related to active M1 family peptidases, is highly unlikely to be catalytically active for peptidase function.
Figure 5. Expression analysis of MNP-1.
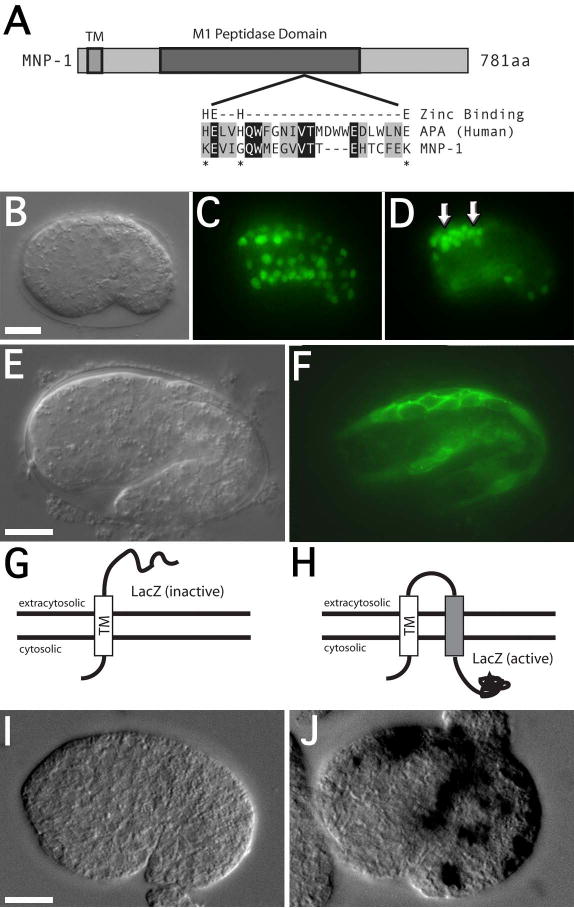
(A) Schematic representation of the MNP-1 domain structure, including an alignment of the predicted zinc-binding domains between aminopeptidase A (APA) and MNP-1. This alignment highlights the lack of conservation for three of the four essential zinc-binding residues in the MNP-1 domain (asterisks). (B) DIC and (C-D) fluorescence images of an mnp-1p::GFP transgenic embryo at the bean stage (25°C; approximately 290 min) showing expression in muscle cells and unidentified cell bodies in the head region (arrows). The identity of the muscle cells is based on position and the relative movements of the GFP expressing nuclei during embryogenesis. (E) DIC and (F) fluorescence images of mnp-1p::mnp-1tm::GFP transgenic embryo at the 1½ fold stage (25°C; approximately 380 minutes), showing localization of GFP proximal to plasma membrane of the muscle cells. (G-H) Schematic drawings indicating the inferred location of the β-gal portion of the hybrid proteins derived from the mnp-1tm::β-gal and mnp-1tm(2x)::β-gal respectively. (I) x-gal staining in a transgenic embryo expressing the mnp-1tm::β-gal hybrid protein. (J) x-gal staining in a transgenic embryo expressing the mnp-1tm(2x)::β-gal hybrid protein. Scale bars represent 10μm.
MNP-1 is expressed at the plasma membrane of migrating muscle cells
To characterize the expression of mnp-1, we performed reporter analysis using an mnp-1p::GFP transgene containing 3 kb of upstream sequence driving a nuclear localized green fluorescent protein (GFP). At the comma stage (approximately 400 cells) and persistent through hatching, GFP expression was apparent in all of the body-wall muscle cells (fig. 5b-d). At these embryonic stages, GFP expression is also seen in the migrating z1 and z4 gonadal precursor cells (not shown) and a number of unidentified cells in the dorsal anterior region (arrows, fig. 5d). These unidentified cells are likely neuronal precursors based on their position, although other cells types are not ruled out. After hatching, GFP expression fades quickly and is typically absent by mid to late L1 larval stage.
To determine the possible sub-cellular localization of MNP-1 we made a reporter expressing the mnp-1 open reading frame, in which we had replaced the putative M1 family peptidase domain with sequence encoding GFP. Expression from this reporter clearly localized GFP proximal to the plasma membrane of embryonic muscle cells (fig. 5e-f), suggesting that the portion of MNP-1 containing the predicted transmembrane domain is sufficient to direct localization to the plasma membrane.
The topology of the MNP-1 transmemebrane domain
We explored the topology of the MNP-1 transmemebrane domain using a β-galactosidase (β-gal) hybrid approach that has been used for deducing the topology of other membrane proteins in C. elegans (Fire et al., 1990; Li and Greenwald, 1996). We constructed transgenes encoding a hybrid MNP-1::β-gal protein, in which β-gal was placed after the predicted transmembrane domain. We also constructed a control transgene encoding the MNP-1::TM::β-gal protein that include a short spacer sequence followed by an additional transmembrane domain, placed between the native MNP-1 trans-membrane domain and β-gal (Fire et al., 1990). These transgenes were expressed from the mnp-1 promoter.
Transgenes expressing fusion proteins in which β-gal is localized to the cytosol should stain positive for β-gal activity. However, transgenes expressing fusion proteins in which β-gal is located in the extra-cellular compartment should not display staining (fig. 5g-h) (Fire et al., 1990; Li and Greenwald, 1996). Assays for β-gal activity of the MNP-1::β-gal and MNP-1::TM::β-gal hybrid transgenic lines indicated the corresponding MNP-1::β-gal hybrid protein did not display staining (fig. 5i). However, the corresponding MNP-1::TM::β-gal protein did display staining (fig. 5j). These observations indicated a topology of the MNP-1 trans-membrane domain that would expose the COOH-terminus and subsequently the M1 family, non-peptidase domain to the extra-cellular space.
VAB-1, INA-1, and MNP-1 function independently and from distinct tissues to influence the notched head phenotype
In order to determine if VAB-1, INA-1, or MNP-1 act in the same or independent pathways we analyzed the severity of the notched head phenotype in single and double mutant combinations. The notched head phenotype was categorized on a scale from zero to three (fig. 6a). The “class three” phenotype represents the most sever anterior morphology defect seen in these strains and is characterized as a thinning of ventral tissue in the isthmus region of the pharynx accompanied by a large accumulation of cells to the anterior (triangles, fig. 6a).
Analysis of the ina-1(gm86);mnp-1(RNAi), vab-1(dx31);mnp-1(RNAi), and vab-1(dx31);ina-1(gm86) strains indicated that each displayed increased severity of the notched head phenotype when compared to mnp-1(RNAi), ina-1(gm86), or vab-1(dx31) single mutants alone (fig. 6b). This is most dramatically displayed when comparing the frequency of the most sever anterior morphology defects (class three) between these strains. Specifically, mnp-1(RNAi), ina-1(gm86), or vab-1(dx31) single mutants alone displayed no more than 6% class three defects (0%, 6% and 6% respectively; n >109). However, ina-1(gm86);mnp-1(RNAi), vab-1(dx31);mnp-1(RNAi), and vab-1(dx31);ina-1(gm86) strains each displayed greater than 53% class three defects (70%, 53% and 87% respectively; n >104). These results were consistent with an additive affect between these mutations and suggested that VAB-1, INA-1 and MNP-1 may each act in predominantly independent pathways.
Previous analysis indicated mutations in vab-1 and vab-2 affect closure of the ventral cleft via movements of surrounding neuronal precursor cells and subsequently affect the organization of the ventral neurons (George et al., 1998; Chin-Sang et al., 1999). Furthermore, tissue specific rescue of vab-1 and vab-2 mutations, using neuronal specific promoters, suggest that the ephrin pathway likely functions within the developing nervous system to influence both ventral cleft closure and the notched head phenotype.
To gain insights into the site of INA-1 action, we asked whether neuronal, epidermal or muscle expression of INA-1 was sufficient to suppress the notched head defects of ina-1(gm86) mutants. We generated animals in which INA-1 was expressed under the control of the ajm-1 promoter (Koppen et al., 2001) and found that this epidermal expression was able to significantly suppress the notched head defect seen in the ina-1(gm86) strain from 85% to 16% (n >239; fig. 6c). In contrast, neither pan-neuronal nor muscle specific expression from the F25B3.3 (Altun-Gultekin et al., 2001) or hlh-1 promoters (Chen et al., 1994) significantly rescued the notched head phenotype of the ina-1(gm86) mutation (81% and 84% respectively, n >237; fig. 6c).
To address the cell specificity of MNP-1 function we restricted mnp-1(RNAi) to embryonic muscle tissue in C. elegans by using the hlh-1 promoter to drive expression of an mnp-1 RNA hairpin (hlh-1p::mnp-1hp). We utilized the sid-1(qt2) mutation to prevent the RNAi effect from spreading between cells and ensure a tissue specific effect (Winston et al., 2002). sid-1(qt2) strains containing control arrays, including an hlh-1 promoter driven GFP and an hlh-1 promoter driven ina-1 RNA hairpin, did not display any morphological defects (not shown). In contrast, sid-1(qt2) L1 larva containing the hlh-1p::mnp-1hp displayed severe body morphology defects including the most sever defect observed in other notched head strains (class three), characterized by the thinning of ventral tissue in the isthmus region of the pharynx and a large accumulation of cells to the anterior (triangles, fig. 6d). We observed acute disruption of the body-wall muscle quadrants, including cells localized to the lateral sides of the worm (arrows, fig. 6e).
Discussion
Three predominantly independent genetic pathways influence the process of embryonic muscle cell migration in C. elegans. We identified four gene products required for proper muscle cell migration: ina-1, vab-1, vab-2, and mnp-1. Each displayed similar defects in both muscle cell migration and anterior body morphology (fig. 2-4). This co-occurrence of muscle cell migration defects and the notched head phenotype suggest a close relationship between these two phenotypes. Furthermore, muscle specific RNAi of mnp-1 resulted in defects in muscle cell positioning, as well as body morphology defects consistent with the notched head phenotype (fig 6d). These results indicated a direct association between the notched head phenotype and muscle cell migration defects. These results also suggested the possibility that the muscle specific defects seen in these strains may be the underlying cause of the notched head phenotype.
Our observations indicated that the α-integrin, INA-1, functions from the neighboring epidermal tissue to influence migration of the muscle cells. We observed that loss of function mutations in ina-1 resulted in misplaced embryonic muscle cells in a manner consistent with failed or aberrant migration events (fig 3a-d). Additionally, we observed that epidermal specific expression of INA-1 was able to rescue the notched head defect of an ina-1 loss of function mutation (fig 6b). This observation was consistent with previous mosaic analysis indicating the loss of ina-1 expression from the epidermal lineage affecting the notched head phenotype (Baum and Garriga, 1997). These observations suggested the possibility that either a signaling event arising from the epidermal cells, the integrity of the epidermal tissue itself, or the architecture of the surrounding ECM may play a specific role during muscle cell migration. The C. elegans epidermal cells themselves migrate as a sheet towards the ventral midline to enclose the embryo (Chisholm and Hardin, 2005). This process of epiboly concludes at approximately the same time as the onset of the muscle cell migrations, indicating there may be an association between these two events. However, our own observations indicated that the overall tissue architecture of the epidermis prior to and during muscle cell migration was unaffected by mutations in ina-1 (unpublished) suggesting that the integrity of the epidermis is unlikely to cause the muscle cell migration defects seen in this strain. A cell non-autonomous role for integrins in cell migration has been observed in Droshoplila embryos (Martin-Bermudo et al., 1999; Boube et al., 2001) and accredited to defects in ECM assembly in amphibian embryos (Skalski et al., 1998). If ina-1 is affecting the assembly of the ECM, these studies may provide an opportunity to dissect this specific integrin function in the C. elegans model.
Several observations have indicated that the Eph receptor, VAB-1, and the ephrin ligand, VAB-2, function from within the neighboring neuronal tissue to influence migration of the muscle cells. We observed that loss of function mutations in either vab-1 or vab-2 resulted in misplaced embryonic muscle cells in a manner consistent with failed or aberrant migration events (fig. 3). In addition, previous observations indicate that VAB-1 and VAB-2 are required for the proper movements of the neuronal precursor cells surrounding the ventral cleft, between 230 and 290 minutes, and mutations in these genes can result in significant disorganization of this tissue (George et al., 1998; Chin-Sang et al., 1999). Furthermore, neuronal specific expression of either VAB-1 or VAB-2 is able to rescue both the embryonic lethality associated with the ventral cleft closure defect and the notched head defect (George et al., 1998; Chin-Sang et al., 1999). Eph receptors and ephrin ligands can directly influence both cell movements and tissue organization in many organisms by their ability to modulate cell affinity (Poliakov et al., 2004). Though our observations do not rule out a direct role for VAB-1 and VAB-2 in controlling muscle cell migrations in C. elegans, the disorder of the underling neuronal tissue in these mutants suggests another possibility. It is plausible that the organization of the underling tissue may provide information essential to promote an appropriate migration path for the muscle cells.
Our work also identified the non-peptidase homologue, MNP-1, as being required for proper muscle cell migration. RNAi against mnp-1 resulted in both misplaced embryonic muscle cells and a notched head phenotype consistent with failed or aberrant migration events (fig. 4d-g). Analysis of reporter transgenes suggested that MNP-1 was expressed in the embryonic muscle cells during migration and was localized to the plasma membrane with the non-peptidase domain exposed to the extra cellular space (fig. 5). Furthermore, muscle specific RNAi against mnp-1 resulted in both misplaced muscle cells and body morphology defects consistent with the notched head phenotype (fig. 6d-e). Together these observations suggested that MNP-1 functions cell autonomously during migration of the embryonic muscle cells and points to a possible role within the surrounding extra-cellular space. The exact role of MNP-1 is unknown, though there are a number of possible roles the non-peptidase domain may play. One possibility is that the non-peptidase domain may function as a novel adhesion molecule. In this case MNP-1 would affect cell migration by directly affecting the adhesion properties of the migrating muscle cells. Another appealing possibility is that the non-peptidase domain may function as a competitive inhibitor of another active peptidase. In this case MNP-1 may promote cell migration by protecting an unidentified factor within the extra cellular space from improper degradation.
Previously, the notched head phenotype has been interpreted as a specific defect of the epidermal cells (Baum and Garriga, 1997; George et al., 1998; Chin-Sang et al., 1999). Though, it's clear that mutations in vab-1 and vab-2/efn-1 result in epidermal enclosure defects, our own observations suggest that defects in epidermal morphogenesis are not present in either ina-1 mutants or mnp-1(RNAi) strains (not shown). To reconcile these observations we propose a simple model: aberrant muscle migrations result in a failure of the ventral muscle quadrants to form the proper connections proximal to the sensory depression. Mechanical forces generated during the process of enlongation lead to a local deformation or buckling of the anterior ventral surface. In turn, this may lead to deformation of the epidermis as seen in ina-1 mutant L1 larva (Baum and Garriga, 1997).
Correct execution of cell migrations is an important developmental phenomenon essential for proper embryogenesis. Our results indicated that ina-1, vab-1, and the mnp-1 genes each act independently to promote the proper positioning of the body-wall muscle cells along the ventral midline. How each of these gene products affects these cell migrations has not yet been revealed. However, the strikingly similar phenotypes between these three genes lead us to speculate that they may converge on a common process such as regulation of cell-ECM interactions. Analysis of the embryonic muscle cell migrations in C. elegans has proven a tractable model for studying cell movements in an intact organism and will advance our understanding of how cells navigate the complex environments encountered during development.
Acknowledgments
We thank the C. elegans Gene Knockout Consortium (Oklahoma Medical Research Foundation) for generating the ok2434 deletion strain and T. Stiernagle at the C. elegans Genetic Center (University of Minnesota) for supplying various strains used in this work. This work was supported by the Howard Hughes Medical Institute, of which M. H. is an investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altun-Gultekin Z, Andachi Y, Tsalik EL, Pilgrim D, Kohara Y, Hobert O. A regulatory cascade of three homeobox genes, ceh-10, ttx-3 and ceh-23, controls cell fate specification of a defined interneuron class in C. elegans. Development. 2001;128:1951–1969. doi: 10.1242/dev.128.11.1951. [DOI] [PubMed] [Google Scholar]
- Arnaout MA, Mahalingam B, Xiong JP. Integrin Structure, Allostery, and Bidirectional Signaling. Annu Rev Cell Dev Biol. 2005;21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- Baum PD, Garriga G. Neuronal migrations and axon fasciculation are disrupted in ina-1 integrin mutants. Neuron. 1997;19:51–62. doi: 10.1016/s0896-6273(00)80347-5. [DOI] [PubMed] [Google Scholar]
- Bökel C, Brown NH. Integrins in Development: Moving on, Responding to, and Sticking to the Extracellular Matrix. Developmental Cell. 2002;3:311–321. doi: 10.1016/s1534-5807(02)00265-4. [DOI] [PubMed] [Google Scholar]
- Boube M, Martin-Bermudo MD, Brown NH, Casanova J. Specific tracheal migration is mediated by complementary expression of cell surface proteins. Genes Dev. 2001;15:1554–6. doi: 10.1101/gad.195501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chen L, Krause M, Sepanski M, Fire A. The Caenorhabditis elegans MYOD homologue HLH-1 is essential for proper muscle function and complete morphogenesis. Development. 1994;120:1631–41. doi: 10.1242/dev.120.6.1631. [DOI] [PubMed] [Google Scholar]
- Chin-Sang ID, George SE, Ding M, Moseley SL, Lynch AS, Chisholm AD. The ephrin VAB-2/EFN-1 functions in neuronal signaling to regulate epidermal morphogenesis in C. elegans. Cell. 1999;99:781–90. doi: 10.1016/s0092-8674(00)81675-x. [DOI] [PubMed] [Google Scholar]
- Chisholm AD, Hardin J. Epidermal morphogenesis. The C. elegans Research Community, WormBook, editor. WormBook. 2005 doi: 10.1895/wormbook.1.35.1. http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Epstein HF, Shakes DC. Methods in Cell Biology: Caenorhabditis elegans: Modern Biological Analysis of an Organism. San Diego: Academic Press, Inc; 1995. [Google Scholar]
- Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- Fire A, Kondo K, Waterston R. Vectors for low copy transformation of C. elegans. Nucleic Acids Res. 1990;18:4269–4270. doi: 10.1093/nar/18.14.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- George SE, Simokat K, Hardin J, Chisholm AD. The VAB-1 Eph receptor tyrosine kinase functions in neural and epithelial morphogenesis in C. elegans. Cell. 1998;92:633–43. doi: 10.1016/s0092-8674(00)81131-9. [DOI] [PubMed] [Google Scholar]
- Hresko MC, Williams BD, Waterston RH. Assembly of body wall muscle and muscle cell attachment structures in Caenorhabditis elegans. J Cell Biol. 1994;124:491–506. doi: 10.1083/jcb.124.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2 doi: 10.1186/gb-2000-2-1-research0002. research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppen M, Simske JS, Sims PA, Firestein BL, Hall DH, Radice AD, Rongo C, Hardin JD. Cooperative regulation of AJM-1 controls junctional integrity in Caenorhabditis elegans epithelia. Nat Cell Biol. 2001;3:983–91. doi: 10.1038/ncb1101-983. [DOI] [PubMed] [Google Scholar]
- Li X, Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron. 1996;17:1015–21. doi: 10.1016/s0896-6273(00)80231-7. [DOI] [PubMed] [Google Scholar]
- Martin-Bermudo MD, Alvarez-Garcia I, Brown NH. Migration of the Drosophila primordial midgut cells requires coordination of diverse PS integrin functions. Development. 1999;126:5161–9. doi: 10.1242/dev.126.22.5161. [DOI] [PubMed] [Google Scholar]
- Meighan CM, Schwarzbauer JE. Control of C. elegans hermaphrodite gonad size and shape by vab-3/Pax6-mediated regulation of integrin receptors. Genes Dev. 2007;21:1615–1620. doi: 10.1101/gad.1534807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman DG, Hutter H, Mullen GP, Schnabel R. Cell autonomous expression of perlecan and plasticity of cell shape in embryonic muscle of Caenorhabditis elegans. Dev Biol. 1996;173:228–42. doi: 10.1006/dbio.1996.0019. [DOI] [PubMed] [Google Scholar]
- Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–64. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigel M, Hooper NM. Families of zinc metalloproteases. FEBS Letters. 1994;354:1–6. doi: 10.1016/0014-5793(94)01079-x. [DOI] [PubMed] [Google Scholar]
- Poliakov A, Cotrina M, Wilkinson DG. Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev Cell. 2004;7:465–80. doi: 10.1016/j.devcel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Sela-Donenfeld D, Wilkinson DG. Eph receptors: two ways to sharpen boundaries. Curr Biol. 2005;15:R210–2. doi: 10.1016/j.cub.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RH. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol. 2002;12:1317–9. doi: 10.1016/s0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
- Skalski M, Alfandari D, Darribere T. A key function for alphav containing integrins in mesodermal cell migration during Pleurodeles waltl gastrulation. Dev Biol. 1998;195:158–73. doi: 10.1006/dbio.1997.8838. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–12. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002;295:2456–9. doi: 10.1126/science.1068836. [DOI] [PubMed] [Google Scholar]
- Yochem J, Gu T, Han M. A new marker for mosaic analysis in Caenorhabditis elegans indicates a fusion between hyp6 and hyp7, two major components of the hypodermis. Genetics. 1998;149:1323–1334. doi: 10.1093/genetics/149.3.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]