

Abstract
Reactive oxygen species (ROS) are produced mainly during oxidative phosphorylation and by activated phagocytic cells during oxidative burst. The excessive production of ROS can damage lipids, protein, membrane and nucleic acids. They also serve as important intracellular signalling that enhances the inflammatory response. Many studies have demonstrated a role of ROS in the pathogenesis of inflammatory chronic arthropathies, such as rheumatoid arthritis. It is known that ROS can function as a second messenger to activate nuclear factor kappa-B, which orchestrates the expression of a spectrum of genes involved in the inflammatory response. Therefore, an understanding of the complex interactions between these pathways might be useful for the development of novel therapeutic strategies for rheumatoid arthritis.
Keywords: anti-oxidant enzyme, inflammation, oxidative stress, rheumatoid arthritis, ROS
Introduction
Reactive oxygen species (ROS) are produced in cells by several physiological and environmental stimulations, such as infections, ultraviolet radiation and pollutants, known collectively as oxidants. Interestingly, ROS have also been considered as risk and enhancer factors for autoimmune diseases [1], as there is a significant relation between the oxidative stress and such diseases [2].
Rheumatoid arthritis (RA) is an inflammatory systemic and autoimmune disease, characterized by chronic, symmetric and erosive synovitis, mainly of peripheral joints. Most patients present rheumatoid factors, which are autoantibodies directed to the Fc fraction of immunoglobulin G, and antibodies reactive with citrullinated peptides [3,4]. RA has a prevalence of approximately 1% in the world population [5].
Rheumatoid arthritis aetiology is basically unknown, but several studies have implicated a combination of a genetic background and environmental triggers, such as infections and smoking, leading to defects in immunoregulation and a host of inflammatory mechanisms involved in joint tissue damage, including a role for oxidative stress [6].
The definition of oxidative stress as ‘a disturbance in the prooxidant-anti-oxidant balance in favour of the former’[7] implies that disturbance because of pro-oxidant conditions can be corrected by the addition of appropriate anti-oxidants. However, redox mechanisms have been shown to influence intracellular signalling, and cells seem to be very sensitive to the loss of these regulatory and control systems. These two concepts have been incorporated recently into a new definition of oxidative stress as ‘an imbalance between oxidants and anti-oxidants in favour of the oxidants, leading to a disruption of redox signalling and control and/or molecular damage’[8–10].
In this review we explore the role of oxidative stress, ROS and redox signalling in the physiopathology of RA.
Generation of ROS
Reactive oxygen species are produced during normal aerobic cell metabolism, have important physiological roles in maintaining cell redox status and are required for normal cellular functions, including cell proliferation, aggregation, chemotaxis and apoptosis, as well as regulation of intracellular signalling pathways and the activity of transcription factors, such as nuclear factor (NF)-κB, activator protein 1 and hypoxia-inducible factor-1α. ROS produced by phagocytes are critical for the protection against invading microorganisms and also seem to have important physiological roles in priming the immune system [11–13]. The functioning of T lymphocytes is influenced markedly by alterations in the intracellular redox balance. Exposure to ROS has been demonstrated to down-regulate the activity of T lymphocytes; ROS produced by phagocytes also seem to have essential physiological roles in priming the immune system as second messengers [9]. Hitchon and El-Gabalawy propose that the physiological production of ROS by phagocytes in response to antigen affects T cell–antigen interactions and possibly induces apoptosis of autoreactive arthritogenic T cells, thereby preventing autoimmune responses [14].
Phagocytic cells, such as macrophages and neutrophils, are known to be activated under oxidative conditions. This activation is mediated by the oxidase nicotinamide adenine dinucleotide phosphate (NADPH) system and results in a noticeable increment in oxygen consumption and consequent superoxide anion production O2•−) [15]. The activation of oxidase NADPH may be induced by lipopolysaccharides, lipoproteins and cytokines, such as interferon (IFN)-γ, interleukin (IL)-1β and tumour necrosis factor (TNF)-α[14–16] (see Fig. 1).
Fig. 1.
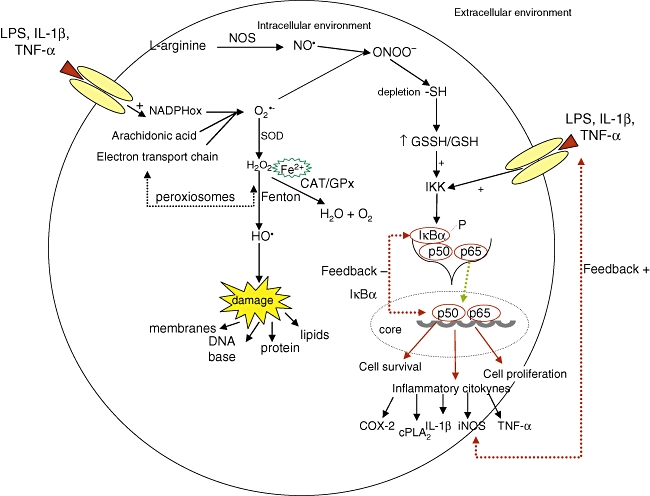
Formation of active oxygen and nitrogen species (upper left corner), targets of these reactive species (lower left corner), relation of reactive oxygen species (ROS) with the activation of NF-κB and transcription of pro-inflammatory cytokines (right). O2•−, superoxide anion radical; H2O2, hydrogen peroxide; HO•, hydroxyl radical; SOD, superoxide dismutase endogenous enzyme; CAT, catalase endogenous enzyme; GPx, glutathione endogenous enzyme; L-arginine, precursory enzyme of nitric oxide; NO, nitric oxide; NOS, nitric oxide synthase; ONOO-, peroxynitrite; -SH, sulphydryl grouping; GSSH/GHS, oxidized/reduced glutathione ratio; IKK, inhibitor kappa kinase; IκBα, inhibitor kappa B; P, phosphorylation; cPLA2, cytosolic phospholipase A2; COX2, cyclooxyganase 2; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharides; TNF-α, tumour necrosis factor alpha; IL-1β, interleukin 1 beta.
In the presence of iron ions (Fe2+) or other transition metals O2•−and hydrogen peroxide (H2O2) are converted, via the Fenton reaction, to highly reactive, aqueous soluble hydroxyl radicals (HO•), that are probably responsible for much of the cell toxicity associated with ROS [11]. As soon as HO• is formed it will react rapidly with the closest molecules, which may be lipids, proteins or DNA bases. It happens because the constant rate of hydroxyl radical reaction is very high if compared with the other reactive species (k > 109 M−1 s−1) [11].
Besides ROS, reactive nitrogen species (RNS), such as the peroxynitrite radical ONOO- generated by the reaction between O2•− and nitric oxide (NO), can also cause oxidative damage [17]. NO is a very reactive inorganic free radical, with a half-life shorter than 10 s because of its rapid oxidation to nitrites [18]. It is produced by the deaimination of L-arginine by NO synthase in the presence of NADPH and O2, producing L-citrulline and NO [17]. The addition of ONOO– to body cells, tissues and fluids leads to fast protonation, which may result in the depletion of –SH groups and other anti-oxidants, oxidation and nitration of lipids, DNA disruption, nitration and deamination of DNA bases (mainly guanine) [11].
Among the oxygen and nitrogen radicals, ONOO- can deplete –SH groupings and consequently change the redox balance in the glutathione towards oxidative stress. This unbalanced glutathione redox status induces, by redox regulation [19], the kappa-B inhibitor (IκB) kinase to phosphorylate IκB, enabling translocation of the transcription factor NF-κB to the nucleus, leading to the transcription of several inflammatory mediators (see Fig. 1).
More recently ROS have been believed to function as second messengers. Typically, second messengers are molecules generated at the time of activation of a receptor, are short-lived and act specifically on effectors to alter their activity transiently. Indeed, ROS and RNS can be generated at the time of receptor activation and are short-lived, as are other second messengers, but the specificity of their action has been more difficult to assess, except for that of NO, which binds specifically to the haem of the regulatory domain of soluble guanylate cyclase, resulting in its activation [8,20].
Oxidant defence mechanisms
Despite important physiological roles, an unbalanced redox status presents potentially destructive effects on cellular biology. For this reason, several enzymatic and non-enzymatic anti-oxidant mechanisms are involved in the protection of cells and organisms in case of eventual damage caused by excessive amounts of such highly reactive mediators [17,21].
Superoxide is converted to H2O2either spontaneously or more rapidly when catalysed by the enzyme superoxide dismutase (SOD) [14]. There are three SOD isoforms: manganese-SOD resides in the mitochondria and is inducible by cytokines through the NF-κB pathway and other co-factors; copper–zinc-SOD is constitutive; and extracellular-SOD (EC-SOD or SOD3) [11].
Glutathione peroxidase (GPx) and catalase (CAT) are responsible for H2O2 degradation [15]. CAT resides in the peroxisome matrix and therefore it can degrade only the H2O2 produced in the matrix and not the H2O2 produced in the peroxisome core. H2O2 produced in the nucleus is transported to cytoplasm by tubules in the nuclear membrane, where GPx will perform the degradation [22]. The H2O2 GPx also degrades other peroxides. This is the first mitochondrial protection from H2O2 and is regulated by p53 and hypoxia [14].
Anti-oxidant mechanisms also have the participation of non-enzymatic anti-oxidants deriving from the diet, which include vitamin E, β-carotene, vitamin C and glutathione; the latter is considered the most important hydrosoluble non-enzymatic anti-oxidant, as it participates in numerous ox-reduction reactions [11,13,21]. Glutathione acts as a co-factor of GPx and other enzymes and is involved in many other metabolic processes, including the metabolism of ascorbate, communication between cells, prevention of oxidation of thiol groups of proteins and radioprotection [11].
Oxidative stress evaluation
Currently, the use of oxidative stress biomarkers can help to explore the relation between oxidative damage to macromolecules (DNA, lipids and proteins) and several diseases. Evaluation, both in vivo and ex vivo, includes measurements of DNA oxidation, lipid peroxidation and protein oxidation [23] (see Table 1).
Table 1.
Biomarkers for oxidative stress status.
| Substrate of damage | Oxidative stress evaluation |
|---|---|
| DNA | 8-hydroxyl-deoxyguanosine (8-OHdG) |
| • The 8-OHdG is the end product of guanine oxidation through hydroxyl radicals [23] | |
| Comet assay | |
| • Very sensitive method for measuring DNA strand breaks in individual cells | |
| • Used in environmental toxicology, cancer research, and radiation biology to assess DNA damage | |
| • Damaged DNA migrates during electrophoresis from the nucleus towards the anode, forming a shape of a ‘comet’ with a head (cell nucleus with intact DNA) and a tail (relaxed and broken DNA) | |
| • The proportion of DNA in the tail is indicative of the frequency of breaks [23,24] | |
| 5-Hydroxyluracil (5-OHUra) | |
| • Presumed to form in DNA via deamination and loss of water from the oxidative DNA lesion cytosine glycol | |
| • A modified pyrimidine, 5-OHUra is a substrate for hNTH1, accounting for an earlier observation of repair of this lesion in human cell extracts [25,26] | |
| Proteins | Protein carbonyls |
| • Conventional assay: colorimetric procedure that involves dinitrophenylhydrazine | |
| • ELISA method: correlates well with the colorimetric assay for quantifying protein carbonyls in plasma samples [27] | |
| Lipids | Thiobarbituric acid-reactive substances (TBA-RS) |
| • Malondialdehyde (MDA) is a secondary product of lipid peroxidation by enzymatic via | |
| • It is a β-rupture byproduct of polyunsaturated fatty acids, such as linoleic, arachidonic and docosahexaenoic acids | |
| • Based on the reaction of TBA with MDA, one of the aldehyde products of lipid peroxidation | |
| • More specific technique for MDA quantification is by high performance liquid chromatography (HPLC), where the particles are separated and only MDA is detected [28] | |
| F2 isoprostanes | |
| • Produced by free radical-related peroxidation of arachidonic acid [16] | |
| • Formed in phospholipids and are then cleaved and released into the circulation before excretion in the urine as free isoprostanes | |
| • Most abundant is 8-isoprostaglandin F2α (8-iso-PGF2α), which has been suggested to be a promising marker for oxidation injury [29] | |
| Reduced/oxidized glutathione (GSH/GSSH) | |
| • Major anti-oxidant in human tissues that provides reducing equivalents for the glutathione peroxidase catalysed reduction of hydrogen peroxide and lipid hydroperoxides to water and the respective alcohol [21] | |
| • During this process GSH becomes oxidized glutathione. The GSSG is then recycled into GSH by gutathione reductase (GR) and NADPH | |
| • Mammalian cells are exposed to increased oxidative stress, the ratio of GSH/GSSG will decrease as a consequence of GSSG accumulation [11] | |
| • Measurement of the GSSG level and determination of the GSH/GSSG ratio are an useful indicators of oxidative stress and can be used to monitor the effectiveness of anti-oxidant intervention strategies [30] |
ELISA, enzyme-linked immunosorbent assay; NADPH, nicotinamide adenine dinucleotide phosphate.
Evidence of oxidative stress in RA
Rheumatoid arthrities is an inflammatory disease characterized by chronic inflammation of the synovial joints associated with proliferation of synovial cells and infiltration of activated immunoinflammatory cells, including memory T cells, macrophages and plasma cells, leading to progressive destruction of cartilage and bone [14,31]. The perpetuation of this inflammatory process is considered to be mediated by a number of cytokines, such as TNF-α, IL-1β, IL-6, IL-8, IL-12, IL-17, IL-18, IL-23 and IFN-γ[32]. Several cytokines, including TNF-α and IL-1β, are known initiators of the NF-κB activation cascade [1] and are under its transcriptional control, constituting a positive feedback loop (see Fig. 1). TNF-α participates positively in the phosphorylation of kinase kappa inhibitor, allowing NF-κB dimers (p50 and p65 portions) to migrate to the nucleus and then bind to promoters of proinflammatory genes [33] and stimulate the oxidase NADPH activation (see Fig. 2). Increased cytokine production driven by NF-κB can enhance expression of vascular adhesion molecules that attract leucocytes into the joint, as well as matrix metalloproteinases that help to degrade the extracellular matrix.
Fig. 2.
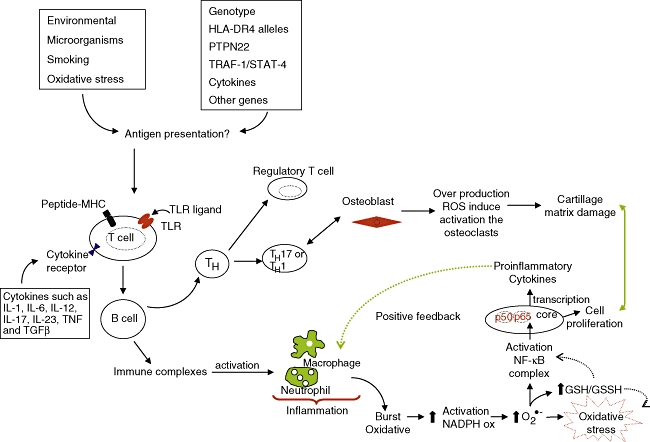
An overview of the signalling T-cells and oxidative stress. See text for details.
Another central feature of RA synovitis is the transformation of fibroblast-like synovial cells into autonomously proliferating cells with a tissue-infiltrating nature, forming hyperplastic tissue with potential for bone erosion and cartilage degradation known as pannus. These cells can proliferate in an anchorage-independent manner, lack contact inhibition and express several oncogenes characteristic of cells that have escaped normal growth-regulatory mechanisms [34]. Because NF-κB activation can induce gene expression of cell growth-promoting factors such as cyclin D1 and c-Myc and physiological inhibitors of apoptosis such as cIAPs, Bcl-X and cFLIP, it can also have a role in RA synovial hyperproliferation, indicating that it is a crucial determinant to the disease pathogenesis [35].
There is a great deal of evidence of the important role of oxidative stress in RA physiopathology.
Several groups have demonstrated increased oxidative enzyme activity, along with decreased anti-oxidant levels in RA sera and synovial fluids (SF). Studies with SF and tissues in RA have demonstrated oxidative damage of hyaluronic acid [36], lipoperoxidation products [37], oxidation of low-density lipoproteins [38] and carbonyl increment by protein oxidation [39]. Evidence of oxidative damage in cartilage, extracellular collagen and DNA has also been reported. Peripheral blood lymphocyte DNA from RA patients contains significantly elevated levels of the promutagenic 8-oxohydrodeoxyguanosine [40]. Moreover, the levels of thioredoxine, a cellular reducing catalyst known to participate in the redox regulation of cellular proteins by reducing the redox-active cysteines through reversible oxidation of the active centre dithiol to disulphide, are higher in SF of RA patients [41]. The production of NO is also up-regulated in RA synovial tissue; there are also increased levels of nitrite in the SF, indicating an increased local production [42].
Jikimoto et al. [43] reported a correlation between the disease activity and the presence of oxidative stress in patients with RA. Other studies have found non-significant correlations [3]. It seems that the strongest correlation is between DNA damage and the oxidative stress index,i.e. the relation between the levels of oxidation and anti-oxidant capacity {OSI = [(TOS, μmol/l)/(TAS, mmol trolox equivalent/l]} × 100 [3].
Epidemiological studies have shown that RA occurs in previously healthy subjects who have low levels of circulating anti-oxidants [44], implying a pathogenic role of increased oxidative stress in the development of RA. Patients with RA have been reported to have lower serum levels of a variety of anti-oxidants, including vitamin E, vitamin C, β-carotene, selenium and zinc, in comparison with controls [45], but these associations may be a result of the disease or its treatment [46]. In two nested case–control studies using serum obtained prior to diagnosis of RA, selenium, α-tocopherol, β-carotene and an overall anti-oxidant index were associated inversely with the later development of RA [47]. In a case–control study assessing dietary intake using a food frequency questionnaire, intake of vitamin C, but not intake of vitamin E, was associated weakly and inversely with RA [48].
Several factors could be involved in the generation of oxidative stress in the joints of RA patients. Intra-articular pressure is much higher in RA joints, probably because of decreased compliance of the joint wall because of synovial membrane swelling and fibrosis of the capsule [49]. Together with the reduced capillary density and elevated tissular metabolic rate, this elevated pressure could decrease capillary flow rates and induce a repetitive ischaemia–reperfusion injury in the joint. In fact, SF pO2levels are frequently below those detected in venous blood [14]. Tissue injury could release iron and copper ions, which are catalytic for free radical reactions. Recently, neutrophils from the SF of RA, but not from other arthritides, were found to be activated and producing ROS intracellularly in RA, probably as a result of active processing of endocytosed material [50].
Environmental factors appear to have a role in the induction, magnitude and rate of progression of the disease, and could also be involved in the generation of oxidative stress. Recent data implicate smoking strongly as an important environmental risk factor for development of disease in human leucocyte antigen D-related 4-positive individuals [51]. Smoking is a well-known source of ROS. Tobacco smoke presents several organic compounds, among which are the quinones, source of the semiquinone radical, which can generate O2- and H2O2[52].
Free radicals have also been considered as mediators of tissue damage in RA, in conjunction with proinflammatory cytokines. The role of ROS and, in particular, of O2- in the degradation of cartilage and bone is not unexpected. Cartilage is the only protein known to be fragmented by the superoxide anion, and SOD inhibits this degradation strongly. ROS degrade SF and depolymerize hyaluronic acid, which leads to a loss of viscosity in the joint, inactivation of anti-proteinases and induction of bone resorption [53]. O2- is produced by osteoclasts during bone resorption, and this formation occurs at the osteoclast–bone surface interface [54]. These effects are amplified several-fold in the presence of cytokines such as IL-1β[55]. Experimentally, it has been verified that excessive production of ROS may lead to an accelerated damage to joint cartilage and osteoclast activation [56,57] (see Fig. 2).
Studies in SF and tissue have demonstrated oxidative damage in hyaluronic acid, which has been shown to induce T cell hyporesponsiveness in RA through effects on proteins and proteosomal degradation with significant decrease of the intracellular reduced glutathione (GSH) levels, has also been correlated with the hyporesponsive state of these cells [58].
As well as the well-established damage to the lipid bilayer caused by free radicals, DNA damage, which is an important target to oxidative injuries, has also been investigated. Studies evaluating the DNA damage through the comet test in RA patients have demonstrated elevated damage levels, which were related to increased oxidative stress and decreased total anti-oxidant capability [3]. ROS-induced genotoxic events have also been linked to mutations of the tumour suppressor gene p53 observed in RA-derived fibroblast-like synoviocytes, which could explain, at least in part, the transformed phenotype of these cells and their inadequate apoptosis [34].
In addition to active oxygen species, active nitrogen species have also been investigated in RA. This link occurs because of the participation of RNS in the activation of NF-κB, as the formation of peroxynitrite interferes in the redox balance of glutathione. Studies indicate that RNS donors caused NF-κB activation and increased activation of proteolytic systems [19,59] (Fig. 1). A positive effect of thioredoxin in NF-κB activation has also been suggested [60], as this transcription factor must be in a reduced state to bind to the κB DNA sequence of the target genes. Therefore, it is very likely that ROS are involved importantly in regulation of the NF-κB signalling.
Some observations derived from manipulation of the oxidative stress in animal models have also pointed to a role of ROS in RA pathogenesis. Administration of vitamin E prevented articular destruction in an animal model of RA, but vitamin E did not change the inflammatory components of the disease (including TNF-α level and arthritis index score) or the oxidation status of the animals [61].
Superoxide dismutase extracellular (SOD3) exert protective effects in animal models of ischaemia and inflammation [14]. In mice that are genetically deficient in SOD3, both the severity of collagen-induced arthritis (CIA) and the production of proinflammatory cytokines are increased. SOD3 gene transfer via the subcutaneous route or into the knee decreases the severity of experimental arthritis in rodents [62,63].
Recently, it has demonstrated that M40403, a new SOD mimetic (SODm) that removes O2- catalytically as effectively as the native enzyme, exerts a beneficial effect in the type II collagen (CII)-CIA, which suggests the possible use of an SODm as a disease-modifying therapeutic agent in chronic diseases such as RA [64].
Studies have shown that the use of alpha-lipoic acid (LA) − a co-factor for mitochondrial α-keto dehydrogenase complexes and which participates in S–O transfer reactions − can attenuate the development of CIA in mice. Lee et al. evaluated clinical, histological and biochemical parameters in mice with arthritis induced by bovine CII [65]. Amelioration of joint disease by LA was associated with reduction in oxidative stress, as well as inhibition of inflammatory cytokine activation and NF-κB DNA binding activity. Moreover, LA inhibited bone destruction in vivo and osteoclastogenesis in vitro[65].
Rheumatoid arthritis pharmacological treatments and oxidative stress
Today, methotrexate, a folate antagonist developed initially to treat malignant neoplasias, is the first-choice drug for RA treatment. In RA, the doses utilized are much lower than oncological doses, and it is not believed that its efficacy in disease control is related to this anti-proliferative action. Other mechanisms have been proposed, including the synthesis inhibition of toxic compounds spermine and spermidine and the extracellular accumulation of adenosine, which has a known anti-inflammatory action mediated by the adenosine receptors [66]. In addition, it has already been demonstrated that methotrexate can suppress directly or indirectly the generation of active oxygen metabolites induced by IL-6, which in turn is produced after stimulation with TNF-α in synovial cells of RA [67], as well as in polymorphonuclear cells [68]. However, studies suggest that low doses of methotrexate induce more accentuated ROS-mediated apoptosis in lineages of lymphocyte T cells than in monocytes [69].
More recently, biological agents (monoclonal antibodies or recombinant proteins) with antagonist action of TNF-α have been shown to be efficacious in the control of phlogistic signs and radiological progression of RA. These agents do not seem to act directly on the production of oxygen radicals, but lead to inhibition of the activation and chemotaxis of neutrophils to the synovial tissue, with consequent reduced generation of such radicals [70].
Studies with TNF-α inhibitors etanercept and infliximab have demonstrated a reduction of oxidative stress markers in patients with RA. This study evaluated 22 patients with RA, besides the oxidative stress parameters, as well as laboratory and clinical parameters. This study demonstrated that etanercept acts as a regulator against pentosidine formation, oxidative DNA damage and lipid peroxidation in RA patients [71].
Recent epidemiological studies have shown an inverse association between dietary intake of anti-oxidants and RA incidence, analysed through a standardized questionnaire including demographic data, reproduction and medical history, utilization of hormonal therapy, smoking and other lifestyle factors. Such remarks strengthen the hypothesis that a balanced diet and anti-oxidant supplementation may protect against disease development or aggravation, as patients with RA have reported lower levels of anti-oxidants, including vitamin C, vitamin E, β-carotene, selenium and zinc compared with control patients [44,45,72].
Conclusion
Chronic inflammatory arthropathies, such as RA, are characterized by a complex progressive and chronic inflammatory process which involves numerous factors of transcription and molecular signallers. Oxygen metabolism has an important role in the pathogenesis of RA.
Several studies have observed that there is a relation between the oxidative damage caused to lipids, proteins and DNA and the maintenance of the inflammatory process in RA, as well as the inhibiting effects of medications with anti-rheumatic action on oxidative processes. However, the nature of this relation, notably whether it represents a direct or indirect effect, has not been well determined and requires additional studies. This information has important potential, as it enables understanding of the action mechanisms of current therapies and in particular the development of new therapeutic strategies.
Acknowledgments
This study was supported by Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior and Fundo de Incentivo a Pesquisa e Eventos.
References
- 1.Okamoto T. Oxidative stress in rheumatoid arthritis. In: Surh Y-J, Packer L, editors. Oxidative stress, inflammation and health. Califórnia, CA: Taylor & Francis; 2005. pp. 245–70. [Google Scholar]
- 2.Avalos I, Chung CP, Oeser A, et al. Oxidative stress in systemic lupus erythematosus: relationship to disease activity and symptoms. Lupus. 2007;16:195–200. doi: 10.1177/0961203306075802. [DOI] [PubMed] [Google Scholar]
- 3.Altindag O, Karakoc M, Kocyigit A, Celik H, Soran N. Increased DNA damage and oxidative stress in patients with rheumatoid arthritis. Clin Biochem. 2007;40:167–71. doi: 10.1016/j.clinbiochem.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Kinne RW, Brauer R, Stuhlmuller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Res. 2000;2:189–202. doi: 10.1186/ar86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senna ER, De Barros AL, Silva EO, et al. Prevalence of rheumatic diseases in Brazil: a study using the COPCORD approach. J Rheumatol. 2004;31:594–7. [PubMed] [Google Scholar]
- 6.Ozkan Y, Yardym-Akaydyn S, Sepici A, Keskin E, Sepici V, Simsek B. Oxidative status in rheumatoid arthritis. Clin Rheumatol. 2007;26:64–8. doi: 10.1007/s10067-006-0244-z. [DOI] [PubMed] [Google Scholar]
- 7.Sies H. Oxidative Stress: Introductory Remarks. London: Academic Press; 1985. [Google Scholar]
- 8.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–56. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 9.Jones DP. Disruption of mitochondrial redox circuitry in oxidative stress. Chem Biol Interact. 2006;163:38–53. doi: 10.1016/j.cbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Roehrs R, Guecheva TN, Saffi J, Henriques JAP. Redox sensitive targets in signaling cascades. In: ULBRA, editor. Free radicals and the cellular response to the oxidative stress. Universidade Luterana do Brasil: Canoas; 2004. pp. 161–84. [Google Scholar]
- 11.Barry Halliwell JG. Free radicals in biology and medicine. 4. New York: Oxford University Press; 2007. [Google Scholar]
- 12.Griffiths HR. ROS as signalling molecules in T cells − evidence for abnormal redox signalling in the autoimmune disease, rheumatoid arthritis. Redox Rep. 2005;10:273–80. doi: 10.1179/135100005X83680. [DOI] [PubMed] [Google Scholar]
- 13.Oktyabrsky ON, Smirnova GV. Redox regulation of cellular functions. Biochemistry (Mosc) 2007;72:132–45. doi: 10.1134/s0006297907020022. [DOI] [PubMed] [Google Scholar]
- 14.Hitchon CA, El-Gabalawy HS. Oxidation in rheumatoid arthritis. Arthritis Res Ther. 2004;6:265–78. doi: 10.1186/ar1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 16.Curi R. In: PIHdB Jr. Arachdonic acid metabolism in sepsis. Manole, editor. São Paulo: SEPSE; 2007. [Google Scholar]
- 17.Soneja A, Drews M, Malinski T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol Rep. 2005;57(Suppl.):108–19. [PubMed] [Google Scholar]
- 18.Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–37. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 19.Bar-Shai M, Reznick AZ. Reactive nitrogen species induce nuclear factor-kappaB-mediated protein degradation in skeletal muscle cells. Free Radic Biol Med. 2006;40:2112–25. doi: 10.1016/j.freeradbiomed.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Ignarro LJ. Heme-dependent activation of soluble guanylate cyclase by nitric oxide: regulation of enzyme activity by porphyrins and metalloporphyrins. Semin Hematol. 1989;26:63–76. [PubMed] [Google Scholar]
- 21.Blair IA. Endogenous glutathione adducts. Curr Drug Metab. 2006;7:853–72. doi: 10.2174/138920006779010601. [DOI] [PubMed] [Google Scholar]
- 22.Fritz R, Bol J, Hebling U, et al. Compartment-dependent management of H(2)O(2) by peroxisomes. Free Radic Biol Med. 2007;42:1119–29. doi: 10.1016/j.freeradbiomed.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Hwang ES, Kim GH. Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research. Toxicology. 2007;229:1–10. doi: 10.1016/j.tox.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Aukrust P, Luna L, Ueland T, et al. Impaired base excision repair and accumulation of oxidative base lesions in CD4+ T cells of HIV-infected patients. Blood. 2005;105:4730–5. doi: 10.1182/blood-2004-11-4272. [DOI] [PubMed] [Google Scholar]
- 25.Asagoshi K, Odawara H, Nakano H, et al. Comparison of substrate specificities of Escherichia coli endonuclease III and its mouse homologue (mNTH1) using defined oligonucleotide substrates. Biochemistry. 2000;39:11389–98. doi: 10.1021/bi000422l. [DOI] [PubMed] [Google Scholar]
- 26.Yang N, Galick H, Wallace SS. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst) 2004;3:1323–34. doi: 10.1016/j.dnarep.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Winterbourn CC, Buss IH. Protein carbonyl measurement by enzyme-linked immunosorbent assay. Methods Enzymol. 1999;300:106–11. doi: 10.1016/s0076-6879(99)00118-4. [DOI] [PubMed] [Google Scholar]
- 28.Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis. 2005;15:316–28. doi: 10.1016/j.numecd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Wong YT, Ruan R, Tay FE. Relationship between levels of oxidative DNA damage, lipid peroxidation and mitochondrial membrane potential in young and old F344 rats. Free Radic Res. 2006;40:393–402. doi: 10.1080/10715760600556074. [DOI] [PubMed] [Google Scholar]
- 30.Hassan MQ, Hadi RA, Al-Rawi ZS, Padron VA, Stohs SJ. The glutathione defense system in the pathogenesis of rheumatoid arthritis. J Appl Toxicol. 2001;21:69–73. doi: 10.1002/jat.736. [DOI] [PubMed] [Google Scholar]
- 31.Andreoli TE, Bennett JC, Carpenter CCJ, Plum F. Medicina interna básica. Livro. 1998;1:568–70. [Google Scholar]
- 32.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–42. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 33.Moynagh PN. The NF-kappaB pathway. J Cell Sci. 2005;118:4589–92. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- 34.Tak PP, Zvaifler NJ, Green DR, Firestein GS. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21:78–82. doi: 10.1016/s0167-5699(99)01552-2. [DOI] [PubMed] [Google Scholar]
- 35.Okamoto T, Sakurada S, Yang JP, Merin JP. Regulation of NF-kappa B and disease control: identification of a novel serine kinase and thioredoxin as effectors for signal transduction pathway for NF-kappa B activation. Curr Top Cell Regul. 1997;35:149–61. doi: 10.1016/s0070-2137(97)80006-4. [DOI] [PubMed] [Google Scholar]
- 36.Grootveld M, Henderson EB, Farrell A, Blake DR, Parkes HG, Haycock P. Oxidative damage to hyaluronate and glucose in synovial fluid during exercise of the inflamed rheumatoid joint. Detection of abnormal low-molecular-mass metabolites by proton-n.m.r. spectroscopy. Biochem J. 1991;273:459–67. doi: 10.1042/bj2730459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taysi S, Polat F, Gul M, Sari RA, Bakan E. Lipid peroxidation, some extracellular anti-oxidants, and anti-oxidant enzymes in serum of patients with rheumatoid arthritis. Rheumatol Int. 2002;21:200–4. doi: 10.1007/s00296-001-0163-x. [DOI] [PubMed] [Google Scholar]
- 38.Dai L, Lamb DJ, Leake DS, et al. Evidence for oxidised low density lipoprotein in synovial fluid from rheumatoid arthritis patients. Free Radic Res. 2000;32:479–86. doi: 10.1080/10715760000300481. [DOI] [PubMed] [Google Scholar]
- 39.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 40.Bashir S, Harris G, Denman MA, Blake DR, Winyard PG. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann Rheum Dis. 1993;52:659–66. doi: 10.1136/ard.52.9.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maurice MM, Nakamura H, Gringhuis S, et al. Expression of the thioredoxin–thioredoxin reductase system in the inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:2430–9. doi: 10.1002/1529-0131(199911)42:11<2430::AID-ANR22>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 42.McInnes IB, Leung BP, Field M, et al. Production of nitric oxide in the synovial membrane of rheumatoid and osteoarthritis patients. J Exp Med. 1996;184:1519–24. doi: 10.1084/jem.184.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jikimoto T, Nishikubo Y, Koshiba M, et al. Thioredoxin as a biomarker for oxidative stress in patients with rheumatoid arthritis. Mol Immunol. 2002;38:765–72. doi: 10.1016/s0161-5890(01)00113-4. [DOI] [PubMed] [Google Scholar]
- 44.Cerhan JR, Saag KG, Merlino LA, Mikuls TR, Criswell LA. Anti-oxidant micronutrients and risk of rheumatoid arthritis in a cohort of older women. Am J Epidemiol. 2003;157:345–54. doi: 10.1093/aje/kwf205. [DOI] [PubMed] [Google Scholar]
- 45.Hagfors L, Leanderson P, Skoldstam L, Andersson J, Johansson G. Anti-oxidant intake, plasma anti-oxidants and oxidative stress in a randomized, controlled, parallel, Mediterranean dietary intervention study on patients with rheumatoid arthritis. Nutr J. 2003;2:5. doi: 10.1186/1475-2891-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aaseth J, Haugen M, Forre O. Rheumatoid arthritis and metal compounds − perspectives on the role of oxygen radical detoxification. Analyst. 1998;123:3–6. doi: 10.1039/a704840h. [DOI] [PubMed] [Google Scholar]
- 47.Comstock GW, Burke AE, Hoffman SC, et al. Serum concentrations of alpha tocopherol, beta carotene, and retinol preceding the diagnosis of rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. 1997;56:323–5. doi: 10.1136/ard.56.5.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shapiro JA, Koepsell TD, Voigt LF, Dugowson CE, Kestin M, Nelson JL. Diet and rheumatoid arthritis in women: a possible protective effect of fish consumption. Epidemiology. 1996;7:256–63. doi: 10.1097/00001648-199605000-00007. [DOI] [PubMed] [Google Scholar]
- 49.Jayson MI, Dixon AS. Intra-articular pressure in rheumatoid arthritis of the knee. 3. Pressure changes during joint use. Ann Rheum Dis. 1970;29:401–8. doi: 10.1136/ard.29.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cedergren J, Forslund T, Sundqvist T, Skogh T. Intracellular oxidative activation in synovial fluid neutrophils from patients with rheumatoid arthritis but not from other arthritis patients. J Rheumatol. 2007;34:2162–70. [PubMed] [Google Scholar]
- 51.Klareskog L, Padyukov L, Alfredsson L. Smoking as a trigger for inflammatory rheumatic diseases. Curr Opin Rheumatol. 2007;19:49–54. doi: 10.1097/BOR.0b013e32801127c8. [DOI] [PubMed] [Google Scholar]
- 52.Garcez M, Bordin D, Peres W, Salvador M. Free radicals and reactive species. In: Ulbra, editor. Free radicals and the cellular response to the oxidative stress. Canoas: Porto Alegre; 2004. pp. 13–34. [Google Scholar]
- 53.McCord JM. Free radicals and inflammation: protection of synovial fluid by superoxide dismutase. Science. 1974;185:529–31. doi: 10.1126/science.185.4150.529. [DOI] [PubMed] [Google Scholar]
- 54.Henrotin YE, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003;11:747–55. doi: 10.1016/s1063-4584(03)00150-x. [DOI] [PubMed] [Google Scholar]
- 55.Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest. 1990;85:632–9. doi: 10.1172/JCI114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldring SR. Pathogenesis of bone erosions in rheumatoid arthritis. Curr Opin Rheumatol. 2002;14:406–10. doi: 10.1097/00002281-200207000-00013. [DOI] [PubMed] [Google Scholar]
- 57.Miossec P. An update on the cytokine network in rheumatoid arthritis. Curr Opin Rheumatol. 2004;16:218–22. doi: 10.1097/00002281-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 58.Gringhuis SI, Leow A, Papendrecht-Van Der Voort EA, Remans PH, Breedveld FC, Verweij CL. Displacement of linker for activation of T cells from the plasma membrane due to redox balance alterations results in hyporesponsiveness of synovial fluid T lymphocytes in rheumatoid arthritis. J Immunol. 2000;164:2170–9. doi: 10.4049/jimmunol.164.4.2170. [DOI] [PubMed] [Google Scholar]
- 59.Bar-Shai M, Reznick AZ. Peroxynitrite induces an alternative NF-kappaB activation pathway in L8 rat myoblasts. Anti-oxid Redox Signal. 2006;8:639–52. doi: 10.1089/ars.2006.8.639. [DOI] [PubMed] [Google Scholar]
- 60.Sakurada S, Kato T, Okamoto T. Induction of cytokines and ICAM-1 by proinflammatory cytokines in primary rheumatoid synovial fibroblasts and inhibition by N-acetyl-L-cysteine and aspirin. Int Immunol. 1996;8:1483–93. doi: 10.1093/intimm/8.10.1483. [DOI] [PubMed] [Google Scholar]
- 61.De Bandt M, Grossin M, Driss F, Pincemail J, Babin-Chevaye C, Pasquier C. Vitamin E uncouples joint destruction and clinical inflammation in a transgenic mouse model of rheumatoid arthritis. Arthritis Rheum. 2002;46:522–32. doi: 10.1002/art.10085. [DOI] [PubMed] [Google Scholar]
- 62.Dai L, Claxson A, Marklund SL, et al. Amelioration of antigen-induced arthritis in rats by transfer of extracellular superoxide dismutase and catalase genes. Gene Ther. 2003;10:550–8. doi: 10.1038/sj.gt.3301916. [DOI] [PubMed] [Google Scholar]
- 63.Iyama S, Okamoto T, Sato T, et al. Treatment of murine collagen-induced arthritis by ex vivo extracellular superoxide dismutase gene transfer. Arthritis Rheum. 2001;44:2160–7. doi: 10.1002/1529-0131(200109)44:9<2160::aid-art369>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 64.Salvemini D, Mazzon E, Dugo L, et al. Amelioration of joint disease in a rat model of collagen-induced arthritis by M40403, a superoxide dismutase mimetic. Arthritis Rheum. 2001;44:2909–21. doi: 10.1002/1529-0131(200112)44:12<2909::aid-art479>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 65.Lee EY, Lee CK, Lee KU, et al. Alpha-lipoic acid suppresses the development of collagen-induced arthritis and protects against bone destruction in mice. Rheumatol Int. 2007;27:225–33. doi: 10.1007/s00296-006-0193-5. [DOI] [PubMed] [Google Scholar]
- 66.Cronstein BN. Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev. 2005;57:163–72. doi: 10.1124/pr.57.2.3. [DOI] [PubMed] [Google Scholar]
- 67.Sung JY, Hong JH, Kang HS, et al. Methotrexate suppresses the interleukin-6 induced generation of reactive oxygen species in the synoviocytes of rheumatoid arthritis. Immunopharmacology. 2000;47:35–44. doi: 10.1016/s0162-3109(99)00185-x. [DOI] [PubMed] [Google Scholar]
- 68.Laurindo IM, Mello SB, Cossermelli W. Influence of low doses of methotrexate on superoxide anion production by polymorphonuclear leukocytes from patients with rheumatoid arthritis. J Rheumatol. 1995;22:633–8. [PubMed] [Google Scholar]
- 69.Herman S, Zurgil N, Deutsch M. Low dose methotrexate induces apoptosis with reactive oxygen species involvement in T lymphocytic cell lines to a greater extent than in monocytic lines. Inflamm Res. 2005;54:273–80. doi: 10.1007/s00011-005-1355-8. [DOI] [PubMed] [Google Scholar]
- 70.den Broeder AA, Wanten GJ, Oyen WJ, Naber T, van Riel PL, Barrera P. Neutrophil migration and production of reactive oxygen species during treatment with a fully human anti-tumor necrosis factor-alpha monoclonal antibody in patients with rheumatoid arthritis. J Rheumatol. 2003;30:232–7. [PubMed] [Google Scholar]
- 71.Kageyama Y, Takahashi M, Nagafusa T, Torikai E, Nagano A. Etanercept reduces the oxidative stress marker levels in patients with rheumatoid arthritis. Rheumatol Int. 2007;28:245–51. doi: 10.1007/s00296-007-0419-1. [DOI] [PubMed] [Google Scholar]
- 72.Mangge H, Hermann J, Schauenstein K. Diet and rheumatoid arthritis − a review. Scand J Rheumatol. 1999;28:201–9. doi: 10.1080/03009749950155553. [DOI] [PubMed] [Google Scholar]