

Abstract
Pemphigus vulgaris (PV) is an autoimmune blistering disease that affects the skin and multiple mucous membranes, and is caused by antibodies to desmoglein (Dsg) 1 and 3. Natural killer (NK) cells have a role in autoimmunity, but their role in PV is not known. NK cells in the peripheral blood leucocytes (PBL) of 15 untreated Caucasian patients with active PV were studied and compared with healthy controls for the expression of major histocompatibility complex (MHC) class II and co-stimulatory molecules. CD56+ CD16- CD3- NK or CD56+ CD16+ CD3- NK cells from the PBL of PV patients co-express MHC class II and co-stimulatory molecule B7-H3 without exogenous stimulation. CD4+ T cells from the PBL and perilesional skin of PV patients were co-cultured with CD56+ CD3- NK cells from the PBL of the same patients; in the presence of Dsg3 peptides underwent statistically significant proliferation, indicating that NK cells functioned as antigen-presenting cells. Supernatants from these co-cultures and serum of the same patients with active PV had statistically significantly elevated levels of interleukin (IL)-6, IL-8 and interferon-γ, compared with controls indicating that the NK cells stimulated CD4+ T cells to produce proinflammatory cytokines. In these experiments, we present preliminary evidence that NK cells may play a role in the pathobiology of PV.
Keywords: B cell, MHC class II, natural killer cells, T lymphocyte
Introduction
Pemphigus vulgaris (PV), is an autoimmune blistering disease which affects the skin and multiple mucous membranes [1,2]. It is mediated by autoantobodies to desmoglein (Dsg) 1 and 3 [3,4]. The transplacental transfer of maternal autoantibodies causes transient blisters in newborns [5]. Sera of PV patients containing these autoantibodies produce blisters when injected into neonatal mice [4]. Hence, it is considered an autoantibody-mediated disease.
The binding of antidesmoglein antibody to Dsg results in biochemical and cellular processes that lead to acantholysis, which manifests as an intraepidermal vesicle or blister [1]. Investigators have shown that CD4+ T cells, in the presence of antigen-presenting cells (APC), undergo stimulation by Dsg3 and/or Dsg3 peptides [6,7], indicating a role for T cells in autoantibody production.
Natural killer (NK) cells are characterized phenotypically as CD56+ CD3–. The majority of NK cells (∼90%) express CD56dim and CD16+ and are known to mediate antibody-dependent cellular cytotoxicity [8,9]. They also express CXCR1, CX3CR1 chemokine receptors and are recruited to sites of inflammation [10]. CD56dim NK express a wider variety of inhibitory NK cell receptors (NKR) and lacks L-selectin and CCR7 [11,12]. The minority of NK cells (∼10%) are phenotypically CD56bright CD16-[10]. CD56bright express the interleukin (IL)-2 receptor and secrete high levels of interferon (IFN)-γ[13], homing molecules such as l-selectin, CXCR3 and CCR7 [11,12] and NKR [14], and have a greater role in cytokine production [13,15].
In this study we report the identification and some of the functional characterizations of both subsets of NK cells in the peripheral blood and cutaneous lesions of patients with PV. These subsets of NK cells expressed major histocompatibility complex (MHC) class II molecules and the co-stimulatory molecule B7-H3. When CD4+ T cells from the peripheral blood and perilesional skin of PV patients are co-cultured with these NK cells in the presence of Dsg3 peptides, cytokines and chemokines are detected in the supernatants. A similar profile of cytokines and chemokines is observed in the sera of PV patients with active disease. In this paper we study the hypothesis that the cellular and molecular interaction of the combination of NK, CD4+ T cells and Dsg1 and 3 influences the microenvironment of the skin and mucosal tissues during the preclinical stage. These cells, in concert, then influence the bone marrow, spleen or lymph nodes, where these responses may be amplified many-fold. Thus these events may be the precursors to the clinical manifestations of PV and may play a role in the subsequent clinical stages.
Materials and methods
Patients
Fifteen Caucasian patients presented in this study were treated at the Center for Blistering Diseases, Boston, MA. The ages ranged from 50 to 70 years (mean 57 years). The diagnosis of PV was based on clinical profile, an intraepidermal vesicle with suprabasilar acantholysis on the histology and deposition of immunoglobulin G (IgG) on the keratinocyte cell surface on direct immunofluorescence. All patients had severe disease. Sera of all the patients had high, but variable, titres of IgG4 antibodies to keratinocyte cell surface using monkey oesophagus as substrate [16] (Table 1). Patients with severe and moderately severe disease were chosen because it was felt that if changes in NK cells were present, they would most probably be seen in such disease severity. The 15 patients presented in the study have not been reported in any previous publication. Controls included 15 age- and sex-matched normal human volunteers who did not have any known autoimmune disease. An institutional review board approved the study and informed written consent was obtained from all the patients.
Table 1.
Clinical and diagnostic characteristics of pemphigus vulgaris patients.
| IIF§ | ELISA¶ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient no. | Sex | Age of onset | Clinical profile | Severity* | Histo† | DIF‡ | IgG | IgG4 | Dsg1 | Dsg3 |
| 001 | M | 50 | Skin, oral, nasal pharyngeal | Severe | + | + | 640 | 1260 | 118 | 410 |
| 002 | M | 52 | Skin, oral, penile, larynx | Severe | + | + | 320 | 1260 | 130 | 359 |
| 003 | F | 69 | Oral, pharyngeal, oesophageal, nail | Severe | + | + | 1260 | 2560 | 55 | 330 |
| 004 | M | 70 | Oral, nasal, larynx | Moderately severe | + | + | 320 | 1280 | 36 | 437 |
| 005 | F | 63 | Skin, oral, oesophageal | Moderately severe | + | + | 160 | 640 | 315 | 279 |
| 006 | F | 53 | Oral, pharyngeal, ocular | Moderately severe | + | + | 160 | 320 | 29 | 283 |
| 007 | F | 54 | Skin, nasal, oesophageal, nail | Severe | + | + | 2560 | 2560 | 310 | 317 |
| 008 | M | 51 | Skin, pharyngeal, anal | Moderately severe | + | + | 640 | 1280 | 258 | 188 |
| 009 | F | 60 | Skin, nasal, oesophageal | Severe | + | + | 640 | 640 | 149 | 210 |
| 010 | M | 54 | Oral, pharyngeal, penile, larynx | Severe | + | + | 320 | 640 | 81 | 286 |
| 011 | F | 53 | Skin, ocular, anal, larynx | Severe | + | + | 640 | 1280 | 338 | 276 |
| 012 | F | 51 | Oral, nasal | Moderately severe | + | + | 320 | 640 | 73 | 345 |
| 013 | F | 55 | Skin, oesophageal, larynx, nail | Severe | + | + | 2560 | 2560 | 301 | 336 |
| 014 | M | 50 | Oral, pharyngeal | Moderately severe | + | + | 160 | 160 | 86 | 377 |
| 015 | M | 66 | Skin, penile, ocular | Severe | + | + | 320 | 1280 | 259 | 162 |
Severity of disease.
Histology, biopsy of lesion demonstrated suprabasilar acantholysis.
Direct immunofluorescence, deposition of immunoglobulin G (IgG) on the keratinocyte cell surface of perilesional tissue.
Prior to initiation of therapy titre of pemphigus autoantibody determined by indirect immunofluorescence using monkey oesophagus. Titre of total IgG and IgG4 reported.
Enzyme-linked immunosorbent assay (ELISA) using recombinant dsg peptide in commercially available plates. Titre for desmoglein 1 (Dsg1) and Dsg3 provided.
Desmoglein 3 peptides
Two Dsg3 peptides used were synthesized by Sigma-Aldrich (St. Louis, MO, USA) to 95% purity: Dsg3 (96–112) [7] and Dsg3 (190–204) [2], with free acids at both the N- and C-termini.
Flow cytometry staining and cell sorting of NK and CD4+ T cells from peripheral blood and skin biopsies of PV patients and control subjects
Peripheral blood leucocytes (PBLs) from PV patients and control subjects were stained with different combinations of anti-human monoclonal antibodies: CD16-Cy-Chrome, CD16-peridinin chlorophyll (PerCP) or CD16-phycoerythrin (PE), CD56-PE-Cy7, CD56-PE-Cy5 or CD56-PE or CD56-Alexa 700, CD20-fluorescein isothiocyanate (FITC) or CD20-APC, CD4-APC, CD4-FITC or CD4-PE, CD3-PerCP, CD3-Cy-Chrome or CD3-PE, CD40-PE or CD40-APC, CD70-PE or CD70-FITC, CD81-PE, CD80-PE, MHC class II antigens [human leucocyte antigen D-related (HLA-DR)-DQ-DP-FITC] and isotype controls for all antibodies (Pharmingen, San Diego, CA, USA). B7-H1, B7-H2, B7-H3 (E-Biosciences, San Diego, CA, USA) and B7-H4 (Pharmingen) anti-mouse monoclonal antibodies were all PE-conjugated. Briefly, antibodies were incubated with cells for 45 min on ice in 1× phosphate-buffered saline (PBS) containing 2% FCS and 0·01 sodium azide (Sigma-Aldrich). Cells were washed and obtained by flow cytometry using a fluorescence activated cell sorter (FACSCalibur) (BD Biosciences, Franklin Lakes, UJ, USA) and analysed by FlowJo software (Treestar, OR, USA). A FACS Aria (BD Biosciences) was used for cell sorting. The purity of sorted cells was 95–99% using FACS sorting. CD11c dendritic cells with CD1 markers were excluded from the population of lymphocytes studied. Cells obtained from the perilesional skin tissues of PV patients with active disease were processed as described below.
Immunofluorescence microscopy of NK cells from patients with PV
CD56bright CD16– CD3– and CD56dim CD16+ CD3– NK cells were sorted (see above) using FACS Aria (BD Biosciences) [17]. The purity of sorted cells was 97–99%. These cells were reacted with CD56-Cy-Chrome and MHC class II-FITC antibodies for 1 h on ice in 1× PBS containing 2% FBS (Invitrogen, Carlsbad, CA, USA) and fixed with 1% paraformaldehyde (Fisher Scientific, Suwanee, GA, USA). 4,6-Diamidino-2-phenylindole staining (Vector Laboratories, Burlingame, CA, USA) was used to visualize nuclear morphology. Samples were mounted on poly l-lysine coated slides (Electron Microscopy Sciences, Hatfield, PA, USA) and imaged using a fluorescent microscope (Nikon Eclipse, Melville, NY, USA).
Proliferation assay of CD56+ NK cells co-cultured with CD4+ T cell lines derived from skin lesions or PBL of patients with PV
For preparation of T cell lines, 4-mm punch biopsies were taken from perilesional skin of three patients with severe active PV, three age- and sex-matched normal healthy volunteers, and one patient with active and severe bullous pemphigoid (BP). Tissues were digested with collagenase (Sigma) in Hank's PBS (Invitrogen) for approximately 2 h. Cells were incubated for 3 weeks and sorted for CD4+ T cells, which were then cultured for an additional week in complete Dulbecco's modified Eagle's medium (Mediatech, Manassas, VA, USA) containing phytohaemagglutinin (1 μg/ml) (Sigma), IL-2 (100 U) (Peprotech, Rocky Hill, NJ, USA), 1% human serum (Invitrogen) and 10% low IgG FBS (Invitrogen). Skin biopsies did not yield sufficient NK cells, and therefore NK cells in these experiments were obtained from PBL from the same patients and normal controls. Low proliferation assay counts may be due to the small amount of blood taken from patients with active PV (approximately 5 ml) or FACS cell sorting. CD56+ CD16– CD3– and CD56+ CD16+ CD3– MHC class II-positive NK cells and CD20+ B cells of PV patients and control subjects were used in proliferation assays. These NK cells and B cells were incubated with 10 μg of either Dsg3 (96–112) or Dsg3 (190–204) peptide for 3 h, and then irradiated (3000 rad). Peptide-loaded NK cells and B cells (5 × 104 per well) were co-cultured with CD4+ T cells (1 × 105) from PBL or CD4+ T cell lines (1 × 105) from perilesional skin for a total of 5 days, using 96-well round-bottomed plates in 200 μl of RPMI-1640, supplemented with 10% low IgG FBS, 1% human serum, l-glutamine (2 mM) (Mediatech), penicillin (100 U/ml) (Mediatech), streptomycin (100 μg/ml) (Mediatech), 2-mercaptoethanol (50 μM) (Invitrogen), non-essential amino acids (Mediatech) and sodium pyruvate (Mediatech). On day 4 of the cultures, 1 μCi of [3H] (Amersham Pharmacia Biotech, Piscataway, NJ, USA) was added to the cultures and cells were harvested 16 h later. The [3H] uptake was measured by a Wallac liquid scintillation counter (Waltham, MA, USA).
Measurements of cytokines and chemokines in supernatants of co-culture experiments by FACS analysis
Supernatants were obtained from CD56+ CD16– CD3– and CD56+ CD16+ CD3– MHC class II-positive NK cells (prepared by sorting) and CD20+ B cells incubated with CD4+ T cells, from the PBL or from the perilesional skin of the same patient, in the presence or absence of Dsg3 peptides were measured for levels of cytokine/chemokine production using both inflammatory and T helper 1 (Th1)/Th2 cytokine bead arrays (CBA) according to the manufacturer's protocol (BD Biosciences). Supernatants were removed from proliferation assays on day 3 of each proliferation experiment and incubated with cytokine-labelled beads for 3 h. Levels of cytokine production were determined by FACSCalibur (BD Biosciences) and analysed by CBA software (BD Biosciences).
Statistical analysis
A non-parametric analysis (Mann–Whitney U-test) using spss version 11·5 was used to analyse statistical significance. Additionally, an unpaired Student's t-test and anova was used to determine the statistical significance for all comparisons (StataCorp, College Station, TX, USA). A value of P < 0·05 was considered statistically significant (*). A value of P > 0·05 was not significant (**).
Results
Expression of MHC class II gene products on NK cells from PV patients using FACS analysis and immunofluorescence microscopy
One to 2·1% (mean 1·7%) of PBL from 15 patients with active PV were CD56brightCD3– NK cells-expressed MHC class II molecules, compared with 0·1–0·4% (mean 0·2%) in 15 normal controls (P < 0·004) by FACS analysis (Fig. 1a and b). This difference represents a 64% increased expression level of MHC class II molecules on these NK cells. Additionally, 3·1% to 5·8% (mean 4·2%) of CD56dim CD3– NK cells expressed MHC class II gene products, compared with 0·4–0·8% (mean 0·7%) in normal controls (P < 0·004) (Fig. 1a and b). Hence it appears that more CD56dim CD16+ NK cells have MHC class II molecules on their cell surface in PV patients. The higher frequency of MHC class II-bearing CD56dim CD3– cells are due to the fact that more NK cells are CD56dim CD3– than CD56bright CD3– in human peripheral blood. Using immunofluorescence analysis in three PV patients it was demonstrated that NK cells, sorted for CD56bright CD16– CD3– and CD56dim CD16+ CD3– markers, co-expressed HLA-DR-DP-DQ molecules on the cell surface (Fig. 1c).
Fig. 1.
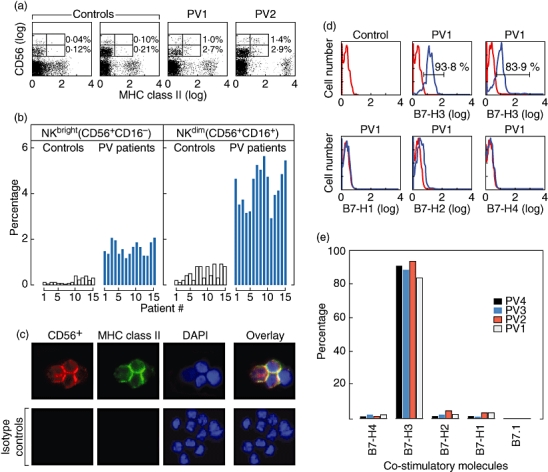
Frequency distribution and detection of major histocompatibility complex (MHC) class II molecules on natural killer (NK) cells in patients with pemphigus vulgaris (PV), compared with normals, by fluorescence activated cell sorter (FACS) and immunofluorescence microscopy. (a) Representative demonstration of presence of MHC class II molecules on NK cells. The FACS analysis of peripheral blood leucocytes (PBLs) from two representative PV patients of 15 with active disease, and two 15 normal controls are presented. NK cells consist of two separate populations, CD56bright CD16– CD3– and CD56dim CD16+ CD3–. Both subsets of CD56+ NK cells demonstrated surface binding with antibodies to MHC class II molecules. The lower quadrant of the grid represents CD56dim NK cells. The frequency of expression of MHC class II molecules is significantly higher in PV patients compared with normal controls (P < 0·001). However, it is similar in both NK cell populations. (b) Histogram representation of the observations in (a). Instead of only using two representative patients' data, the FACS analysis of PBL from all 15 PV patients and all 15 normal controls are presented. The y-axis represents the percentage of MHC class II molecule bearing cells from both the CD56bright CD16– and CD56dim CD16+ NK cells. The percentage of MHC class II-bearing cells are higher in CD56dim CD16– cell compared with CD56bright cell populations (P < 0·004). The frequency of the CD56dim NK cells expressing MHC class II is statistically significantly higher in PV patients. In (c), CD56+ CD16– CD3– cells were sorted to demonstrate expression of MHC class II molecules using immunofluorescence microscopy. The red stain represents CD56+ CD16– CD3– NK cells. The green stain identified the MHC class II molecules. Nuclear staining is represented with 4,6-diamidino-2-phenylindole shown in blue. CD56dim CD16+ CD3– cells stained similarly (data not shown). Thus NK cells in PV patients with active disease express MHC class II molecules. Photograph magnification is 100× using an oil emersion objective lens (1·4 NA, Plan Apo). (d) Demonstrating the presence of co-stimulatory molecule B7 on the cell surface of CD56+ CD3– NK cells that co-express MHC class II molecules. Approximately 84% and 94% these NK cells with MHC class II molecules from two PV patients with active disease co-expressed the co-stimulatory molecule B7-H3. The co-stimulatory molecules B7-H1, B7-H2 and B7-H4 were not co-expressed. (e) Histogram representation of (d). More than 80% of the CD56+ CD3– NK cells co-expressed MHC class II molecules. In addition they express the co-stimulatory molecule B7-H3 in four representative PV patients. The co-stimulatory molecule B7-H1, B7-H2 and B7-H4 were not co-expressed.
Co-expression of B7 co-stimulatory molecules on the cell surface of CD56+ CD3– NK cells that express MHC class II molecules from PV patients by FACS
In patients with active PV, CD56+ CD3– NK cells from PBL that co-expressed MHC class II molecules also express the co-stimulatory molecule B7-H3 (Fig. 1d). Two representative examples are shown in Fig. 1d. Co-expression was approximately 84–94% for B7-H3 molecules in four representative PV patients (Fig. 1e). CD56+ CD3– NK cells from PV patients that co-expressed MHC class II molecules did not express B7·1, B7-H1, B7-H2 and B7-H4. The co-expression of these molecules was not tested in normal controls, because normal individuals do not express MHC class II molecules on NK cells.
Peptide presentation by CD56+ CD16– CD3– and CD56+ CD16+ CD3– MHC class II-positive, NK cells and B cells from peripheral blood of PV patients
In these experiments we demonstrated that both subsets of NK cells, without the exogenous addition of cytokines, were capable of presenting antigen (Dsg3) to CD4+ T cells. CD56bright CD16– CD3– and CD56dim CD16+ CD3– MHC class II-positive NK cells and CD20+ B cells were separated from PBL of four untreated patients with active and severe PV by FACS sorting. Significant proliferation was observed in the NK cells and B cells of patients mixed with CD4+ T cells of the same patient, in the presence of Dsg3 peptides, compared with control CD4+ T cells (P < 0·01). CD56bright CD16– CD3– and CD56dim CD16+ CD3– NK cells and CD20+ B cell-stimulated CD4+ T cells from PV patients only in the presence of Dsg3 peptides (Fig. 2a). When co-cultured, CD56bright CD16– CD3–, CD56dim CD16+ CD3– NK cells and CD4+ T cells isolated from normal healthy individuals did not proliferate, even after incubation with Dsg3 peptides. Controls consisted of APCs (NK and CD20+ B cells) co-cultured with CD4+ T cells without Dsg3 peptide and NK, B cells and T cells alone with and without Dsg3 peptide (Fig. 2a). The proliferative response of NK cells was similar to that of the proliferation of CD20+ B cells. The B cells are known to present antigen to T cells. The same response in NK cells would suggest that MHC Class II molecules on these NK cells also present Dsg3 to the CD4+ T cells.
Fig. 2.
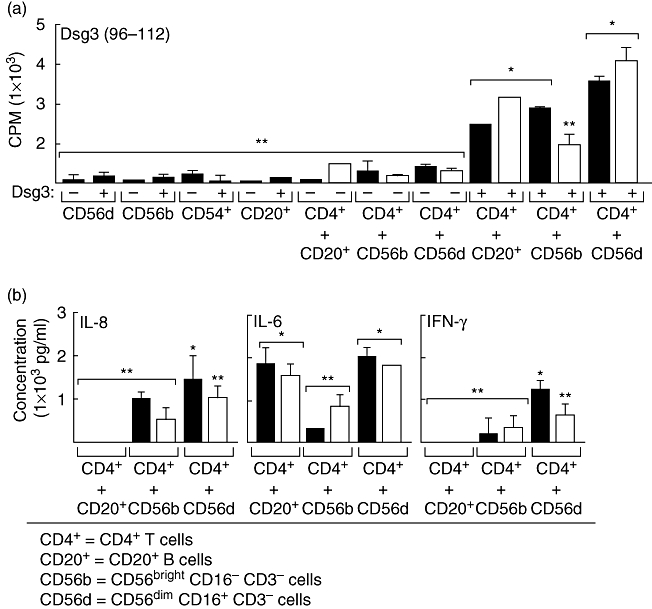
Peptide presentation by CD56+ major histocompatibility complex (MHC) class II-bearing natural killer (NK) cells and cytokine profile of peripheral blood leucocytes of pemphigus vulgaris (PV) patients. CD56+ NK cells that expressed MHC class II molecules were obtained from the peripheral blood of two PV patients, and mixed with CD4+ T cells from the same patients and with desmoglein 3 (Dsg3) peptides. The controls included CD4+ T cells alone, CD20+ and CD56+ cells alone. (a) CD4+ T cell proliferation using [3H] incorporation is presented, after removing the corresponding control measured as counts per minute. T cell proliferation is statistically significantly higher when NK CD56+ cells are treated with Dsg3 peptides compared with absence of peptides (P < 0·002). (b) Profile of cytokine production by CD4+ T cells obtained from peripheral blood of PV patients when interacted with CD56+ MHC class II-positive NK cells and Dsg3 peptides, after removing control background. CD4+ T cells of PV patients produced high to moderate levels of interleukin (IL)-8, IL-6 and interferon-γ compared with controls. The two patients with active PV studied in this experiment are presented as black and white bars. *P < 0·05; **statistically not significant.
Levels of IL-8, IL-6 and IFN-γ were increased in the supernatants of cultures of NK CD56+ CD16– CD3– or CD56+ CD16+ CD3– cells and CD4+ T cells from PBL of PV patients in the presence of Dsg3 (Fig. 2b). Only IL-6 was increased in the supernatants of co-cultures of CD20+ B cells and CD4+ T cells from the PBLs of the same patients. The IL-6 produced was at approximately the same level by the CD20+ B cells, and the CD56+ NK cells from PBL indicates that the NK cells with MHC class II molecules produced a B cell-stimulating cytokine. Not surprisingly, CD20+ B cells did not produce IL-8 or IFN-γ. However, CD56+ NK cells did produce the proinflammatory cytokines IL-8 and IFN-γ at statistically significant high levels, indicating that the NK cells from the PBLs produce proinflammatory cytokines in PV patients (Fig. 2b). Other cytokines tested (IL-2, IL-4, IL-10 and TNF-α) were not detected in the supernatants.
Presentation of Dsg3 peptides using NK cells from peripheral blood and CD4+ T cell lines obtained from skin lesions of PV patients
The experiments consisted of perilesional skin biopsies from three patients with active untreated PV, one patient with severe, active BP and two normal healthy age- and sex-matched human volunteers. CD4+ T cells from skin lesions of PV patients showed proliferation after incubation with CD56+ CD3– NK cells with two different Dsg3 peptides. A statistically significantly increased response to Dsg3 peptide (96–112) was observed when compared with the response to Dsg3 peptide (190–204) (P < 0·03). This response to the two different peptides was measured in counts/min when CD4+ T cells and CD56+ T cells were cultured with these two different peptides (Fig. 3a). None the less, the response to both Dsg3 peptides was significantly higher than that observed in the controls (Fig. 3a). CD4+ T cells cultured from perilesional skin of a patient with BP or from normal controls did not proliferate in response to Dsg3 peptides (Fig. 3a). CD56+ CD3- NK cells from PBL incubated with CD4+ T cells from the skin of the same PV patients, in the presence of Dsg3 peptide (96–112), produced levels of IL-8, IL-6 and IFN-γ that were statistically significantly higher than controls (Fig. 3b). Levels of IL-8 were greater than IL-6 (P < 0·02) or IFN-γ. The levels of IL-6 were higher in cultures of CD4+ T cells from the skin (Fig. 3b) compared with PBL, which are presented in Fig. 2b (P < 0·03). The levels of IL-8 and IFN-γ produced were similar by CD4+ T cells from PBL and the skin (Figs 2b and 3b) The levels of IFN-γ, while lower than IL-8 and IL-6, were similar in cultures of CD4+ T cells from PBL and the skin (Figs 2b and 3b), but were higher than those produced by normal controls (Fig. 3b). CD4+ T cells alone, CD56+ CD3– cells alone with or without Dsg3 peptides, NK cells from PBL of a BP patient or normal control with CD4+ T cells and Dsg3 peptide did not produce detectable levels of IL-8, IL-6 or IFN-γ. Other cytokines tested, including IL-2, IL-4, IL-10, IL-1β, TNF-α and IL-12, were not detectable (data not shown). These experiments demonstrate that the CD4+ T cells from PV patients, in the microcirculation of the skin, respond in a manner similar to that observed in the peripheral blood. They interact with Dsg3 peptides only in the presence of CD56+ NK cells, as demonstrated by the observation that they proliferate and produce statistically significant elevated levels of IL-6, IL-8 and IFN-γ.
Fig. 3.
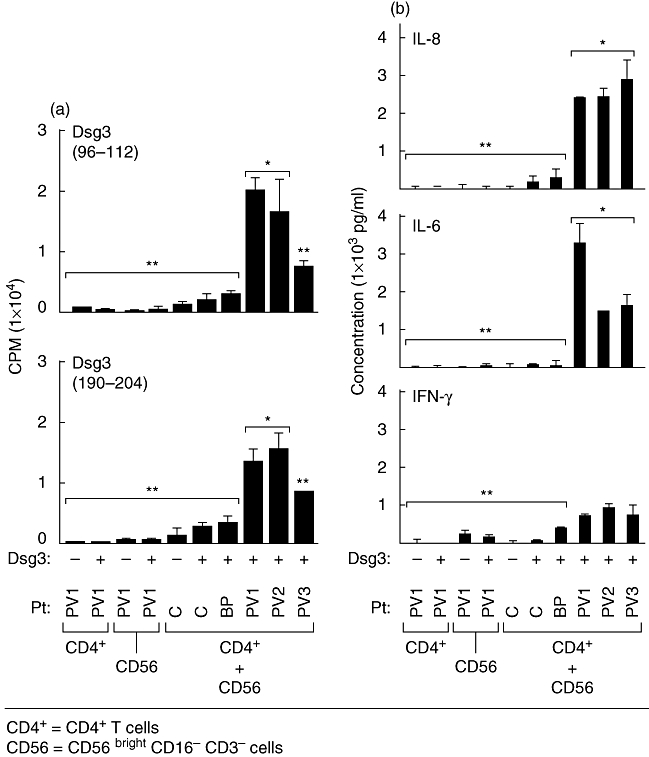
Specific presentation of desmoglein 3 (Dsg3) peptides to CD4+ T cells obtained from skin lesions of pemphigus vulgaris (PV) patients by CD56+ major histocompatibility complex (MHC) class II-positive natural killer (NK) cells. CD4+ T cells were obtained from perilesional skin of three untreated PV patients with active disease and interacted with CD56+ MHC cells II-positive NK cells of the same patient, in the presence of two different Dsg3 peptides. (a) Dsg3 (a.a. 96–112) and Dsg3 (a.a. 190–204) were used. T cell proliferation was statistically significantly higher in CD56+ NK cells compared with controls (P < 0·02). Responses to Dsg3 (96–112) were statistically significantly higher than the response to Dsg3 (190–204) (P < 0·03). (b) Supernatants collected from these cultures are presented. Levels of cytokines produced in response to both Dsg3 peptides were similar. The levels of interleukin (IL)-8 are statistically significantly higher than levels of IL-6 (P < 0·02). Interferon-γ levels were also increased but not statistically significant. Controls included CD4+ T cells from skin biopsies of two normal controls and one patient with bullous pemphigoid. Bars represent the standard deviation. *P < 0·05; **statistically not significant.
Discussion
The histopathological hallmark of PV is acantholysis or loss of adhesion between epithelial cells. Acantholysis is the end-product of a series of preceding cellular and molecular events that involve the immune and inflammatory system [18]. In this study the principal focus was to study some of the participants in the mechanism of pathogenesis by characterizing some of the participants and their functions. Thus we are studying some of the processes that may define and characterize events in the microenvironment of the skin and mucosal tissues, and those present in the central immune system and are reflected in the peripheral circulation.
Pemphigus vulgaris is a potentially fatal rare disease. The authors recognize that a limited number of patients have been studied. They also recognize that a longitudinal follow-up of the same patients will provide more insight and understanding, particularly studying the patients when they go into a prolonged clinical remission. Hence, these observations are perceived to be preliminary. A larger cohort of patients studied will provide a clearer and better analysis of these events.
The results of the study demonstrate that a combination interaction of CD56+ CD3– NK cells and CD4+ T cells in concert produce a profile of cytokines and chemokines that may explain partially the inflammation observed in the diseased tissues and the production of specific pathogenic autoantibody. Dendritic cells (CD11c+ and CD1+) were removed from the PBL used in proliferation assays and therefore could not have played a direct role in these processes.
CD4+ T cell lines from perilesional skin of PV patients co-cultured with CD56+ CD3– NK cells expressing MHC class II molecules, in the presence of Dsg3, secreted statistically significant levels of IL-6 and IL-8 (Fig. 3b). Levels of IFN-γ were elevated but not statistically significant and TNF-α was not detected. CD4+ T cells from PBL of patients with active disease co-cultured with CD56+ CD3– NK cells expressing MHC class II molecules, in the presence of Dsg3, without the addition of exogenous cytokines, produced similar profiles (Fig. 2).
In several experiments, the NK cells have been partially characterized. These NK cells that are CD56+ CD3– express MHC class II molecules and the co-stimulatory molecule B7-H3. Professional APCs express B7-H3 and have been shown to enhance the induction of cytotoxic T cells and stimulate IFN-γ[19]. Recently it has been reported that NK cells expressing MHC class II molecules, following activation with cytokines, have APC-like properties and regulate T cell activation, using a peptide of the influenza virus [20]. However, our studies demonstrate high levels of in vivo expression MHC class II molecules on these NK cells, without the addition of exogenous cytokines.
We studied mainly the CD56+ NK cells because they have different cytotoxic and regulatory functions [10]. However, in our studies both subsets co-cultured with CD4+ T cells, in the presence of Dsg3, produced a similar cytokine profile; CD4+ T cells from skin lesions and peripheral blood of PV patients demonstrated specific proliferation when cultured with CD56+ NK (CD56dim or CD56bright) and the relevant peptides of Dsg3 and produced IL-6, IL-8 and IFN-γ (Figs 2 and 3). In addition, it has been shown that CD56brigtht NK cells are enriched at sites of inflammation and together with monocytes are involved in maintaining the inflammation [21]. However, such studies did not investigate the role of CD56dim NK cells, and the expression of MHC class II molecules on these cells, within the inflammatory site or the influence of these cells in the circulation. The cumulative observations in this study would suggest that these subsets of CD56+ CD3– NK cells may hone into specific sites within the skin and mucosal tissues of PV patients. Thereafter it is possible that this combination of MHC class II molecules bearing NK cells and CD4+ T cells in the microenvironment of disease-specific tissue and circulation facilitate and amplify the local and systemic production of an inflammatory process and eventually the production of high titres of pathogenic autoantibodies.
In the microenvironment of the diseased tissues they function as APCs and present Dsg3 peptides to resident and circulating CD4+ T cells. These T cells proliferate and produce various cytokines that exert their effect locally and are absorbed in the circulation. These in vivo-activated NK cells may travel to the lymph nodes, spleen and bone marrow, where a higher proportion of antibody-producing B cells are present, and may respond to their stimulation, resulting in high levels of IgG4 pathogenic autoantibodies.
B cells co-cultured with CD4+ T cells from PV patients in the presence of Dsg3 peptides produced significantly high levels of IL-6. In addition, we observed that both subsets of NK cell with in-vivo expression of MHC class II molecules co-cultured with CD4+ T cells and Dsg3 peptides produce high levels of IL-6. These two sources of IL-6 augment further the production of pathogenic autoantibody and may expand the population of B cells producing them [22,23].
Other peptide-presenting cells may also participate in the pathogenesis of PV, but the possible cross-talk among them in co-cultures with CD4+ T cells will need to be investigated [24–26]. For example, dendritic cells, plasmocytoid cells and monocytes may be necessary to produce the required processing of Dsg and equally importantly in the presentation of peptides [15,25].
Based on the observations in this report and the literature, we present a hypothetical role of NK cells in the pathogenesis of PV (Fig. 4). Resident Langherhans cells in skin and mucosal tissue produce cytokines that recruit NK cells into the site of pathology [27]. Langerhans cells may also process the Dsg3 available from the surrounding keratinocytes to make it available to other cells in the microenvironment of the skin and mucosal tissues. Along with CD4+ T cells, NK cells as APCs then facilitate the local production of inflammatory cytokines and other mediators of inflammation. These NK cells may act as APCs and present Dsg3 peptides to CD4+ T cells. These proliferating CD4+ T cells secrete IL-6, which may stimulate resident B cells to produce anti-Dsg3 antibodies. These T cells, B cells and NK cells and the cytokines and the chemokines they produce are probably absorbed. Upon arrival in the bone marrow, spleen or lymph nodes, they amplify this autoimmune response several-fold. Once it reaches a critical level, clinical disease is produced.
Fig. 4.
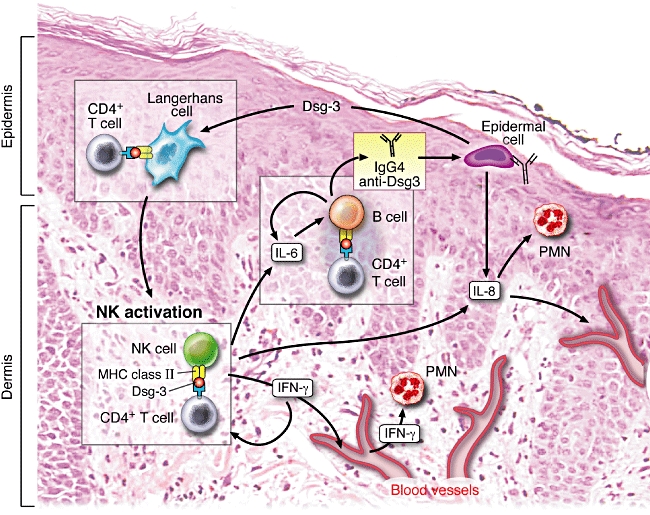
Representation of the possible role of natural killer (NK) cells in pemphigus vulgaris (PV) − a hypothetical model for pathogenesis of clinical disease. NK cells acting as antigen-presenting cells present desmoglein 3 (Dsg3) peptides to CD4+ T cells, both of which hone into the perilesional skin and oral mucosa, from the peripheral blood. The proliferating T cells secrete interleukin (IL)-6, which stimulates resident B cells to produce anti-Dsg3 antibodies. NK cells produce interferon (IFN)-γ, which then further up-regulates major histocompatibility complex (MHC) class II expression and simultaneously attracts other NK cells to the site of pathology. IL-8 secreted by activated NK cells will stimulate resident polymorphonuclear cell (PMN) and monocytes. These NK cells, IL-8, IL-6, IFN-γ and other cytokines, chemokines and inflammatory mediators are absorbed into the blood vessels of the skin and mucosal tissues and carried to lymph nodes, spleen and bone marrow. Here the inflammatory response and the levels of autoantibody production may be amplified significantly. Once substantial levels of the autoantibody and inflammatory chemokines and cytokines reach the target tissue, the adhesions between the epithelial cells in the stratum malphigii are lost and clinical disease is observed. The simultaneous critical role of the central immune system, perhaps in the bone marrow, spleen or lymph nodes, and the essential role of the microenvironment of the skin and mucosal tissues are emphasized in this model.
Interleukin-6 is a pleiotropic cytokine important in the regulation of the immune response, acute phase reactions and haematopoiesis and it is involved in the pathophysiology of autoimmune disorders, lymphoid malignancies and inflammation [28]. Interestingly, animal models of lupus, using BALB/c IL-6-deficient mice, have demonstrated that IL-6 plays a role in the production of anti-DNA and chromatin autoantibodies [29]. Studies have shown that IL-6 plays a role in subclass-switching mechanisms of IgG [30]. These studies would suggest that IL-6 in the microenvironment of the skin, mucosal tissue and bone marrow of PV patients may play a role in the production of class-specific pathogenic anti-Dsg antibodies.
Experiments in these studies demonstrate that NK cells co-cultured with CD4+ T cells from the perilesional skin and PBL of patients, in the presence of Dsg3, produced high levels of IL-8. Studies show that keratinocytes from PV patients also produce high levels of IL-8 in vitro when incubated with anti-Dsg3 antibodies [31]. Thus, it is possible that in the microenvironment of the skin and mucosa of PV patients, locally increased levels of IL-8 may play a role in the recruitment and activation of polymorphonuclear cells (PMNs) to the site of inflammation [32,33].
It is also known that IL-8 and other inflammatory chemokines play an important role in organ-specific and systemic autoimmune disorders through leucocyte recruitment and trafficking of PMN to affected tissues [28,34,35]. Increased levels of IL-8 and other inflammatory chemokines have been found in the plasma of systemic lupus erythematosus patients [36] and in the serum of multiple sclerosis patients [37], and have been correlated with disease activity. In patients with multiple sclerosis, high levels of IL-8 in cerebrospinal fluid are present during the relapses [37]. IL-8 and NK cells have been shown to be involved in the pathogenesis of ankylosing spondilitis [38].
Although statistically not significant, the increased levels of IFN-γ could maintain the up-regulation of MHC class II molecules on these NK cells and other APCs in the microenvironment and thus amplify the local autoimmune and inflammatory processes [24–26,28]. Other cytokines and chemokines, not studied or assayed, may also play an important role in the various stages of immune disregulation and inflammation observed in PV.
In a recent study, role of NK cells were studied in PV patients. These investigators observed that CD56+ CD3– NK cells were increased in patients with active PV [39]. Using gene expression analysis they demonstrated increased expression of IL-10 and decreased expression of IL-12Rβ2, perforin and granzyme B and increased expression of IL-5 [39]. The authors conclude that NK cells contribute to Th2-based immune responses through IL-12Rβ2 signalling impairment and up-regulation of IL-5 and Il-10. While this study does not address the above issue, it helps to validate further a complex multi-dimensional and vital role for NK cells in the pathogenesis of PV.
The importance of the observations in this study, are many-fold and multi-faceted. These experiments may demonstrate the importance of the various cells and their products in the microenvironment of the disease tissue, and in the process of blister formation. Pemphigus is known to be chronic and characterized by relapses and remissions. While current therapies may target and influence acantholysis and blister formation, they produce temporary remissions, as the underlying processes that lead to the generation of pathogenic B cells are not reversed. In the future, identifying cells, molecular and multi-step molecular processes that precede the production and induction of pathogenic autoantibody would help in designing therapeutics that prevent or abort these processes. Thus a long-term sustained remission should be the goal of the treatment of pemphigus.
Acknowledgments
We are very grateful to David Hamrock MD for assistance in obtaining the skin biopsies, Phillip Chandler DPhil for advice with experimental procedures and Hakan M. Gurcan MD for editing this manuscript. E. J. Y. was supported by the Public Health Service grants HL29583 and from the National Heart, Lung and Blood Institute of the National Institute of Health HL59838. N. B. was supported by a grant from the Rockefeller Brother's Foundation.
References
- 1.Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429–52. [PubMed] [Google Scholar]
- 2.Wucherpfennig KW, Yu B, Bhol K, et al. Structural basis for major histocompatibility complex (MHC)-linked susceptibility to autoimmunity: charged residues of a single MHC binding pocket confer selective presentation of self-peptides in pemphigus vulgaris. Proc Natl Acad Sci USA. 1995;92:11935–9. doi: 10.1073/pnas.92.25.11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denning MF, Guy SG, Ellerbroek SM, Norvell SM, Kowalczyk AP, Green KJ. The expression of desmoglein isoforms in cultured human keratinocytes is regulated by calcium, serum, and protein kinase C. Exp Cell Res. 1998;239:50–9. doi: 10.1006/excr.1997.3890. [DOI] [PubMed] [Google Scholar]
- 4.Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med. 1982;306:1189–96. doi: 10.1056/NEJM198205203062001. [DOI] [PubMed] [Google Scholar]
- 5.Storer JS, Galen WK, Nesbitt LT, DeLeo VA. Neonatal pemphigus vulgaris. J Am Acad Dermatol. 1982;6:929–32. doi: 10.1016/s0190-9622(82)80127-8. [DOI] [PubMed] [Google Scholar]
- 6.Hertl M, Karr RW, Amagai M, Katz SI. Heterogeneous MHC II restriction pattern of autoreactive desmoglein 3 specific T cell responses in pemphigus vulgaris patients and normals. J Invest Dermatol. 1998;110:388–92. doi: 10.1046/j.1523-1747.1998.00156.x. [DOI] [PubMed] [Google Scholar]
- 7.Veldman CM, Gebhard KL, Uter W, et al. T cell recognition of desmoglein 3 peptides in patients with pemphigus vulgaris and healthy individuals. J Immunol. 2004;172:3883–92. doi: 10.4049/jimmunol.172.6.3883. [DOI] [PubMed] [Google Scholar]
- 8.Cooper MA, Fehniger TA, Turner SC, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56 (bright) subset. Blood. 2001;97:3146–51. doi: 10.1182/blood.v97.10.3146. [DOI] [PubMed] [Google Scholar]
- 9.Orange JS, Fassett MS, Koopman LA, Boyson JE, Strominger JL. Viral evasion of natural killer cells. Nat Immunol. 2002;3:1006–12. doi: 10.1038/ni1102-1006. [DOI] [PubMed] [Google Scholar]
- 10.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–40. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 11.Campbell JJ, Qin S, Unutmaz D, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol. 2001;166:6477–82. doi: 10.4049/jimmunol.166.11.6477. [DOI] [PubMed] [Google Scholar]
- 12.Frey M, Packianathan NB, Fehniger TA, et al. Differential expression and function of 1-selectin on CD56bright and CD56dim natural killer cell subsets. J Immunol. 1998;161:400–8. [PubMed] [Google Scholar]
- 13.Fehniger TA, Cooper MA, Nuovo GJ, et al. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood. 2003;101:3052–7. doi: 10.1182/blood-2002-09-2876. [DOI] [PubMed] [Google Scholar]
- 14.Jacobs R, Hintzen G, Kemper A, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol. 2001;31:3121–7. doi: 10.1002/1521-4141(2001010)31:10<3121::aid-immu3121>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Cooper MA, Fehniger TA, Fuchs A, Colonna M, Caligiuri MA. NK cell and DC interactions. Trends Immunol. 2004;25:47–52. doi: 10.1016/j.it.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Bhol K, Natarajan K, Nagarwalla N, Mohimen A, Aoki V, Ahmed AR. Correlation of peptide specificity and IgG subclass with pathogenic and nonpathogenic auto antibodies in pemphigus vulgaris: a model for autoimmunity. Proc Natl Acad Sci USA. 1995;92:5239–43. doi: 10.1073/pnas.92.11.5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhol KC, Desai A, Kumari S, Colon JE, Ahmed AR. Pemphigus vulgaris: the role of IL-1 and IL-1 receptor antagonist in pathogenesis and effects of intravenous immunoglobulin on their production. Clin Immunol. 2001;100:172–80. doi: 10.1006/clim.2001.5061. [DOI] [PubMed] [Google Scholar]
- 18.Amagai M, Ahmed AR, Kitajima Y, et al. Are desmoglein auto antibodies essential for the immunopathogenesis of pemphigus vulgaris, or just ‘witnesses of disease’? Exp Dermatol. 2006;15:815–31. doi: 10.1111/j.1600-0625.2006.00499_1.x. [DOI] [PubMed] [Google Scholar]
- 19.Chapoval AI, Ni J, Lau JS, et al. B7–H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2:269–74. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- 20.Hanna J, Gonen-Gross T, Fitchett J, et al. Novel APC-like properties of human NK cells directly regulate T cell activation. J Clin Invest. 2004;114:1612–23. doi: 10.1172/JCI22787. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol. 2004;173:6418–26. doi: 10.4049/jimmunol.173.10.6418. [DOI] [PubMed] [Google Scholar]
- 22.Hilbert DM, Cancro MP, Scherle PA, et al. T cell derived IL-6 is differentially required for antigen-specific antibody secretion by primary and secondary B cells. J Immunol. 1989;143:4019–24. [PubMed] [Google Scholar]
- 23.Freeman GJ, Freedman AS, Rabinowe SN, et al. Interleukin 6 gene expression in normal and neoplastic B cells. J Clin Invest. 1989;83:1512–8. doi: 10.1172/JCI114046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–26. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 25.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–8. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 26.Mellman I, Turley SJ, Steinman RM. Antigen processing for amateurs and professionals. Trends Cell Biol. 1998;8:231–7. doi: 10.1016/s0962-8924(98)01276-8. [DOI] [PubMed] [Google Scholar]
- 27.Munz C, Dao T, Ferlazzo G, de Cos MA, Goodman K, Young JW. Mature myeloid dendritic cell subsets have distinct roles for activation and viability of circulating human natural killer cells. Blood. 2005;105:266–73. doi: 10.1182/blood-2004-06-2492. [DOI] [PubMed] [Google Scholar]
- 28.Nishimoto N. Cytokine signal regulation and autoimmune disorders. Autoimmunity. 2005;38:359–67. doi: 10.1080/08916930500124106. [DOI] [PubMed] [Google Scholar]
- 29.Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188:985–90. doi: 10.1084/jem.188.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawano Y, Noma T, Kou K, Yoshizawa I, Yata J. Regulation of human IgG subclass production by cytokines: human IgG subclass production enhanced differentially by interleukin-6. Immunology. 1995;84:278–84. [PMC free article] [PubMed] [Google Scholar]
- 31.O'Toole EA, Mak LL, Guitart J, et al. Induction of keratinocyte IL-8 expression and secretion by IgG auto antibodies as a novel mechanism of epidermal neutrophil recruitment in a pemphigus variant. Clin Exp Immunol. 2000;119:217–24. doi: 10.1046/j.1365-2249.2000.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brandt E, Muller-Alouf H, Desreumaux P, Woerly G, Colombel JF, Capron M. Circulating growth-regulator oncogene alpha contributes to neutrophil priming and interleukin-8-directed mucosal recruitment into chronic lesions of patients with Crohn's disease. Eur Cytokine Netw. 1998;9:647–53. [PubMed] [Google Scholar]
- 33.Gilli UO, Schneider MK, Loetscher P, Seebach JD. Human polymorphonuclear neutrophils are recruited by porcine chemokines acting on CXC chemokine receptor 2, and platelet-activating factor. Transplantation. 2005;79:1324–31. doi: 10.1097/01.tp.0000155429.44902.44. [DOI] [PubMed] [Google Scholar]
- 34.Wilkinson JE, Smith CA, Suter MM, Falchek W, Lewis RM. Role of plasminogen activator in pemphigus vulgaris. Am J Pathol. 1989;134:561–9. [PMC free article] [PubMed] [Google Scholar]
- 35.Theilgaard-Monch K, Knudsen S, Follin P, Borregaard N. The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol. 2004;172:7684–93. doi: 10.4049/jimmunol.172.12.7684. [DOI] [PubMed] [Google Scholar]
- 36.Lit LC, Wong CK, Tam LS, Li EK, Lam CW. Raised plasma concentration and ex vivo production of inflammatory chemokines in patients with systemic lupus erythematosus. Ann Rheum Dis. 2006;65:209–15. doi: 10.1136/ard.2005.038315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartosik-Psujek H, Stelmasiak Z. The levels of chemokines CXCL8, CCL2 and CCL5 in multiple sclerosis patients are linked to the activity of the disease. Eur J Neurol. 2005;12:49–54. doi: 10.1111/j.1468-1331.2004.00951.x. [DOI] [PubMed] [Google Scholar]
- 38.Azuz-Lieberman N, Markel G, Mizrahi S, et al. The involvement of NK cells in ankylosing spondylitis. Int Immunol. 2005;17:837–45. doi: 10.1093/intimm/dxh270. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi H, Amagai M, Tanikawa A, et al. T helper type 2-biased natural killer cell phenotype in patients with pemphigus vulgaris. J Invest Dermatol. 2007;127:324–30. doi: 10.1038/sj.jid.5700527. [DOI] [PubMed] [Google Scholar]