

Abstract
Varicella zoster viru (VZV)-specific T cell responses are believed to be vital in recovery from primary VZV infection and also in the prevention of viral reactivation. While glycoprotein E (gE) is the most abundant and one of the most immunogenic proteins of the virus, there are no data addressing potential T cell epitopes within gE, nor the phenotype of specific T cells. Using interferon gamma enzyme-linked immunospot assays and intracellular cytokine assays, we identified gE-specific immune responses in 20 adult healthy immune donors which were found to be dominated by the CD4+ subset of T cells. We characterized three immune dominant epitopes within gE restricted through DRB1*1501, DRB1*07 and DRB4*01, and used DRB1*1501 class II tetrameric complexes to determine the ex vivo frequency and phenotype of specific T cells. In healthy immune donors, the cells were largely positive for CCR7, CD28 and CD27, but expressed variable CD62L and low levels of cutaneous lymphocyte associated antigen with evidence of recent activation. In summary, we show that circulating gE-specific CD4+ T cells are detected at a relatively high frequency in healthy immune donors and show evidence of recent activation and mixed central and effector memory phenotype. These data would be compatible with frequent exposure to replicative cycle antigens in healthy donors and are consistent with a role for gE-specific CD4+ T cells in the control of viral replication.
Keywords: CD4+ T cells, glycoprotein E, varicella zoster virus
Introduction
Varicella zoster viru (VZV) is an alpha herpes virus which causes chickenpox during primary infection and herpes zoster during reactivation. VZV-specific T cell responses are believed to be vital in the recovery from primary VZV infection and also in the prevention of virus reactivation [1,2]. Early development of VZV-specific proliferative T cell responses has been shown to be associated with milder disease [3]. Proliferative and cytotoxic memory T cell responses to several viral glycoproteins and tegument proteins have been demonstrated following natural infection and vaccination [4–7]. Recently, glycoprotein I (gI) and immediate early (IE)4-specific CD4+ T cells were shown to circulate at persistently high frequencies in the peripheral blood of healthy immune donors without any history of reactivation [8,9].
Varicella zoster virus has a double-stranded DNA genome consisting of more than 70 open reading frames (ORF) [10]. VZV genes are transcribed in a temporally controlled manner and accordingly called IE, early and late proteins, depending on temporal transcription during the virus replicative cycle [10]. The VZV genome encodes seven glycoproteins, gB, gC, gE, gH, gI, gK and gH, which are thought to mediate cellular attachment, penetration and fusion of the infected cell membrane and also cell-to-cell spread of the virus [11]. gE is the most abundant glycoprotein and is encoded by ORF 68 [12]. It is thought to be an essential glycoprotein for the virus, as attempts to generate gE deletion mutants have so far been unsuccessful [13]. It is expressed on the plasma membrane and in the cytoplasm of infected cells and is thought to have multiple functions, such as cell-to-cell spread of the virus and skin tropism [14], and it is also essential for the formation of infectious virions and viral replication [12]. Recently, it was shown that gE interacts with the insulin-degradation enzyme which was identified as the cellular receptor for both cell-free and cell-associated VZV [15]. gE is thought to be an important T cell target, as the gE vaccinia virus recombinants induced the highest proliferative T cell responses when compared with other VZV glycoprotein vaccinia recombinants in guinea pigs [16]. Furthermore, guinea pigs immunized with recombinant VZV gE and gI developed protective antibody responses and were able to clear the virus efficiently [9]. However, there are no data addressing potential human T cell epitopes within gE.
Expression of different co-stimulatory and lymph node homing molecules on virus-specific T cells is thought to correlate with differentiation and functional characteristics [17]. Recently, IE63-specific CD4+ T cells have been shown to express markers of early intermediate differentiation and up to 30% showed evidence of recent activation [18]. This could be explained by frequent re-exposure or reactivation or, alternatively, that as IE63 is expressed during latency, it could reflect T cell exposure to latently expressed protein. Distinguishing these possibilities is important in the understanding of disease pathogenesis and the critical components of immune control of latency. In order to address these possibilities, we sought to identify T cell epitopes within gE, a VZV protein that is not expressed during latency, and then use these to determine the functional and differentiation phenotype of the gE-specific T cells using class II tetrameric complexes.
Methods
Subjects
The initial study participants consisted of 20 healthy seropositive adult individuals with a history of primary VZV infection but no clinical reactivation and eight seronegative donors. The mean age of the seropositive donors was 34·4 years (range 26–63 years), with an average age of primary infection at 6·4 years (range 3–15 years). We also recruited eight seropositive DRB1*1501-positive individuals and three seronegative DRB1*1501-positive individuals for further DRB1*1501 tetramer analysis. Ethical approval was granted by the Oxfordshire ethics committee. Peripheral blood mononuclear cells (PBMC) were obtained from fresh heparinized blood by Ficoll-Hypaque density gradient centrifugation. They were then resuspended in RPMI-1640 plus 10% fetal calf serum (FCS) for ex vivo enzyme-linked immunospot (ELISPOT) assays and ex vivo intracellular cytokine staining (ICS) assays and in RPMI-1640 plus 10% human serum for cell cultures.
Peptides
Synthetic 20 mer peptides overlapping by 10 amino acids which spanned the whole length of the gE protein were synthesized in house in an automated synthesizer using 9-fluorenylmethoxycarbonyl chemistry. The purity of the peptides was determined to be greater than 90% by high-pressure liquid chromatography analysis and mass spectrometry.
Ex vivo ELISPOT assays
Ex vivo ELISPOT assays were performed as described previously [19]. Briefly, ELISPOT plates (Millipore Corp., Bedford, MA, USA) were coated with anti-human interferon (IFN)-γ antibody overnight (Mabtech AB, Nacka, Sweden). The plates were washed six times with RPMI-1640 and incubated for 1 h with RPMI-1640 and 10% FCS; 0·1 × 106 PBMC were added to a final volume of 200 μl and peptide was added at a final concentration of 10 μM. The 62 20 mer overlapping gE peptides were made into 20 pools, each containing three 20 mer peptides (the last pool consisted of five 20 mer peptides). PBMC from each individual donor were tested in duplicate with the 20 peptide pools. Phytohaemagglutinin was included as a positive control and an irrelevant peptide was included as a negative control.
The plates were incubated overnight at 37°C and 5% CO2 and developed using 1 μg/ml of biotin-linked anti-IFN-γ monoclonal antibody (mAb) (mAb 7-B6-1-biotin; Mabtech AB) as a detection antibody, which was conjugated subsequently to streptavidin alkaline phosphatase (AP) (Mabtech AB), and visualized using an AP conjugate substrate kit (Biorad, Hercules, CA, USA; 170–643). The spots were enumerated using an automated ELISPOT reader. Background (cells plus media) was subtracted and data expressed as number of spot-forming units (SFU) per 106 PBMC. All peptides that induced an IFN-γ response of more than mean ± 3 standard deviations (s.d.) of the irrelevant peptide were considered positive.
Cultured ELISPOT assays
Peripheral blood mononuclear cells from each donor were incubated with the gE peptide pool consisting of all overlapping peptides. Briefly, 5·0 × 106 PBMCs were incubated for 10 days with 200 μl of 40 μM peptide pool in a 24-well plate. Interleukin-2 was added on days 3 and 7 at a concentration of 100 units/ml. All cell lines were maintained routinely in RPMI-1640 supplemented with 2 mM L-glutamine, 100 IU/ml penicillin and 100 μg/ml plus 10% human serum at 37°C, in 5% CO2. T cell lines were tested after 10 days' culture against the individual peptides included in the pools in a 96-well ELISPOT plate. All peptides that induced an IFN-γ response of more than mean ± 3 s.d. of the irrelevant peptide were considered positive.
Intracellular cytokine secreting assay
To determine IFN-γ production, ex vivo PBMC or T cell lines were stimulated with either culture medium alone, phorbol myristate acetate (50 ng/ml) and ionomycin (1 mM), peptide (5 μM) or pooled peptides (each peptide 1 μM) for 16 h at 37°C and 5% CO2 with the addition of GolgiStop [Becton Dickinson (BD) Biosciences, Oxford, UK; 554715] after 2 h of incubation. Cells were washed and stained with anti-CD3 [fluorescein isothiocyanate (FITC)], anti-CD4 [peridinin chlorophyll (PerCP)] (BD Biosciences) and anti-CD8 [phycoerythrin (PE)]. Cells were then permeabilized and fixed with Cytofix/Cytoperm (BD Biosciences) and stained for intracellular IFN-γ[allophycocyanin (APC)], according to the manufacturer's instructions and analysed by flow cytometry.
Maintenance of cell lines
Epstein–Barr virus (EBV)-transformed B cell lines were maintained routinely in RPMI-1640 supplemented with 2 mM L-glutamine, 100 IU/ml penicillin and 100 μg/ml plus 10% FCS at 37°C, in 5% CO2. Three established keratinocyte lines were used: HaCat cells (a gift from Dr N. Fusenig) were developed spontaneously from adult epidermal keratinocytes, natural killer and nuclear factor K cells are human papilloma virus-16 immortalized keratinocytes (a gift from Dr E. O'Toole). Keratinocytes were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% FCS, 2 mM L-glutamine, 50 U/ml penicillin and 50 μg/ml streptomycin. Murine fibroblast cell lines transfected with HLA-DRB1*15 (kindly supplied by Professor Lars Fugger) were maintained in DMEM/10% FCS at 37°C with 5% CO2.
Major histocompatibility complex class II HLA restriction
All major histocompatibility complex (MHC) class II HLA restrictions were performed in triplicate. Cells from short-term cultures were incubated with 10 μl of monoclonal antibodies at 0·2 mg/ml specific for HLA-DR (L243), HLA-DQ (SPV-L3) (kindly supplied by Professor Lars Fugger) and HLA-DP (Leinco Technologies, Ballwin, MO, USA; H127) at 37°C for 1 h before addition of peptides. B cells partially matched or mismatched to the testing HLA molecule were pulsed initially with 25 μl of 40 μM peptide for 1 h at 37°C in 5% CO2. They were then washed three times in RPMI-1640 plus 10% FCS and used as APCs to washed T cells harvested from cell cultures. Following overnight incubation with IFN-γ, keratinocytes matched and mismatched for the HLA type of the T cells were pulsed initially with 100 μl of 100 μM peptide for 1 h at 37°C, in 5% CO2. Once pulsed they were washed three times in RPMI-1640 plus 10% FCS and used as APCs to washed T cells harvested from cell cultures. VZV-infected cell extract (ABI, Maryland, USA; 10-514-001) was used at 5 μg/ml and Vero-uninfected cell extract (ABI, 10-508-001) was used at 5·4 μg/ml. The live attenuated VZV (Varilirix Middlesex, UK; GlaxoSmithKline) was added to a final concentration of 10 × 4 plaque-forming units/ml.
Tetramer and phenotypic staining and flow cytometry
DRB1*1501 iTAg MHCII tetramer was purchased from Beckman Coulter (Hialeah, FL, USA). DRB1*1501 tetramer was complexed to VZV gE peptide 54 (aa531-545; TSPLLRYAAWTGGLA). Unless stated otherwise, cell lines and PBMC were incubated with 2 μg/ml HLA class II tetramer for 60 min at 37°C in RPMI-1640 and 10% human serum. We analysed the tetramer expression within the CD4+ T cell subset by gating on the lymphocytes and excluding B cells, monocytes and dead cells (via probe positive population) routinely using 5 million PBMC per stain The anti-PE tetramer enrichment was performed as described previously [18].
The cell surface marker antibodies CD4-pacific blue (Biolegend, San Diego, CA, USA), CD14-PerCP, C19-PerCP and 7-aminoactinomycin D (7-AAD) (all BD Pharmingen, Oxford, UK) were added for 20 min at room temperature. For phenotypic analysis of tetramer-positive CD4+ cells, antibodies to CD27 (FITC); CD28 (allophycocyanin, APC); CD38 (APC); CD45RO (APC); CD62L (APC); CD56 (Pe-Cy7); CCR7 (Pe-Cy7); cutaneous lymphocyte associated antigen (CLA; FITC); PD1 (FITC) and perforin (FITC) were added with the other surface antibodies. Stained cells were washed with phosphate-buffered saline (PBS) and fixed in 0·5% PBS/formaldehyde. Cells were acquired on a CyAn™ (DakoCytomation, Glostrup, Denmark) and analysed using FlowJo software.
Results
Overall gE-specific T cell responses ex vivo
Using the 20 peptide pools (each consisting of three 20 mer peptides) encompassing the gE protein, we screened the ex vivo ELISPOT responses with PBMC derived from 20 healthy immune donors with a history of VZV infection and positive VZV serology (Fig. 1a). The ex vivo PBMC responses specific for these peptide pools ranged from 0 to 465 SFU/million cells. Seronegative individuals (n = 8) made no responses to any of the gE peptide pools ex vivo (data not shown). There was no correlation of ex vivo gE-specific T cell responses with age of the donors or with time as primary infection. As we had previously studied ex vivo responses to the gI protein [8,9] we then went on to compare ex vivo immune responses of overlapping gI peptides with gE peptides. We found that gE-specific IFN-γ responses, in the majority of donors, were present at a significantly higher level than gI-specific immune responses (P < 0·001). Overall these data showed that gE-specific T cell responses could be identified ex vivo and are present at a higher level than those of gI in healthy immune donors.
Fig. 1.
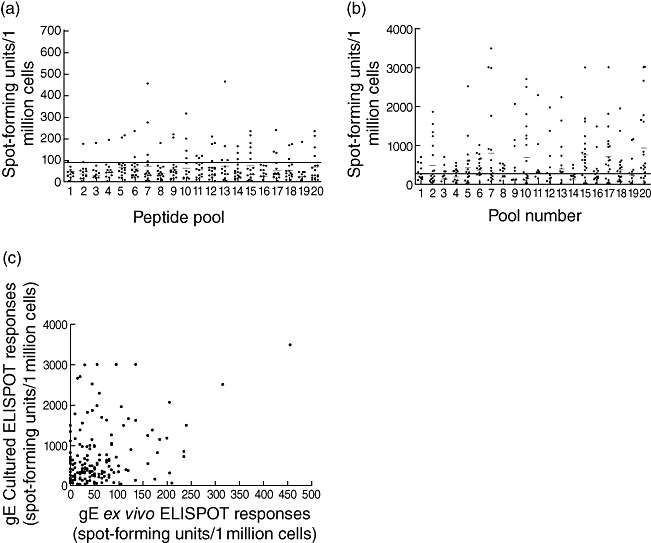
(a) Ex vivo interferon (IFN)-γ enzyme-linked immunospot (ELISPOT) responses to overlapping glycoprotein E (gE) peptide pools in the cohort of healthy immune donors with a history of primary varicella zoster virus (VZV) but no clinical reactivation. The horizontal bar shows the mean ± 3 standard deviations (s.d.) of the irrelevant peptide control. (b) Cultured IFN-γ ELISPOT responses to overlapping gE peptide pools in cohort of healthy immune donors with a history of primary VZV but no reactivation. The horizontal bar shows the mean ± 3 s.d. of the irrelevant peptide control. (c) Correlation between ex vivo and cultured IFN-γ ELISPOT responses for overlapping gE peptide pools in a cohort of healthy immune donors (P < 0·0001, r = 0·3016).
We then proceeded to confirm the responses detected ex vivo by expanding peptide-specific cells in vitro. We observed high levels of gE-specific T cells after 10 days of culture, reaching a maximum of 3488 IFN-γ-producing cells per million total cells (Fig. 1b). The cultured ELISPOT responses showed that 14 of 20 individuals responded to pool 20, 15 of 20 responded to pool 15, 14 of 20 responded to pool 17, nine of 20 to pool 10 and nine of 20 responded to pool 7. After further testing the individuals for each 20 mer peptide included in these peptide pools, we found that the majority of the responding individuals reacted to peptide 20 in pool 7, peptides 58 and 59 in pool 20 and peptide 29 in pool 10 (data not shown). We observed that there was a significant positive correlation between the ex vivo and cultured gE peptide pool-specific responses, confirming that the cultured approach was expanding cells carrying an overlapping specificity to those detected ex vivo (P < 0·0001, r = 0·3016, Fig. 1c).
Intracellular cytokine staining was used to investigate the responding T cell subsets and showed that responses to gE peptides were mediated by CD4+ T cells (data not shown). Indeed, we tested all immune dominant pools and found that responses to all immune-dominant peptide pools and 20 mer peptides were mediated exclusively by CD4+ T cells. Our earlier studies have shown a similar CD4+ T cell dominance of responding T cells both ex vivo and cultured using the viral lysate and vaccine as stimulatory antigens [20]. In these experiments we considered that the use of the peptides and lysate might bias the assay towards stimulation of CD4+ T cells and therefore repeated the assay using live viral vaccine as stimulation. However, we found that the live vaccine also induces a gE-specific cellular immune response which is mediated by CD4+ T cells (Fig. 2). Overall, these data show that it is possible to expand gE-specific T cells in vitro and that such responses are dominated by CD4+ T cells.
Fig. 2.
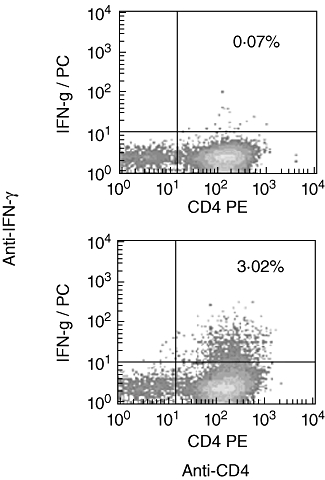
Peripheral blood mononuclear cells cultured with the live varicella zoster virus vaccine for 10 days and tested with overlapping glycoprotein E (gE) pools of peptides. Cells are gated on CD3. The top panel shows the unstimulated control cells and in the lower panel the cells are stimulated with overlapping gE peptides. IFN, interferon; PE, phycoerythrin.
Identification of a DRB1*1501-restricted epitope within gE
Peripheral blood mononuclear cells were cultured with pools of gE 20 mer peptide (10 20 mer peptides in each pool named A–F) and then tested for IFN-γ production using peptide-pulsed DRB1*1501 expressing transfected L cells for antigen presentation. Figure 3a shows the results from the constituent peptides from pool F, which confirmed that peptide 54 contained a DRB1*1501-resticted T cell epitope. To determine the minimum length of the epitope, T cells cultured with the 20 mer peptide were tested by ELISPOT assays using sequential C and N terminal peptide truncations of the 20 mer index peptide. We found the minimum peptide to be a 12 mer (Fig. 3b) and using peptide dose titration have found that the optimum peptide is a 15 mer (TSPLLRYAAWTGGLA) (Fig. 3c).
Fig. 3.
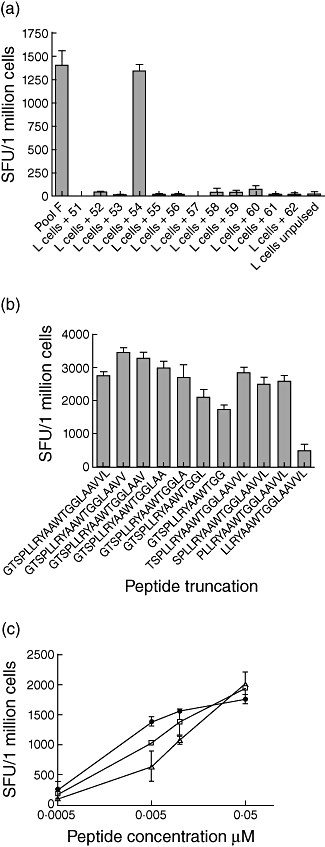
(a) Interferon (IFN)-γ enzyme-linked immunospot (ELISPOT) responses using the glycoprotein E (gE) pool F-specific line, incubated with pool F peptide directly added to T cells and DRB1*1501-expressing L cells pulsed with each of the 20 mer peptides in pool F and used as antigen-presenting cells to the T cell line. (b) IFN-γ ELISPOT responses to truncated peptides from the 20 mer peptide 54 of gE using a peptide 54-specific CD4+ T cell line. The spot-forming units (SFU) of the negative control were 110 per million. (c) IFN-γ ELISPOT responses to truncated peptides from the 20 mer peptide 54 of gE using dose titration with peptide 54-specific CD4+ T cell line. Closed circles represent TSPLLRYAAWTGGLA; open triangles represent GTSPLLRYAAWTGGLAA; open squares represent GTSPLLRYAAWTGGLAAV.
Characterization of other epitopes within gE
As we found that T cell responses to peptide 20 (gE pool 7) and peptide 29 (pool 10) were derived from CD4+ T cells, we proceeded to determine the HLA class II restriction of these epitopes. We initially used DR, DQ and DP blocking antibodies and showed that the DR molecules were involved in the presentation of both epitopes (data not shown). To determine which of the DR molecules presented the epitope within peptide 20, keratinocyte lines (both matched and mismatched to the testing HLA molecule) were used as APCs to short-term T cell lines. This showed that the epitope in peptide 20 was presented by the DRB1*07 molecule (Fig. 4a). To determine the minimum length of the epitope, T cells cultured with the 20 mer peptide were tested by ELISPOT assays using peptide truncations of the 20 mer index peptide. By testing the individuals who responded to gE peptide 20, we found the minimum peptide to be a 9 mer and the optimum epitope to be a 14 mer (GVRYTETWSFLPSL, data not shown). Ex vivo ELISPOT responses to this epitope were found to be 65–925 (mean 292, s.d. ± 356·6) SFU/million cells among healthy seropositive donors.
Fig. 4.
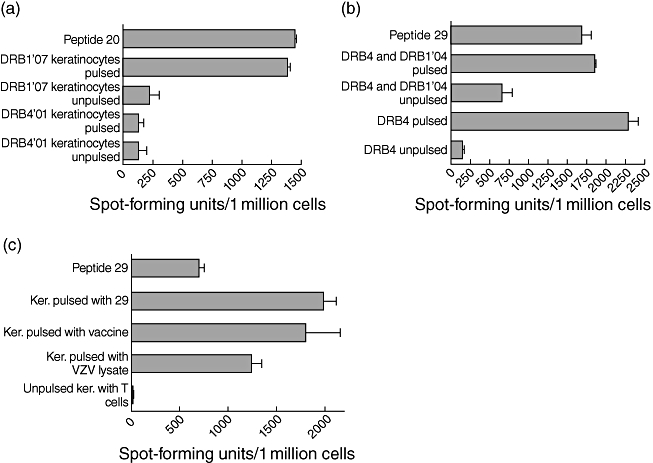
(a) Interferon (IFN)-γ enzyme-linked immunospot (ELISPOT) responses using peptide 20-specific line incubated with peptide directly added to T cells or on DRB4 and DRB1*07-matched or -mismatched and peptide-pulsed or unpulsed keratinocytes. (b) IFN-γ ELISPOT responses using peptide 29-specific line incubated with peptide added directly to T cells or on DRB4 and DRB1*04-matched or -mismatched and peptide-pulsed or -unpulsed Epstein–Barr virus-transformed lymphoblastoid cell lines. (c) IFN-γ ELISPOT responses using a peptide 29-specific line incubated with peptide added directly to T cells or on DRB4*01-matched peptide-pulsed, live varicella zoster virus (VZV) vaccine-pulsed, VZV lysate-pulsed and -unpulsed keratinocytes.
To determine which of the DR molecules presented the epitope within peptide 29, EBV-transformed lymphoblastoid B cell lines, both matched and mismatched, for the presenting HLA molecule were used. Using this method we found that the epitope within peptide 29 was presented by HLA DRB4*01 (Fig. 4b). Truncations of the 20 mer peptide did not enhance the response significantly, and therefore we subsequently used the complete 20 mer in later experiments (IEPGVLKVLRTEKQYLGVYI).
Processing and presentation of gE epitopes by keratinocytes
Keratinocytes are major targets for VZV replication [8,9]. Therefore, we proceeded to investigate if the gE epitopes were processed naturally by keratinocytes and presented to T cells. Following pulsing of DRB4*01- and DRB1*1501-restricted keratinocytes with live VZV vaccine or VZV lysate we used them as APCs to T cell lines specific for gE DRB4*01 peptide 29 (IEPGVLKVLRTEKQYLGVYI) and gE DRB1*1501 peptide 54 (TSPLLRYAAWTGGLA). We found that the keratinocytes were very efficient in presenting the epitopes to specific T cell lines, which suggests that these epitopes were processed naturally by keratinocytes (Fig. 4c shows data for peptide 29).
Analysis of the frequency and phenotype of DRB1*1501 tetramer-specific CD4+ T cells
We initially determined the ex vivo frequency of tetramer positive T cells in VZV-immune healthy immune donors. We also analysed ex vivo tetramer-positive responses in six seropositive non-DRB1*1501 individuals and three seronegative DRB1*1501-positive individuals to exclude any non-specific binding. As shown in Fig. 5a, tetramer-positive CD4+ T cell responses were seen at a relatively low frequency in DRB1*1501 individuals, but such levels are similar to those observed in previous studies of chronic viral infections, such as cytomegalovirus (CMV) and human immunodeficiency virus (HIV). Although two donors (who had never had clinical reactivation) had tetramer-positive responses > 0·03% of CD4+ T cells, the mean ex vivo level of tetramer binding cells was 0·009% ± 0·016 (Fig. 5b). However, DRB1*1501 individuals had significantly higher tetramer-positive T cells when compared with non-DRB1*1501 individuals (P < 0·0001). Using tetramer enrichment strategies, we have shown previously that such frequencies of tetramer-binding cells ex vivo are robust and can be expanded from within the gates used (ViaProbe–, CD14–, CD19–) to generate T cell lines and clones [18,21].
Fig. 5.
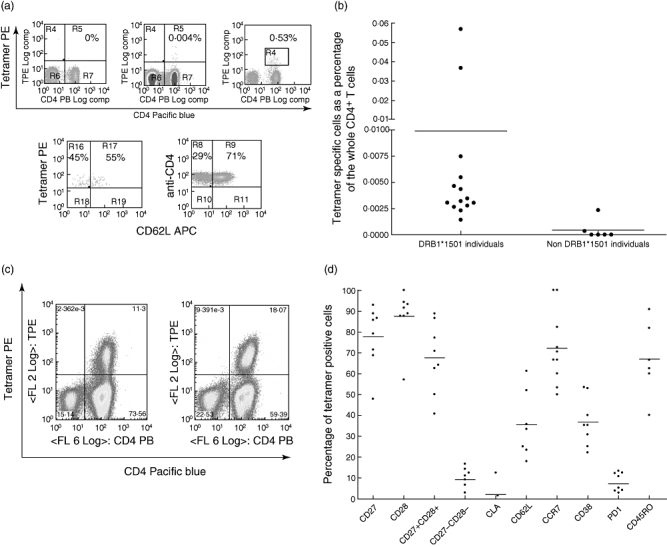
(a) Examples of tetramer staining in a human leucocyte antigen (HLA)-DRB1*1501-negative (upper left panel) individual ex vivo and a HLA-DRB1*1501-positive individual ex vivo (upper middle panel); and after anti-phycoerythrin enrichment (upper right panel), with anti-CD62L phenotyping post-enrichment, gating on the tetramer-positive population (left lower panel) and the total CD4+ population (lower right panel). Cells are gated on CD14-, CD19-, via probe- cells. (b) Percentage of tetramer+ cells (as a proportion of CD4+ T cells) ex vivo in HLA-DRB1*1501-positive and -negative individuals (P < 0·0001). Cells are gated on CD4+ tetramer+ and CD14-, CD19-, via probe- cells. (c) Examples of tetramer staining in HLA-DRB1*1501-positive individuals after short-term culture. Ex vivo frequencies were 0·0026% and 0·057% of CD4+ T cells for the left- and right-hand individuals respectively. Cells are gated on CD14-, CD19-, via probe- cells. (d) Ex vivo percentage of co-stimulatory, activation, skin and lymph node homing marker expression on tetramer-specific T cells. Cells are gated on CD4+ tetramer+ and CD14-, CD19-, via probe- cells. CLA, cutaneous lymphocyte associated antigen; APC, allophycocyanin; PE, phycoerythrin.
Having identified tetramer-positive T cells ex vivo, we then went on to investigate the proliferative capacity of peptide 54-specific T cells. Tetramer-positive T cells were seen at a high frequency (mean 9·3%, ± 10·3) in short-term T cell cultures of PBMCs from DRB1*1501 individuals (Fig. 5c), showing that peptide 54-specific T cells have good proliferative potential.
We next analysed the ex vivo phenotype of tetramer-positive cells of eight VZV-immune individuals (Fig. 5d). The majority of the tetramer-positive cells expressed CD28 (87·53% ± 12·9) and CD27 (77·28% ± 14·3). Although not statistically significant, CD27 expression on tetramer-positive cells was lower than CD28. The expression of CD27 on tetramer-specific T cells was significantly lower (P < 0·05) than expression of this molecule on the whole CD4+ T cell population. However, the expression of CD28 was similar on both the tetramer-specific T cell population (87·5% ± 12·9) and the whole CD4+ T cell population (88·67 ± 15·8). In addition, 67·6% of tetramer-positive cells expressed both CD27 and CD28 while 9·2% of cells did not express either co-stimulatory molecule.
Of the cells ex vivo, 72·23% (s.d. ± 17·4) expressed CCR7 while the expression of CD62L was more variable (mean 35·5%, s.d. ± 15·8) (Fig. 5d). The level of CD45RO expression (52·6 ± 21·08) was similar to that seen on the total CD4+ T cell population. Interestingly, CD62L expression was lower in the individuals who also had low expression of CD27 and CD28, suggesting that in these individuals there was more progression along the putative T cell differentiation pathway. CLA, which is considered to be expressed on T cell homing to the skin, was present only on 2·0% (s.d. ± 4·7) of tetramer-positive cells, suggesting that in healthy immune donors, the gE-specific CD4+ T cells might not provide rapid control of cutaneous antigen. Five of the eight individuals had absent CLA expression in their tetramer-positive CD4 T cells (Fig. 5d). Although the overall expression of CLA on tetramer-positive cells was higher than the total CD4+ T cell population (0·9 ± 0·61), this difference was not significant.
CD38 was expressed in 36·86% (s.d. ± 11·57) tetramer-positive T cells and PD1 on 7·2% (s.d. ± 4·9) tetramer-positive cells ex vivo. PD1 expression was significantly higher in tetramer-positive cells than the CD4+ population (0·51%, s.d. ± 0·4). These data suggest that a proportion of the tetramer-positive cells had recently encountered antigen. Taken with the memory markers described above, the gE-specific CD4+ T cells expressed a mixed central and effector memory phenotype with evidence of recent activation.
Discussion
Our results show that CD4+ T cells specific for gE peptides circulate at high frequencies in healthy VZV immune donors. We mapped novel gE-specific T cell epitopes and used class II DRB1*1501 tetrameric complexes to document the frequency of viral-specific CD4+ T cells, which were found to be similar to antigen-specific CD4+ T cell responses observed with other infections and allergens [22–25].
We have documented previously that IE63-specific CD4+ T cells express mixed early and intermediate levels of differentiation in the context of moderate activation [18]. As IE63 is expressed during latency, we were not able to interpret whether such IE63-specific T cell activation was related to latent expression or to frequent reactivation or re-exposure [26]. In contrast, gE is expressed only during the replicative cycle and therefore the presence of activated T cells with effector memory differentiation would argue in favour of frequent VZV reactivation or re-exposure. Comparison of longitudinal responses to an IE and a late protein in CMV in healthy donors has shown that while T cell responses to the IE protein increased over time, responses to the late protein did not [27]. It will now be important to determine the longitudinal changes in T cell responses to the most abundant IE, early and late proteins during primary infection, latency and reactivation of VZV.
Virus-specific memory T cells vary in the expression of phenotypic markers, which are thought to reflect different functional properties required by these cells in order to control viral replication [17,28,29]. According to this classification, gE-specific T cells appear to be at the early/intermediate stage of differentiation. Although 77·28% and 87·88% of tetramer-positive cells expressed CD27 and CD28, respectively, 9·2% did not express either marker. While 72·23% of the cells expressed CCR7, the expression of CD62L was lower (mean 35·5%), suggesting that while the majority of cells would fit into a central memory phenotype, a significant subset of gE-specific CD4+ T cells showed a phenotype more compatible with effector memory status.
Cutaneous lymphocyte associated antigen is expressed on T cells homing to the skin and is expressed in less than 5% of circulating CD4+ T cells from healthy individuals [25], but in 85% of T cells in inflammatory skin lesions [30]. Interestingly, only 2·0% of gE tetramer-specific T cells expressed CLA. The highest expression of CLA (12·6% of tetramer-positive cells) was seen in the individual who had the highest frequency of tetramer-positive T cells (0·057% of the whole CD4+ T cells). Although CLA expression by tetramer-specific T cells was significantly higher than expression in the total CD4+ T cell population, it was significantly less when compared with expression in herpes simplex virus (HSV) virus-specific T cells. For instance, 50–70% of HSV-specific CD8+ T cells and 15–20% CD4+ T cells [31] have been shown to express CLA. However, CLA expression in these studies were determined following antigen stimulation which could overestimate the expression of CLA by HSV-specific CD4+ T cells, as CLA expression has been shown to be up-regulated with stimulation [32].
On antigen withdrawal, CD38 expression by antigen-specific T cells is lost with a half-life of several weeks [33]. CD38 was expressed by 36·86% of gE tetramer-binding T cells. This is suggestive, therefore, of activation of gE-specific T cells within the preceding few weeks. Overall, the differentiation and activation marker expression would support a mixed central and effector memory pattern with evidence of recent activation in approximately one-third of the gE-specific CD4+ T cells. These would be compatible with ongoing replicative cycle antigen exposure and frequent reactivation or re-exposure. Boosts of glycoprotein-specific antibody titres [34] would also fit with replicative cycle antigen exposure, but the significant expression of CD38 and PD-1 by gE-specific T cells argues that such exposure occurs very frequently within a population not covered by universal vaccination. Indeed, the recent detection of viraemia within 9% of healthy asymptomatic UK blood donors would be compatible with our immunological findings [35].
PD-1 expression was significantly higher among tetramer-positive cells than the whole CD4+ T cell population. PD-1 has been shown to be expressed at moderately high levels in chronic viral infections such as CMV, EBV, HIV and chronic hepatitis C infection, and its expression is thought to be associated with T cell dysfunction because of exhaustion [36–38]. However, it was shown recently that expression of PD-1 on viral-specific CD8+ T cells was related to activation and an earlier stage of differentiation, with its expression having no effect on the functional capacity of HIV-specific CD8+ T cells. These authors showed that CD8+ T cells that were in the early/intermediate stage of differentiation expressed PD-1, while expression was not seen in naive T cells or CD8+ T cells in later stages of differentiation. In addition, activation of T cells was shown further to up-regulate expression of PD-1 [39]. Therefore, the moderately high expression of PD-1 on VZV gE tetramer-specific T cells could be due to recent encounter with the virus rather than exhaustion of tetramer-specific T cells. However, it would be worth investigating the relative expression of this molecule in longitudinal studies of elderly and younger individuals to determine if PD-1 expression was solely a marker of activation and differentiation or if it indeed reflected viral-specific CD4+ T cell dysfunction.
Similarities and differences have been observed for the phenotype of memory T cell responses to different proteins in persistent virus infections. Therefore, we compared the phenotype of gE tetramer-specific T cells with those of IE63-specific T cells. Although the phenotypes of tetramer-specific T cells appeared to be similar overall, there were some differences. For instance, although CD27 and CD28 expression IE63-specific T cells were somewhat similar, IE63-specific T cells appeared to lose CD28 expression before CD27 expression, which was in contrast to what was seen with gE. In addition, the expression of CD45RO was higher in IE63-specific T cells. It will clearly be important to extend these findings to different epitopes within different proteins and to examine longitudinal changes during the course of natural infection.
Glycoprotein E-specific T cell responses in our donors were mediated by CD4+ T cells. This was similar to what was observed by us previously with VZV gI, IE4 [8,9] and IE63 [18], and is also consistent with other reports [40]. Although we expected some CD8+ T cell responses to this protein, similar observations have been made by others, who have shown that T cell immune responses directed at glycoproteins derived from other herpes viruses are predominantly from CD4+ T cells [41]. As with all herpes viruses, VZV viral assembly occurs in the trans-Golgi network. It is believed that, in CMV infection, during viral assembly structural proteins are delivered to the endosomal compartment where class II antigen presentation occurs. Class II presentation of endogenous viral glycoproteins of CMV is thought to be an important mechanism where the host is able to respond to the virus despite MHC class I down-regulation by the virus [42]. Therefore, it is likely that a similar mechanism occurs in the processing of VZV glycoproteins, which would explain the predominance of the VZV gE-specific CD4+ response. Alternatively, autophagy of VZV-infected cells may lead to class II processing and presentation of gE peptides. This has been shown to occur with the EBV nuclear antigen 1, which is a dominant CD4+ T cell target [43].
Varicella zoster virus infections can be prevented largely by the highly effective live attenuated VZV vaccine. However, although rare, it has caused serious illness and even death in some immune-suppressed individuals [44–48]. Furthermore, the vaccine virus establishes latency and may later reactivate, causing severe disease in such individuals [47]. Therefore, it will be important to continue the development of new alternative, safer vaccination strategies for use in immune-suppressed individuals, who are arguably among those with most to gain from protection. We hope that by mapping the first T cell epitopes within gE and characterizing the phenotype of specific T cells in individuals with good clinical control that the data will be of value for such strategies.
Acknowledgments
We are grateful to the Medical Research Council, Commonwealth Commission, Oxford Biomedical Research Centre and Misses Barrie Charitable Trust for their support and to all the donors for their contribution of blood samples.
References
- 1.Levin MJ, Smith JG, Kaufhold RM, et al. Decline in varicella-zoster virus (VZV)-specific cell-mediated immunity with increasing age and boosting with a high-hose VZV vaccine. J Infect Dis. 2003;188:1336–44. doi: 10.1086/379048. [DOI] [PubMed] [Google Scholar]
- 2.Park HB, Kim KC, Park JH, et al. Association of reduced CD4 T cell responses specific to varicella zoster virus with high incidence of herpes zoster in patients with systemic lupus erythematosus. J Rheumatol. 2004;31:2151–5. [PubMed] [Google Scholar]
- 3.Arvin AM, Koropchak CM, Williams BR, et al. Early immune response in healthy and immunocompromised subjects with primary varicella-zoster virus infection. J Infect Dis. 1986;154:422–9. doi: 10.1093/infdis/154.3.422. [DOI] [PubMed] [Google Scholar]
- 4.Diaz PS, Smith S, Hunter E, et al. T lymphocyte cytotoxicity with natural varicella-zoster virus infection and after immunization with live attenuated varicella vaccine. J Immunol. 1989;142:636–41. [PubMed] [Google Scholar]
- 5.Sharp M, Terada K, Wilson A, et al. Kinetics and viral protein specificity of the cytotoxic T lymphocyte response in healthy adults immunized with live attenuated varicella vaccine. J Infect Dis. 1992;165:852–8. doi: 10.1093/infdis/165.5.852. [DOI] [PubMed] [Google Scholar]
- 6.Arvin AM, Sharp M, Smith S, et al. Equivalent recognition of a varicella-zoster virus immediate early protein (IE62) and glycoprotein I by cytotoxic T lymphocytes of either CD4+ or CD8+ phenotype. J Immunol. 1991;146:257–64. [PubMed] [Google Scholar]
- 7.Arvin AM, Sharp M, Moir M, et al. Memory cytotoxic T cell responses to viral tegument and regulatory proteins encoded by open reading frames 4, 10, 29, and 62 of varicella-zoster virus. Viral Immunol. 2002;15:507–16. doi: 10.1089/088282402760312377. [DOI] [PubMed] [Google Scholar]
- 8.Jones L, Black AP, Malavige GN, et al. Persistent high frequencies of varicella-zoster virus ORF4 protein-specific CD4+ T cells after primary infection. J Virol. 2006;80:9772–8. doi: 10.1128/JVI.00564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malavige GN, Jones L, Black AP, et al. Rapid effector function of varicella-zoster virus glycoprotein I-specific CD4+ T cells many decades after primary infection. J Infect Dis. 2007;195:660–4. doi: 10.1086/511274. [DOI] [PubMed] [Google Scholar]
- 10.Abendroth A, Arvin AM. Immune evasion as a pathogenic mechanism of varicella zoster virus. Semin Immunol. 2001;13:27–39. doi: 10.1006/smim.2001.0293. [DOI] [PubMed] [Google Scholar]
- 11.Rahaus M, Wolff MH. Analyses of the transcriptional pattern of glycoproteins E and I of Varicella-zoster virus and evidence for a monocistronic transcription. J Med Virol. 2003;70(Suppl. 1):S51–5. doi: 10.1002/jmv.10321. [DOI] [PubMed] [Google Scholar]
- 12.Mo C, Lee J, Sommer M, et al. The requirement of varicella zoster virus glycoprotein E (gE) for viral replication and effects of glycoprotein I on gE in melanoma cells. Virology. 2002;304:176–86. doi: 10.1006/viro.2002.1556. [DOI] [PubMed] [Google Scholar]
- 13.Berarducci B, Ikoma M, Stamatis S, et al. Essential functions of the unique N-terminal region of the varicella-zoster virus glycoprotein E ectodomain in viral replication and in the pathogenesis of skin infection. J Virol. 2006;80:9481–96. doi: 10.1128/JVI.00533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Ali MA, Cohen JI. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell. 2006;127:305–16. doi: 10.1016/j.cell.2006.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowry PW, Solem S, Watson BN, et al. Immunity in strain 2 guinea-pigs inoculated with vaccinia virus recombinants expressing varicella-zoster virus glycoproteins I, IV, V or the protein product of the immediate early gene 62. J Gen Virol. 1992;73:811–19. doi: 10.1099/0022-1317-73-4-811. [DOI] [PubMed] [Google Scholar]
- 16.Kimura H, Wang Y, Pesnicak L, et al. Recombinant varicella-zoster virus glycoproteins E and I: immunologic responses and clearance of virus in a guinea pig model of chronic uveitis. J Infect Dis. 1998;178:310–17. doi: 10.1086/515638. [DOI] [PubMed] [Google Scholar]
- 17.Appay V, Dunbar PR, Callan M, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–85. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 18.Jones L, Black AP, Malavige GN, Ogg GS. Phenotypic analysis of human CD4+ T cells specific for immediate early protein IE63 of varicella zoster virus. Eur J Immunol. 2007;37:3393–403. doi: 10.1002/eji.200737648. [DOI] [PubMed] [Google Scholar]
- 19.Bunce M, O'Neill CM, Barnardo MC, et al. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP) Tissue Antigens. 1995;46:355–67. doi: 10.1111/j.1399-0039.1995.tb03127.x. [DOI] [PubMed] [Google Scholar]
- 20.Taylor SL, Moffat JF. Replication of varicella-zoster virus in human skin organ culture. J Virol. 2005;79:11501–6. doi: 10.1128/JVI.79.17.11501-11506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ardern-Jones MR, Black AP, Bateman EA, et al. Bacterial superantigen facilitates epithelial presentation of allergen to T helper 2 cells. Proc Natl Acad Sci USA. 2007;104:5557–62. doi: 10.1073/pnas.0700733104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer AL, Trollmo C, Crawford F, et al. Direct enumeration of Borrelia-reactive CD4 T cells ex vivo by using MHC class II tetramers. Proc Natl Acad Sci USA. 2000;97:11433–8. doi: 10.1073/pnas.190335897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day CL, Seth NP, Lucas M, et al. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest. 2003;112:831–42. doi: 10.1172/JCI18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucas M, Day CL, Wyer JR, et al. Ex vivo phenotype and frequency of influenza virus-specific CD4 memory T cells. J Virol. 2004;78:7284–7. doi: 10.1128/JVI.78.13.7284-7287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bateman EA, Ardern-Jones MR, Ogg GS. Persistent central memory phenotype of circulating Fel d 1 peptide/DRB1*0101 tetramer-binding CD4+ T cells. J Allergy Clin Immunol. 2006;118:1350–6. doi: 10.1016/j.jaci.2006.07.040. [DOI] [PubMed] [Google Scholar]
- 26.Cohrs RJ, Gilden DH. Prevalence and abundance of latently transcribed varicella-zoster virus genes in human ganglia. J Virol. 2007;81:2950–6. doi: 10.1128/JVI.02745-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan N, Best D, Bruton R, et al. T cell recognition patterns of immunodominant cytomegalovirus antigens in primary and persistent infection. J Immunol. 2007;178:4455–65. doi: 10.4049/jimmunol.178.7.4455. [DOI] [PubMed] [Google Scholar]
- 28.Appay V, Rowland-Jones SL. Lessons from the study of T-cell differentiation in persistent human virus infection. Semin Immunol. 2004;16:205–12. doi: 10.1016/j.smim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Appay V. The physiological role of cytotoxic CD4(+) T-cells: the holy grail? Clin Exp Immunol. 2004;138:10–13. doi: 10.1111/j.1365-2249.2004.02605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollo P, Marschalko M, Temesvari E, et al. Follow-up analysis of circulating mononuclear cell CLA expression in patients with psoriasis. J Dermatol Sci. 2005;39:131–3. doi: 10.1016/j.jdermsci.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Koelle DM, Gonzalez JC, Johnson AS. Homing in on the cellular immune response to HSV-2 in humans. Am J Reprod Immunol. 2005;53:172–81. doi: 10.1111/j.1600-0897.2005.00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy M, Davis C, Wong J, et al. Cutaneous lymphocyte antigen expression on activated lymphocytes and its association with IL-12R (beta1 and beta2), IL-2Ralpha, and CXCR3. Cell Immunol. 2005;236:131–9. doi: 10.1016/j.cellimm.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 33.Ogg GS, Jin X, Bonhoeffer S, et al. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science. 1998;279:2103–6. doi: 10.1126/science.279.5359.2103. [DOI] [PubMed] [Google Scholar]
- 34.Krause PR, Klinman DM. Varicella vaccination: evidence for frequent reactivation of the vaccine strain in healthy children. Nat Med. 2000;6:451–4. doi: 10.1038/74715. [DOI] [PubMed] [Google Scholar]
- 35.Quinlivan ML, Ayres K, Ran H, et al. Effect of viral load on the outcome of Herpes Zoster. J Clin Microbiol. 2007;45:3909–14. doi: 10.1128/JCM.00874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golden-Mason L, Palmer B, Klarquist J, et al. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–58. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Souza M, Fontenot AP, Mack DG, et al. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J Immunol. 2007;179:1979–87. doi: 10.4049/jimmunol.179.3.1979. [DOI] [PubMed] [Google Scholar]
- 38.Freeman GJ, Wherry EJ, Ahmed R, et al. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med. 2006;203:2223–7. doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sauce D, Almeida JR, Larsen M, et al. PD-1 expression on human CD8 T cells depends on both state of differentiation and activation status. Aids. 2007;21:2005–13. doi: 10.1097/QAD.0b013e3282eee548. [DOI] [PubMed] [Google Scholar]
- 40.Asanuma H, Sharp M, Maecker HT, et al. Frequencies of memory T cells specific for varicella-zoster virus, herpes simplex virus, and cytomegalovirus by intracellular detection of cytokine expression. J Infect Dis. 2000;181:859–66. doi: 10.1086/315347. [DOI] [PubMed] [Google Scholar]
- 41.Elkington R, Shoukry NH, Walker S, et al. Cross-reactive recognition of human and primate cytomegalovirus sequences by human CD4 cytotoxic T lymphocytes specific for glycoprotein B and H. Eur J Immunol. 2004;34:3216–26. doi: 10.1002/eji.200425203. [DOI] [PubMed] [Google Scholar]
- 42.Hegde NR, Dunn C, Lewinsohn DM, et al. Endogenous human cytomegalovirus gB is presented efficiently by MHC class II molecules to CD4+ CTL. J Exp Med. 2005;202:1109–19. doi: 10.1084/jem.20050162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paludan C, Schmid D, Landthaler M, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–6. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 44.Smith JG, Liu X, Kaufhold RM, et al. Development and validation of a gamma interferon ELISPOT assay for quantitation of cellular immune responses to varicella-zoster virus. Clin Diagn Lab Immunol. 2001;8:871–9. doi: 10.1128/CDLI.8.5.871-879.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghaffar F, Carrick K, Rogers BB, et al. Disseminated infection with varicella-zoster virus vaccine strain presenting as hepatitis in a child with adenosine deaminase deficiency. Pediatr Infect Dis J. 2000;19:764–6. doi: 10.1097/00006454-200008000-00022. [DOI] [PubMed] [Google Scholar]
- 46.Levy O, Orange JS, Hibberd P, et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J Infect Dis. 2003;188:948–53. doi: 10.1086/378503. [DOI] [PubMed] [Google Scholar]
- 47.Kramer JM, LaRussa P, Tsai WC, et al. Disseminated vaccine strain varicella as the acquired immunodeficiency syndrome-defining illness in a previously undiagnosed child. Pediatrics. 2001;108:E39. doi: 10.1542/peds.108.2.e39. [DOI] [PubMed] [Google Scholar]
- 48.Kraft JN, Shaw JC. Varicella infection caused by Oka strain vaccine in a heart transplant recipient. Arch Dermatol. 2006;142:943–5. doi: 10.1001/archderm.142.7.943. [DOI] [PubMed] [Google Scholar]