

Abstract
Aberrant T cell responses during T cell activation and immunological synapse (IS) formation have been described in systemic lupus erythematosus (SLE). Kv1.3 potassium channels are expressed in T cells where they compartmentalize at the IS and play a key role in T cell activation by modulating Ca2+ influx. Although Kv1.3 channels have such an important role in T cell function, their potential involvement in the etiology and progression of SLE remains unknown. This study compares the K channel phenotype and the dynamics of Kv1.3 compartmentalization in the IS of normal and SLE human T cells. IS formation was induced by 1–30 min exposure to either anti-CD3/CD28 antibody-coated beads or EBV-infected B cells. We found that, while the level of Kv1.3 channel expression and their activity in SLE T cells are similar to normal resting T cells, the kinetics of Kv1.3 compartmentalization in the IS are markedly different. In healthy resting T cells, Kv1.3 channels are progressively recruited and maintained in the IS for at least 30 min from synapse formation. In contrast, SLE, but not rheumatoid arthritis, T cells show faster kinetics with maximum Kv1.3 recruitment at 1 min and movement out of the IS by 15 min after activation. These kinetics resemble pre-activated healthy T cells, but the K channel phenotype of SLE T cells is identical to resting T cells, where Kv1.3 constitutes the dominant K conductance. The defective temporal and spatial Kv1.3 distribution that we observed may contribute to the abnormal functions of SLE T cells.
Keywords: human, T cells, autoimmunity, systemic lupus erythematosus
INTRODUCTION
Systemic lupus erythematosus (SLE) is a rheumatic autoimmune disease characterized by abnormal T cell function (1–3). A variety of signaling alterations have been identified in SLE T cells (2). In particular, T cells from SLE patients, but not patients with other autoimmune diseases, display an exaggerated response to antigen stimulation (2). The hallmark of this T cell “hyperactivity” is a more pronounced and more sustained increase in intracellular Ca2+ levels ([Ca2+]i) following T cell receptor (TCR) ligation as compared to healthy T cells (4–6). This sustained influx of Ca2+ is essential for the activation of downstream signaling events and ultimately T cell function (7). Thus, impaired regulation of [Ca2+]i appears to contribute to altered T cell functions in SLE T cells. However, the precise mechanisms underlying this aberrant Ca2+ response in SLE T lymphocytes have not yet been identified. Although the increase in [Ca2+]i has been attributed in part to an increased release of Ca2+ from intracellular stores, membrane related processes have also been implicated (5, 6).
The TCR mediated influx of Ca2+ in T cells occurs through Ca2+ release activated Ca2+ (CRAC) channels and is regulated by various membrane channels and signaling molecules (8, 9). Briefly, engagement of the antigen to the TCR activates phospholipase Cγ (PLCγ) and induces the release of Ca2+ from intracellular stores. Depletion of Ca2+ from intracellular stores causes the CRAC channels to open and Ca2+ to flow into the cells. This sustained influx of Ca2+ is essential to activate T cells, regulating both proliferation and cytokine production. The necessary electrochemical driving force for Ca2+ influx is provided by the cation efflux through K channels. The two major K channels expressed in T cells are the voltage-dependent Kv1.3 channel and the Ca2+-activated K channel KCa3.1. Along with allowing initiation of the Ca2+ influx, the crosstalk between these and other channels shapes the overall Ca2+ response, i.e. amplitude and frequency of Ca2+ oscillations which can determine specificity of gene expression (10). Recently, it has been shown that the expression of these K channels depends on the immune cell activation state (11, 12). Kv1.3 channels constitute the predominant K conductance and regulate Ca2+ influx in resting naïve and central memory (TCM) as well as resting and activated effector memory (TEM) T cells. KCa3.1 are instead upregulated when naïve and TCM cells are activated and control Ca2+ influx in these cells (13).
Very recently, a limited number of studies have shown that Kv1.3 and KCa3.1 channels redistribute in the immunological synapse (IS) during TCR engagement (14–17). The IS is a tight and highly organized interactive signaling zone localized at the point of contact between T cell and antigen presenting cell (APC) and it contains membrane molecules (e.g. TCR, CD3 and CD28) as well as signaling components (e.g. Lck and PKCθ) (18). Functionally, the process of IS formation is thought to facilitate signaling through the TCR and to fine-tune the ultimate outcome of TCR engagement. The structure of the IS is very dynamic, with molecules entering and leaving at different times. However, the process of Kv1.3 channel re-localization in the IS is not yet understood. Furthermore, no information is available on potential alterations in Kv1.3 channel redistribution at the IS in pathological conditions. SLE T cells display certain features that can affect the formation of the IS: SLE T cells possess greater capacity to generate lipid rafts than normal T cells in response to activation, faster kinetics of lipid raft clustering and polarization, and faster kinetics of actin polymerization and depolymerization (6). In particular, it has been shown that cross-linking of lipid rafts evokes faster and more pronounced Ca2+ response in SLE T cells, indicating that early structural rearrangements in the T cell membrane contribute to the increased activity of SLE T cells.
The purpose of our study was to investigate whether the expression and activity of key regulators of Ca2+ homeostasis, such as Kv1.3 channels, is altered in SLE T cells. Furthermore, we have investigated whether abnormalities in the process of translocation of these channels in the IS that forms upon TCR binding occur in SLE T cells. Our results indicate that, while the biophysical and pharmacological properties of Kv1.3 channels in SLE T cells are identical to normal T cells, the dynamics of Kv1.3 channel compartmentalization in the IS of SLE T cells are altered. These alterations in TCR activated membrane rearrangements might underlie the downstream functional abnormalities of SLE T cells.
MATERIAL AND METHODS
Human subjects
Twenty patients with SLE fulfilling at least four of the American College of Rheumatology classification criteria for SLE were included in this study: 3 males and 17 females, 4 Caucasian (C), 14 African American (AA) and 2 Hispanic, age 24–68 years (19, 20). Eleven had lupus nephritis of whom two required dialysis. In our cohort, 17 patients had a SLE Disease Activity Index (SLEDAI) >3, indicative of active disease, and 18 were being treated with immunosuppressive therapy (21). Control groups consisted of 5 patients with Rheumatoid Arthritis (RA) who fulfilled the American College of Rheumatology classification criteria for RA and 26 healthy individuals. The RA group consisted of 5 females, 2C and 3AA, with age range of 40–68 years. The healthy control group consisted of 6 males, 17 females and 3 unknown, 21C, 2AA and 3 unknown, age 30–54 years. The study was approved by the University of Cincinnati Institutional Review Board.
Cells
Peripheral blood mononuclear cells (PBMC), CD3+, CD4+ and CD8+ lymphocytes were isolated from venous blood collected from consenting donors by Ficoll-Paque density gradient centrifugation (ICN Biomedicals, Aurora, OH, USA) and E-rosetting (StemCell Tech., Vancouver, Canada) as previously described (22). The homogeneity of the T cell populations was determined by FACS (22). Cells were maintained in RPMI medium supplemented with 10% pooled male human AB serum (Intergen, Milford, MA, USA), 200 U/ml penicillin, 200 μg/ml streptomycin, 1 mM Hepes. Pre-activated T cells were obtained by exposure to 4 μg/ml PHA (Sigma-Aldrich, St. Louis, MO) for 48–72 hrs in the presence of autologous PBMCs. Epstein-Barr Virus (EBV) infected-B cells were cultured in RPMI 1640 supplemented with 20% fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml of penicillin and 100 μg/ml of streptomycin.
Flow Cytometry
Freshly obtained peripheral blood was stained with the following antibodies: CD3-FITC, CD4-PerCP, CD8-APC-Cy7, CD45RA-PE-Cy7 (BD Biosciences, Mountain View, CA), CCR7-PE and CD45RO-APC (Pharmingen, San Jose, CA). The cells were stained for 20 min at room temperature followed by red cell lysis with FACSlyse solution (BD Biosciences, Mountain View, CA) for 10 min. The resultant white cell pellet was washed with PBS and fixed in 1% paraformaldehyde (PFA) prior to analysis by 4- or 6-color flow cytometry (FACSCalibur or FACSCanto flow cytometer, Becton Dickinson, San Jose, CA). Side scatter, CD3 and CD4 staining were used to distinguish for CD4+ and CD4− populations, which were then used as gates for an analysis of CCR7 vs CD45RO or CD45RA staining. Lymphocyte subsets were analyzed using MultiSet reagent cocktail (BD Biosciences).
T Cell Stimulation and Immunocytochemistry
T cells were stimulated using either anti-CD3/CD28 or anti-CD19 antibody-coated beads (Dynal Biotech, Lake Success, NY) (23). Alternatively, T cells were stimulated with EBV-B cells pre-pulsed with 7 μg/ml staphylococcal enterotoxin B (SEB) (Sigma-Aldrich), for 2 hr at 37°C and labeled with 5 μM cell tracker blue CMAC (Molecular Probes, Inc., Eugene, OR). T cells were mixed with either beads or B cells at a ratio of 1:1.5 and spun briefly at 100 g. After stimulation, they were maintained in a humidified incubator at 37°C for 1–30 min and plated onto poly-L-lysine coated coverslips. Attached cells were fixed with 4% PFA for 20 min, blocked using 10% normal goat serum or horse serum, permeabilized with 0.2% Triton X-100, and incubated overnight with primary antibodies followed by the appropriate fluorescent secondary antibodies (Molecular Probes Inc.). The primary antibodies used for detecting Kv1.3 proteins were either a rabbit polyclonal anti-Kv1.3 antibody against an epitope on the C-terminal domain of the protein (Alomone, Jerusalem, Israel) or an extracellular epitope (Sigma-Aldrich). The latter was used for labeling “live” Kv1.3 channels in T lymphocytes before interaction with the EBV-B cells. F-actin and GM1 were stained using Alexa Fluor 546 phalloidin and Alexa Fluor 555 cholera toxin B, respectively (Molecular Probes Inc.) and CD3ε was stained with a goat anti-CD3ε antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Fluorescence and Confocal Microscopy
Protein accumulation was detected by fluorescence microscopy using either a Nikon Microphot FXA or a Zeiss Axioplan Imaging 2 infinity-corrected upright scope coupled to an Orca-ER cooled camera (Axioscope, Carl Zeiss, Microimaging Inc.), Plan-Apochromat 60X-100X oil immersion objectives and the appropriate filters. For co-localization studies a Zeiss LSM510 laser scanning confocal microscope (Axioscope) equipped with an Ar ion laser, a HeNe laser and a Plan-Apochromat 63X oil immersion objective was used. The “Multi Track” option of the microscope was used to exclude cross-talk between detection channels.
Quantitation of fluorescence images
Kv1.3 accumulation at the bead/T cell point of contact was analyzed as previously described (24). Briefly, boxes of equal area were drawn around the IS and in the area most representative of the membrane outside the IS. The mean fluorescence ratio (MFR), indicative of protein recruitment, was calculated as follows: MFR = [Mean Fluoresce Intensity (MFI) at the IS–background]/[MFI outside the IS–background]. More than 50 T/bead conjugates were analyzed for each donor at each time point except for one RA patient for which 33 conjugates were analyzed for the 5 min time point. For the analysis of activated cells we used the increase in cell size as a marker of activation and excluded those cells that showed a resting phenotype (diameter .5 ≤ μm). To determine colocalization confocal image stacks of 0.8–1.5 μm thick optical slices were collected and single optical slices of doubly labeled cells were then evaluated. For quantitation of polarization in B/T cell conjugates a region was drawn around the T/B cell contact area and another region was drawn around the entire T cell. The fluorescence intensity was calculated for both regions. If the contact fluorescence was ≥50% of the total the T/B cell conjugate was scored as positive for protein recruitment into the IS (25). The data were analyzed using the Metamorph computer software.
Electrophysiology
K currents were recorded in whole-cell configuration. The external solution for activating and recording KCa3.1 currents had the following composition (in mM): 160 NaCl, 4.5 KCl, 2.0 CaCl2, 1.0 MgCl2, and 10 HEPES, pH 7.4. The pipette solution was composed of (mM): 145 K-Aspartate, 8.5 CaCl2, 10 K2EGTA, 2.0 MgCl2, and 10 HEPES, pH 7.2, with an estimated free [Ca2+] of 1 μM (26). KCa3.1 current was measured in voltage-clamp mode by ramp depolarization from −120 mV to +40 mV, 200 ms duration, every 10 s, −80 mV holding potential (HP). Data were corrected for a liquid junction potential of −10 mV (22). The slope conductance of the KCa3.1 current was measured between −100 mV and −60 mV. Kv1.3 currents were induced by depolarizing voltage steps from −80 mV HP and applied every 30 s, unless otherwise indicated. The external solution for recording of Kv1.3 currents had the following composition (in mM): 150 NaCl, 5 KCl, 2.5 CaCl2, 1.0 MgCl2, 10 glucose and 10 HEPES, pH 7.4. The pipette solution was composed of (mM): 134 KCl, 1 CaCl2, 10 EGTA, 2 MgCl2, 5 ATP-sodium, and 10 HEPES, pH 7.4 (estimated free Ca2+ concentration 10 nM) (27). The number of Kv1.3 and KCa3.1 channels per cell was determined by dividing the channel maximum conductances for their corresponding single channel conductances. The Kv1.3 single channel conductance was determined by us to be 11 pS (28). For KCa3.1 we used the single channel conductance determined by Grissmer et al. and used by others to calculate the number of KCa3.1 channels in T cells in similar experimental conditions (11, 29). Membrane potential was measured by current-clamp with the same solutions used to record Kv1.3 currents (22). The cell surface area was determined from the cell capacitance based on the approximation that 1 pF = 100 μm2 (13). Data were collected using the Axopatch200A amplifier and analyzed with pClamp8 software (Axon Instruments, Foster City, CA, USA).
Statistical Analysis
All data are presented as means ± SEM, unless otherwise indicated. Statistical analyses were performed using Student’s t-test (paired or unpaired); p≤0.05 was defined as significant.
RESULTS
Kv1.3 channels in T lymphocytes from patients with SLE display biophysical and pharmacological properties similar to those in healthy T cells
We have performed comparative studies aimed to identifying differences in the expression and activity of K channels in normal T lymphocytes and T lymphocytes from patients with SLE that could explain the enhanced Ca2+ response of the latter cells. The T cell phenotype of the SLE patients enrolled in this study was analyzed by flow cytometry. SLE patients displayed a significant reduction in CD4:CD8 ratio (Fig. 1A) as compared to healthy donors and RA patients, attributed to a significant decrease in CD4+ and an increase in CD8+ cells (Fig. 1B) and in agreement with previous reports (30, 31). Furthermore, SLE patients also display a significant increase in CD4+ Tem (CCR7−CD45RO+) cells but a decrease in CD8+ Tem cells (Fig. 1C). This demonstrates that CD4+ cells exist in a more active state in SLE patients as previously reported (31, 32).
Figure 1. Expression of T cell subsets in SLE, RA and normal donors.

(A) Flow cytometry analysis using anti-CD4 and anti-CD8 antibodies as gating antibodies of SLE patients (n=18), healthy controls (n=16) and RA patients (n=4). CD4/CD8 ratio indicates the relative proportions of T cells expressing CD4 or CD8. (B) Levels of expression of CD4 and CD8 in the 3 populations. C. Relative levels of naïve, Tcm and Tem cells in CD4+ (left) and CD8+ (right) lineages for SLE (n=18) and normal (n=16) individuals. Normal and SLE T cell populations were further characterized as follows: naïve (CCR7+ CD45RO−), Tcm (CCR7+ CD45RO+), and Tem (CCR7− CD45RO+). There was an increase in CD4+ Tem cells accompanied by a decrease in CD8+ Tem populations in SLE as compared to healthy donors. The levels of significance within the T cell subsets are indicated at the bottom.
Currently nothing is known about the expression and activity of ion channels in T cells from patients with SLE. Electrophysiological experiments were thus performed to characterize Kv1.3 channels in ex vivo SLE T cells. Throughout this manuscript we report data collected from SLE T cells within 24 hr from isolation from the blood. The time interval between blood collection and analysis is critical since it has been shown that after 24 hr in culture SLE T cells lose their peculiar characteristics (abnormalities in Lck, CD45 and lipid rafts) and revert to a normal T cell phenotype (30). Since the addition of patients’ serum during the in vitro incubation did not prevent this reversion of cell phenotype, it was suggested that interaction with other cell types might be responsible for the alteration in proximal signaling pathways (30). We found that Kv1.3 currents in SLE T cells were voltage-dependent with a V1/2 (voltage at which half of the channels are activated) of −27 ± 1 mV (n = 18) similar to that of normal resting T cells (−25 ± 1 mV, n = 6, p = 0.27) (12, 33) (Fig. 2A). The activation and inactivation time constants of Kv1.3 currents measured at +50 mV in SLE T cells were also similar to those of Kv1.3 current in healthy resting T cells. The activation time constants were 163 ± 13 ms (n = 52) in healthy T cells and 186 ± 29 ms (n = 20; p = 0.5) in SLE T cells. The time constants of inactivation in healthy and SLE resting T cells were 207 ± 19 ms (n = 52) and 247 ± 16 ms (n = 20, p = 0.1), respectively. Furthermore, Kv1.3 channels in SLE T cells display cumulative inactivation, a characteristic property of these channels, as indicated by the progressive decrease of the maximal current amplitude upon application of consecutive depolarizing pulses every second (Fig. 2B) (8). Additional studies established that the Kv1.3 currents recorded in SLE T cells were completely and reversibility blocked by the selective Kv1.3 inhibitor ShK-Dap22 (10 nM, Fig. 2C) and this concentration of ShK-Dap22 induced a 23 ± 4 mV (n = 4, Fig. 2D) membrane depolarization in these cells (34). We have also estimated whether T lymphocytes from SLE patients expressed the same number of Kv1.3 channels as healthy resting T cells. The total number of Kv1.3 channels/cell was determined by dividing the channel maximum conductance for its corresponding single channel conductance (11 pS) (28). SLE T cell express on average 349 ± 30 channels/cell (n = 20), ranging between 178 and 675 channels/cell. This is similar to the number of channels expressed by normal resting T cells (308 ± 16, n = 52; p = 0.2) (Table I).
Figure 2. Electrophysiological and pharmacological properties of Kv1.3 channels in SLE T cells.

A. Kv1.3 currents were recorded in whole-cell configuration and were elicited by depolarizing voltage steps from −60 mV to +40 mV (10 mV increments) from −80 mV HP every 30 sec. The conductance-voltage curve (constructed from current amplitudes such as those shown) was fitted to a Boltzman function and the voltage at which half of the channels are activated (V1/2) calculated. B. Kv1.3 channels’ cumulative inactivation was induced by consecutive depolarizing pulses applied every second. The maximal current amplitude progressively decreased with each successive pulse (indicated by progressive numbers). C. Effect of Shk-Dap22 on Kv1.3 current in SLE T cells. Currents were recorded in whole cell configuration in physiologic solution before application of Shk-Dap22 (-ShK-Dap22), after Shk-Dap22 (10 nM) inhibition and after drug wash-out (wo). D. Membrane potential (MP) measured by current-clamp before and after ShK-Dap22 (10 nM) addition. The time of ShK-Dap22 introduction is indicated by an arrow. The SLEDAI of the patients for this study ranged from 2–12.
Table I.
K channel expression in SLE and normal T lymphocytes
| Normal | SLE | |||
|---|---|---|---|---|
| Resting | Activated | |||
| Capacitance (pF)
n |
1.01 ± 0.04
62 |
4.46 ± 0.19***
55 |
1.49 ± 0.08***,$$$
40 |
|
| Kv1.3 | current density (pA/pF)
total channels (#channels/cell) channel density (#channels/μm2) n |
501 ± 36
308 ± 16 3.50 ± 0.25 52 |
263 ± 27***
786 ± 83*** 1.84 ± 0.19*** 23 |
416 ± 40$
349 ± 30$$$ 2.91 ± 0.28$ 20 |
| KCa3.1 | conductance (nS)
total channels (# channels/cell) channel density (#channels/μm2) n |
0.33 ± 0.03
29.7 ± 3.3 0.24 ± 0.03 10 |
2.55 ± 0.27***
232.0 ± 24.9*** 0.57 ± 0.07** 32 |
0.34 ± 0.03$$$
30.7 ± 2.5$$$ 0.21 ± 0.02$$$ 20 |
p<0.01 vs resting
p<0.0001 vs resting
p<0.005 vs activated
p<0.0001 vs activated
Taken together these data demonstrate that SLE T cells expressed the same number of Kv1.3 channels as resting T cells from healthy donors and that these channels share identical biophysical and pharmacological properties with their healthy counterparts. Moreover, Kv1.3 channels control the membrane potential in SLE T cells as indicated by the depolarization induced by Kv1.3 channel blockade.
Native Kv1.3 channels are recruited in the immunological synapse upon activation of healthy and SLE T cells
Since the biophysical properties of the channel remained unaltered we wanted to investigate whether other alterations in the Kv1.3 channel behavior might be encountered in SLE T cells. Previous studies have shown that recombinant Kv1.3 channels are recruited in the IS (14–16). However, the process by which native Kv1.3 channels transition into the IS is still to be defined. Furthermore, possible alterations of this process in diseased T cells have never been investigated. To address this question, we first investigated Kv1.3 channel polarization to the synapse in human CD3+ T cells from healthy donors. To induce T cell activation, and synapse formation, we used anti-CD3/CD28 antibody coated beads as surrogate APCs. This is a well validated system to study membrane reorganization and downstream functional events triggered by TCR binding (22, 23). Our results indicate that upon stimulation with CD3/CD28 beads, Kv1.3 channels partition to the T cell/bead contact area and colocalize extensively with F-actin and the glycosphingolipid GM1, a marker of lipid rafts (Fig. 3A bottom panels). Both F-actin and GM1 are known to reorganize and accumulate at the IS (18, 35). In contrast, Kv1.3 channels are evenly distributed on the membrane of resting T cells not exposed to beads (Fig. 3A top panels). In the same way, SLE and RA T cells recruit Kv1.3 channels in the cell/bead contact interface upon activation with the CD3/CD28 beads (Fig. 3B–C, lower panels) while the channels remain evenly distributed in the absence of beads (Fig. 3B–C, upper panels). To exclude that Kv1.3 channel relocalization occurs because of simple cell to bead contact, to establish the variability of our technique and to determine the threshold for a significant Kv1.3 channel accumulation in the synapse, we performed identical experiments using beads coated with an antibody against CD19 (a component of the B cell receptor complex) (23). In contrast to CD3/CD28 beads, CD19-coated beads did not have a significant effect on Kv1.3 or F-actin localization to the cell/bead contact interface (Fig. 3D). The degree of protein accumulation at the IS was indicated by the mean fluorescence ratio (MFR), calculated as described in the method section. The distributions of the MFRs in T cells exposed to CD3/CD28 and CD19 coated beads are reported in Fig. 3E. The cells stimulated with CD19 and CD3/CD28 beads had a MFR of 1.04 (SD 0.20, n = 49) and 1.78 (SD 0.24, n = 126), respectively. As a result, T cell/bead conjugates that displayed a MFR >1.5 (>2 folds the SD of the average MFR in CD19 experiments) were scored positive for Kv1.3 channel polarization in the IS. Based on these results we were able to study the kinetics of Kv1.3 accumulation in the IS. The process of IS formation is quite dynamic with different proteins transition in the synapse at different times. Thus specific kinetics of a protein transitioning in the IS might guarantee its coming in contact with signaling molecules present at the IS and thus its proper regulation and function. The time frame of Kv1.3 compartmentalization in the IS is not known in either normal or SLE T cells.
Figure 3. Kv1.3 channels are recruited at the interface between CD3/CD28 beads and T cells.
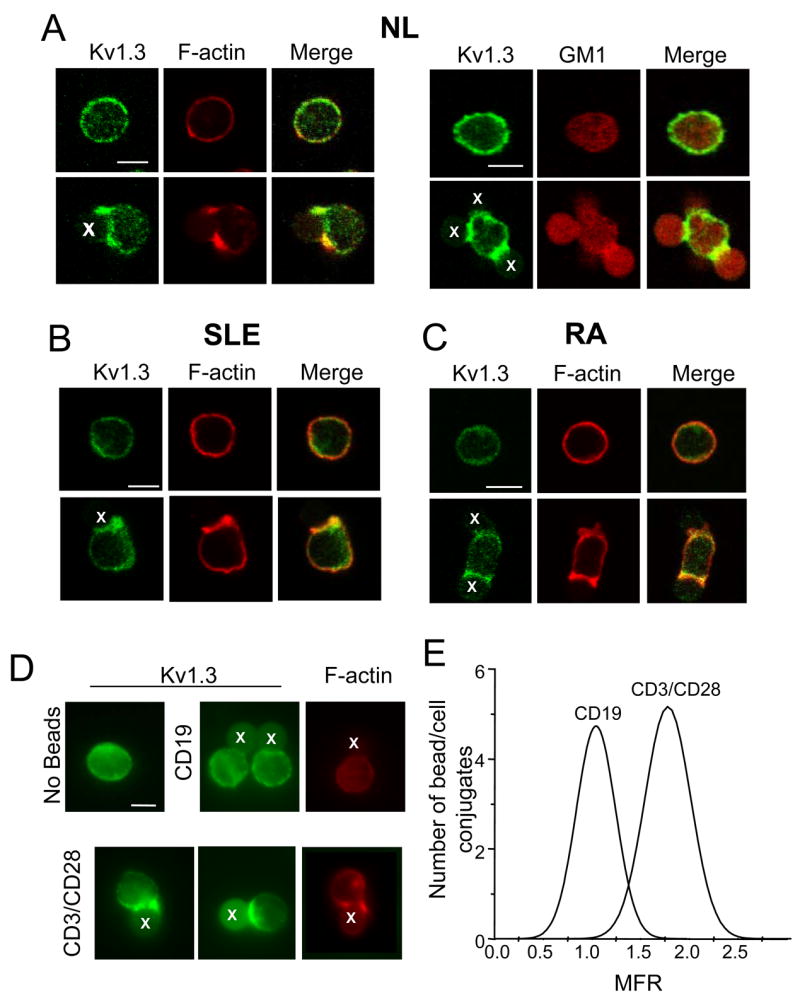
A–C. Normal (A), SLE (B) and RA (C) human CD3+ cells were stimulated with CD3/CD28 beads for 5–15 min at 37°C. Resting (non exposed to beads, top panels) or bead activated T cells (bottom panels) were fixed, permeabilized, immunolabelled for Kv1.3 (green) and either F-actin (A, left panel, B and C) or GM1 (A, right panel) and visualized with confocal microscopy. F-actin and GM1 were identified by fluorescence-labeled phalloidin and fluorescence-labeled cholera toxin B, respectively (red). Areas of co-localization of Kv1.3 channels and F-actin or GM1 are shown in yellow in the right panels (merge). D. T cells were exposed to either CD3/CD28 or CD19 beads for 15 min at 37°C. Resting (non exposed to beads, top left panel), CD19 exposed T cells (top middle and right panels) or CD3/CD28 activated T cells (bottom panels) were fixed, permeabilized, immunolabelled for Kv1.3 (green) and F-actin (red) and visualized with fluorescence microscopy. Beads are marked with an X. Scale bar = 5 μm. E. Distribution of the mean fluorescence ratio (MFR) in T/bead conjugates that form in presence of either CD19 or CD3/CD28 coated beads.
Kv1.3 channel compartmentalization in the immunological synapse is altered in SLE T cells
We studied the process of Kv1.3 channel translocation in the IS in SLE, RA and normal donors. Fourteen SLE patients were included in the following microscopy studies: 11 females and 3 males, 10AA and 4C, age, 38.0 ± 3.1 years (p = 0.3 vs. healthy individuals); range 24–67 years. These patients’ SLEDAI ranged from 2–12. As controls we used nine normal subjects: 5 females, 2 males and two unknown, 2AA, 5C and 2 unknown, age 43.3 ± 3.1 years; range 33–53 years and five RA patients: 5 female, 3AA and 2C, age 57 ± 6.0 and one unknown years; range 40–68.
We first examined the kinetics of Kv1.3 channel recruitment into the IS in resting T cells from healthy individuals by exposing them to CD3/CD28 beads for 1, 5, 15 and 30 min. Cell conjugates formed between CD3/CD28 beads and T cells were then fixed and immunostained with anti-Kv1.3 antibody. The assessment of the time-dependent distribution of Kv1.3 channels in the IS was done by establishing the number of T cell/bead conjugates with polarized Kv1.3 proteins over the total number of conjugates for each time point. Fig. 4A indicates that Kv1.3 channel redistribution in the IS of resting healthy T cells occurs after only 1 min of exposure to the beads and progressively increases overtime. Overall Kv1.3 recruitment in the IS is maintained for at least 30 min from synapse formation. Still at 1 hr there was sustained recruitment. The percentage of Kv1.3 polarized conjugates at 1 hr was 53 ± 5% (n = 2) (data not shown). By 2–5 hr the Kv1.3 channel was removed from the synapse with only 25 ± 4% conjugates showing Kv1.3 recruitment (n = 2) (data not shown). Similar experiments were performed with SLE T cells and we observed that in 7 out of eight patients the kinetics of Kv1.3 channel compartmentalization in the synapse are quite different (Fig. 4B). Specifically, Kv1.3 polarization in primary SLE T cells is maximal at 1 min after TCR engagement and progressively declines over time, indicating that Kv1.3 channels either re-distribute on the plasma membrane outside the IS or that they are internalized and degraded. This defect appears to be restricted to SLE as it was not observed in RA patients (Fig. 4C). Although some degree of variability was observed in individual RA patients, we never encountered Kv1.3 kinetics matching SLE T cells and on average, Kv1.3 channels are recruited in the IS of RA T cells within 1 min and are maintained there for at least 30 min.
Figure 4. Differential kinetics of Kv1.3 channel reorganization in the IS.

Left panels: Representative fluorescent images for resting normal (A) SLE (B) and RA (C) T cells after 1–30 min activation with CD3/CD28 beads. T cells were activated with CD3/CD28 beads for 1, 5, 15 and 30 min, fixed, permeabilized and stained with phalloidin conjugated to Alexa fluor 546 (to visualize F-actin) and Kv1.3 antibody followed by a fluorescent secondary antibody. Scale bar = 5 μm. Right panels: Quantitative analysis of Kv1.3 channel recruitment in the IS was performed as described in the methods section. The % of cells that display Kv1.3 accumulation in the IS at different times of exposure to beads is reported as % of cells with Kv1.3 polarized at the site of contact relative to the number of cells that made contact with beads. The data shown are the average responses collected from 6 healthy individuals (2 AA and 4 C, n=4 donors for 1min), 7 SLE patients (7 AA, SLEDAI 2–12) (n=6 for 1 min) and 5 RA patients (3AA and 2C).
This differential kinetics of Kv1.3 translocation into the IS in healthy and SLE T cells was also observed to occur at the interface between T cells and APCs (Fig. 5). T cells were incubated with EBV-infected B cells in the presence or absence of SEB for 1 and 30 min and the accumulation of Kv1.3 and CD3ε at the IS was determined. Control experiments were performed using EBV-infected B cells in the absence of SEB. To study the compartmentalization of Kv1.3 channels in the contact area between T cells and SEB-pulsed EBV-infected B cells we labeled the T cells “live” with an anti-Kv1.3 antibody against an extracellular (EC) epitope of the Kv1.3 channel protein before encounter with the APCs. This allowed selective labeling of the Kv1.3 channels in the T cell membrane and not those expressed in the B cells (36). This antibody is specific for Kv1.3 channels as determined by the lack of fluorescence signal after pre-adsorption of the antibody to the corresponding antigen (data not shown) and it can be used alternatively to the antibody for an intracellular (IC) epitope of the Kv1.3 channel that we have used in T cell/bead experiments. Similar results were obtained using the two antibodies (Fig. 5A). The accumulation of Kv1.3 channels in the IS was determined as described in the method section and B/T cell conjugates that displayed a fluorescence at the synapse ≥ 50% of the total fluorescence were defined as polarized Kv1.3 conjugates. Our results indicate that in the absence of SEB, Kv1.3 and CD3ε were evenly distributed on the plasma membrane of healthy T cells in the majority of the conjugates while in the presence of SEB Kv1.3 and CD3ε concentrated at the IS (Fig. 5B). Overall, normal resting T cells showed Kv1.3 polarization to the IS at 1 min and the channels were maintained in the IS for at least 30 min (Fig. 5B and D). All the cells that recruited Kv1.3 also recruited CD3ε. A different pattern of translocation into the IS that forms with APCs was instead observed with SLE T cells (Fig. 5C and E). Kv1.3 polarization in SLE T cells is maximal at 1 min after TCR engagement and is decreased by 30 min. These results substantiate, in a more physiological model of T/APC interaction, the observations made with the CD3/CD28 coated beads.
Figure 5. APC-T cell activation induces differential reorganization of Kv1.3 channels in the IS formed with resting healthy and SLE T cells.
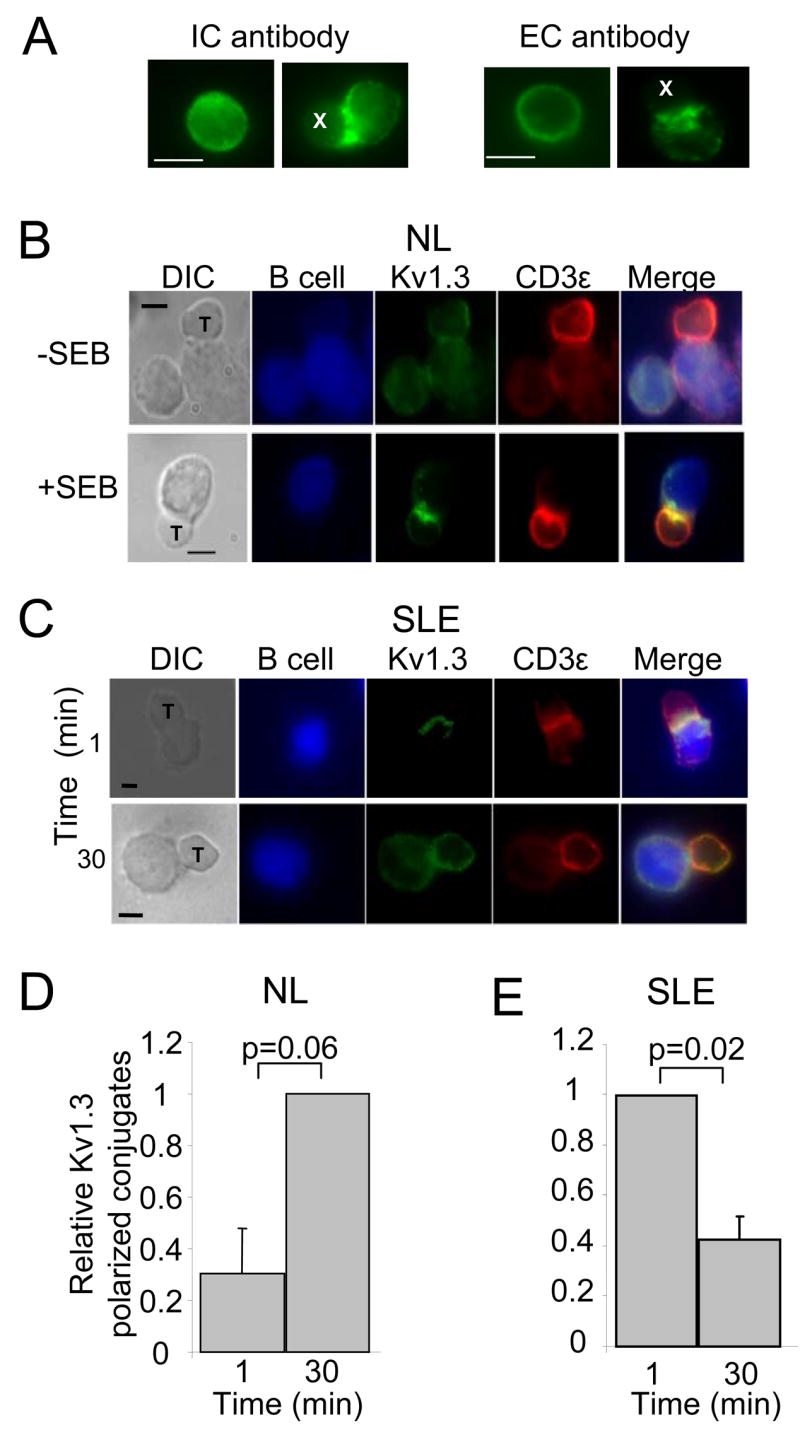
A. Comparable effectiveness of anti-Kv1.3 antibodies against an intracellular epitope (IC antibody) and an extracellular epitope (EC antibody) in detecting Kv1.3 channel accumulation in the IS. Resting T cells and T cells activated with CD3/CD28 beads are shown for each antibody (left and right images, respectively). T cells were either mixed with the beads and immunolabeled, after fixation, with anti-Kv1.3 IC antibody or labeled live with the anti-Kv1.3 EC before interaction with the beads. CD3/CD28 beads are marked by an X. B. Accumulation of Kv1.3 and CD3ε in the IS. Healthy resting T cells were incubated with EBV-infected B cells that had been exposed to medium with (bottom panels) or without (top panels) SEB. B cells were labeled with CMAC cell tracker blue. C. Accumulation of Kv1.3 and CD3ε in the IS formed between APCs and SLE T cells after 1 and 30 min interaction. Scale bar = 5μm. D–E. Time-dependent recruitment of Kv1.3 channels in the IS in healthy and SLE T cells. T/B cell conjugates were quantitatively evaluated for the recruitment of Kv1.3 channels as described in the methods section. The data are reported as average of the relative percentage of Kv1.3 polarized conjugates (normalized for the maximum recruitment of Kv1.3 polarized conjugates). The histograms represent 3 healthy donors (3 C) and 3 SLE patients (1 AA and 2 C, SLEDAI range: 2–8). At least 10 T/APC conjugates were evaluated for each donor per time point.
The kinetics of Kv1.3 redistribution in the immunological synapse of SLE T cells resemble those of pre-activated normal T cells
It is generally believed that SLE T cells exist in an active state (37). Accordingly, it is possible that the different dynamics of Kv1.3 compartmentalization in SLE T cells might reflect a more activated T cell phenotype. We have thus studied the process of Kv1.3 compartmentalization upon TCR-engagement in PHA pre-activated healthy T cells (Fig. 6). While resting T cells display a long-lasting recruitment of Kv1.3 channels in the IS (Figs. 4 and 5), pre-activated T cells display a different time-course: Kv1.3 channels moved rapidly to the IS with maximal recruitment at 1 min and progressively moved out of the synapse by 30 min (Fig. 6). Instead, in the absence of stimulation, Kv1.3 channels remained evenly distributed around the membrane. Consistent results were obtained using either CD3/CD28 beads or SEB-pulsed B cells as APCs. Overall, the dynamics of Kv1.3 compartmentalization in healthy activated T cells parallel the Kv1.3 recruitment observed in resting SLE T cells.
Figure 6. Kv1.3 channel recruitment in the IS in activated healthy T cells parallels SLE T lymphocytes.

A. T cells were pre-activated by exposure to PHA (4μg/ml) in the presence of autologous PBMCs for 72 hr. Pre-activated T cells were or were not stimulated with CD3/CD28 beads for 1–30 min. Left panels: representative photomicrographs. Right Panels: Quantitative analysis of Kv1.3 channel recruitment in the IS of activated T cells was performed as described in the methods section. The histogram shows the percentage of cells showing Kv1.3 accumulation at the IS at different time points (1–30 min). The data are the average of >50 cells/donor from seven healthy donors except 5 min, six donors. B. Activated healthy T cells were incubated with SEB-pulsed EBV B cells for 1 or 30 min. T/B cell conjugates were quantitatively evaluated for the recruitment of Kv1.3 channels as described in the methods section. The histograms represent an average of the relative percentage of Kv1.3 polarized conjugates (normalized for the maximum recruitment of Kv1.3 polarized conjugates) in 3 healthy donors. At least 10 T/APC conjugates were evaluated for each donor per time point.
The process of Kv1.3 channel translocation in the IS during activation with CD3/CD28 beads was quantitatively summarized by determining the rate of change in number of Kv1.3 polarized conjugates over time using a linear regression model. The “slope” of the model, indicative of the rate of formation of Kv1.3 polarized conjugates, was compared in SLE patients and normal controls (Fig. 7A). Overall we observed similar negative slopes in 7 out of 8 SLE patients, indicating that in these patients the localization of Kv1.3 in the IS is short-lived. Similar slopes were observed in activated healthy T cells and they were significantly different from those determined in healthy resting T cells. This behavior appeared to be unrelated to the disease activity and immunosuppressive regime. When we grouped all the SLE patients that were used in the microscopy studies, using either CD3/CD28 coated beads or APCs, we observed this Kv1.3 mobility defect in all except one patient. The outlier (SLE patient #8, Table II) displayed kinetics similar to normal resting T cells. This patient also showed a particularly immunosuppressed T cell phenotype as indicated by a percentage of naïve CD4+ and CD8+ cells well above healthy controls (70% and 84%, respectively). This raised the possibility that the abnormal Kv1.3 behavior in SLE T cells might be determined by the more activated state of their CD4 lineage (Fig. 1). If this is the case, we would expect that CD4+, and not CD8+, display these characteristic kinetics. Experiments were thus performed to compare the rate of Kv1.3 polarized conjugate formation in CD4+ and CD8+ cells from 4 SLE patients. We observed, on average, no differences between these T cell subsets (Fig. 7B).
Figure 7. Comparison of the rates of Kv1.3 channel compartmentalization in the IS in normal and SLE T cells.
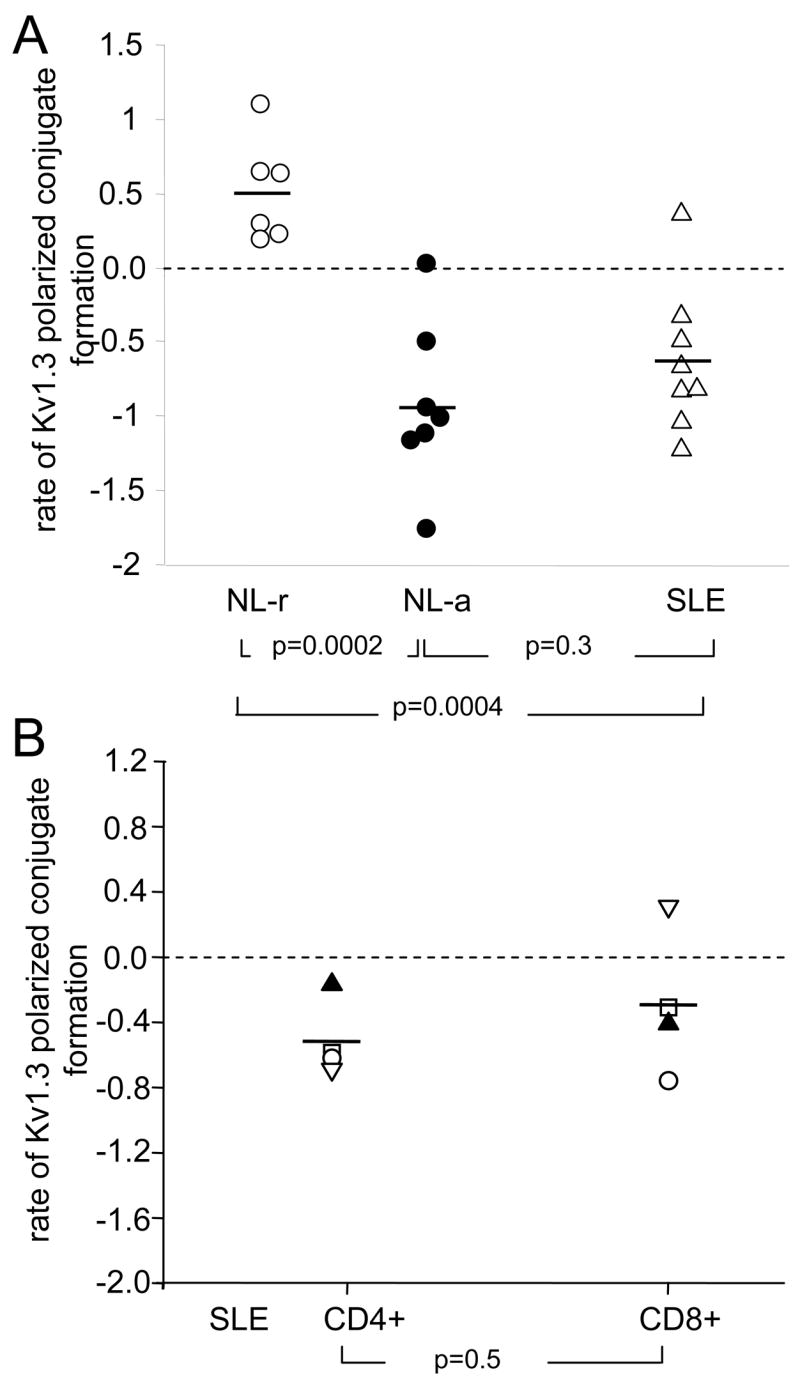
A. The rate of Kv1.3 polarized conjugate formation induced by activation with CD3/CD28 beads was determined in normal and SLE T lymphocytes by linear fitting of the time courses shown in figs 4C–D and 6B. The slope of the model is plotted for each group: normal lymphocytes (NL), resting (○) and activated (●), and SLE (△) T cells. A negative slope is indicative of rapid Kv1.3 channel redistribution outside the IS. B. Rate of Kv1.3 polarized conjugate formation in CD4+ and CD8+ cells from SLE patients. CD4+ and CD8+ cells were separated from the same individual and studied in parallel. A total of 4 SLE patients were studied: 3AA and 1C, SLEDAI range 4–10. T cells were activated by exposure to CD3/CD28 beads for 1 and 30 min. The number of Kv1.3 polarized conjugates for each time point was determined as described in the legend of Fig. 4 and plotted against time. The slope obtained by linear fitting of the time-course is reported. The symbols indicating each donor are conserved among the two groups.
Table II.
Details of SLE and RA patients in the microscopy studies
| Pt # | SLEDAI | MEDICATIONS |
|---|---|---|
| SLE patients | ||
| 1 | 12 | HCQ + MMF + Pred.* |
| 2 | 12 | Azathioprine + Pred.* |
| 3 | 5 | HCQ + Pred.* |
| 4 | 10 | Pred. |
| 5 | 4 | HCQ + Pred.* |
| 6 | 3 | HCQ |
| 7 | 2 | HCQ + MMF |
| 8 | 10 | HCQ + Pred. + MMF |
| 9 | 8 | Pred. + MMF |
| 10 | 2 | None |
| 11 | 4 | None |
| 12 | 4 | Pred.* |
| 13 | 10 | HCQ + Pred. |
| 14 | 4 | HCQ + Pred. + Azathioprine |
| RA patients | ||
| 1 | Pred. | |
| 2 | HCQ + Pred. * | |
| 3 | HCQ + MTX. | |
| 4 | HCQ | |
| 5 | HCQ + MTX + Pred.* | |
HCQ = Hydroxychloroquine (Plaquenil); MMF = Mycophenolate Mofetil (CellCept); MTX = Methotrexate; Pred.= Prednisone,
≤10mg.
Overall, these results establish that the general SLE T cell population displays faster kinetics of Kv1.3 channel translocation in and out of the IS as compared to healthy resting T cells. This defect occurs independently of disease activity as it was observed in patients with a SLEDAI ranging from 2–12. It also occurs independently of immunosuppressive therapy as 3 SLE patients who were not under immunosuppressive therapy (allow < 10mg Prednisone/day) display this defect. Moreover, freshly-isolated SLE T cells behave like healthy blast T cells in regards to Kv1.3 transitioning into the IS.
It has been shown that KCa3.1 channels, and not Kv1.3 channels, control Ca2+ homeostasis in activated cells (26). So it is possible that the rapid dynamics of the Kv1.3 translocation into the IS in pre-activated T cells are compensated by the presence of KCa3.1 channels. The expression of KCa3.1 channels in SLE T cells has yet to be determined. Experiments were thus performed to determine whether the K channel expression in SLE T cells matches pre-activated healthy T cells.
T lymphocytes from patients with SLE display a K channel phenotype similar to healthy resting T cells
Whole-cell voltage-clamp experiments were performed to determine the expression of KCa3.1 channels in SLE T cells and compare it to that of healthy T cells (Fig. 8 and Table I). The healthy T cells studied consisted of both resting (freshly-isolated) and mitogen pre-activated T cells obtained by prolonged exposure to PHA. This intervention has been shown to activate human T cells and increase their cell capacitance, a measure of cell size, and KCa3.1 conductance (26, 38). These cells also showed a faster Kv1.3 channel compartmentalization upon TCR activation (Fig. 6). Membrane capacitance measurements indicated that the activated T cells we studied were indeed activated. The membrane capacitances of mitogen pre-activated and resting (freshly-isolated) CD3+ cells were 4.46 ± 0.19 pF (n = 55) and 1.01 ± 0.04 pF (n = 62; p<0.001), respectively (Table I). Similar capacitance values have been reported for quiescent and pre-activated human T cells (39). Interestingly, we found that resting SLE T cells have membrane capacitance higher than healthy resting T cells, but less than pre-activated T cells. This indicates that resting SLE T cells are bigger than resting healthy T cells with an average cell surface area of 149 μm2 and 110 μm2, respectively (Fig. 8B). The cell surface area was determined from the cell capacitance based on the approximation that 1 pF = 100 μm2. This might indicate that SLE T cells are partially activated or are ‘frozen’ in an early stage of activation as previously suggested (37). Yet, the KCa3.1 conductance in SLE T cells is identical to normal resting T cells, suggesting that the number of channels is the same (Table I). Indeed, the KCa3.1 channel number/cell in SLE T cells is similar to that of primary resting T cells (Fig. 8C and table I). In contrast, healthy pre-activated T cells have an eightfold increase in KCa3.1 conductance which translates to an eightfold increase in channel numbers. When normalized for cell size, SLE T cells have KCa3.1 channel density similar to resting T cells and significantly lower than healthy pre-activated T cells (Table I). Similarly, the Kv1.3 channel density in SLE T cells is comparable to healthy primary T cells. The Kv1.3 and KCa3.1 channel composition in the mixed population of normal (resting and activated) and SLE T cells is summarized in Table I. These results indicate that the number of functional Kv1.3 and KCa3.1 channels expressed in SLE T cells is similar to that of healthy resting T cells. Thus, Kv1.3 channels constitute the main K conductance in SLE T cells and as such are the main regulators of membrane potential and Ca2+ homeostasis in these cells.
Figure 8. The expression of KCa3.1 channels in SLE T cells is comparable to that in resting healthy T cells.

A. KCa3.1 channel currents were recorded in whole cell configuration, voltage-clamp mode by ramp depolarization from −120 mV to +40mV (200 ms duration, every 10 sec, HP= −80mV). Characteristic traces are shown for resting SLE (left), normal resting (middle) and activated (right) T lymphocytes (NL). B. Cell surface area for SLE (n=40) and healthy, resting (n=62) and activated (n=55) T cells. C. Expression levels of KCa3.1 channels in SLE and healthy (resting and activated) T cells. The total number of KCa3.1 channel/cell was calculated by dividing the KCa3.1 maximum conductance (determined by fitting of the linear portion of the KCa3.1 current measured between −100 mV and −60 mV) for the KCa3.1 unitary conductance. The SLEDAI of the patients for this study ranged from 2–12.
DISCUSSION
In this study we examine for the first time K channels in SLE T cells and provide evidence of significant differences in Kv1.3 channel dynamics of translocation in the IS between resting healthy and SLE T lymphocytes. We also find that in SLE T cells the movement of the Kv1.3 channel on the plasma membrane during presentation of the antigen and formation of the IS resembles that of healthy activated T cells. Despite that, SLE T cells lack the corresponding K channel make-up that is integral in regulating the activation response in normal activated T cells. These discrepancies might account for the hyperactivity and exaggerated response of SLE T cells to antigen presentation.
K channels have been shown to be key regulators of T cell activation as they control the membrane potential and the influx of Ca2+ triggered by antigen presentation (8). As such K channels could play a role in the abnormal Ca2+ response to antigen stimulation that has been reported to occur in human SLE T lymphocytes (5, 6). Nevertheless, their activity and function in SLE T cells has never been investigated. We have conducted various studies aimed at dissecting the properties of K channels in SLE T cells. These studies were conducted on a cohort of SLE patients with a T cell phenotype characteristic of this disease as indicated by the low CD4/CD8 ratio (30, 31). This low CD4/CD8 ratio might have been exacerbated by the presence of patients with lupus nephritis and patients under corticosteroid treatment (40). Furthermore, the SLE patients display an activated CD4+ memory phenotype with CD4+ TEM levels higher than healthy individuals as previously described (41). This is accompanied by a decreased expression of CD8+ TEM cells and it is in agreement with the common consent that the altered immune response in SLE is mediated by an imbalance in the functions of T cell subsets: exaggerated activity of CD4+ helper cells and diminished function of CD8+ suppressor/cytotoxic cells (3).
When we analyzed this mixed population of SLE T cells for the expression of K channels we found that SLE T cells display a K channel phenotype similar to normal resting T cells with Kv1.3 channels constituting the main K conductance and controlling the membrane potential. Although we showed no differences in the biophysical and pharmacological properties of Kv1.3 channels in SLE T cells as well as in their number as compared to normal resting T cells, our data indicate that there are fundamental differences in the process of Kv1.3 channel translocation in the IS. The accumulation of Kv1.3 channels in the IS in healthy resting T cells occurs progressively and it is sustained for a long time, well beyond the onset of signal transduction (i.e. the onset of Ca2+ influx) (42). This is consistent with the long time necessary for resting T cells to form mature synapses (43). In contrast, SLE T cells show a faster recruitment of Kv1.3 channels into the IS and redistribution outside the synapse. Interestingly, the process of Kv1.3 recruitment in SLE T cells parallels the process observed in healthy pre-activated T cells. Indeed it has been shown that synapse maturation occurred much faster in T cell blasts than resting T cells (43). This behavior of SLE T cells is consistent with the view that T cells from SLE patients resemble activated T cells (37). T cells from patients with SLE display various characteristics of activated T cells: they exhibit a loss of CD3ζ chain which is replaced by FcRγ chain and Syk recruitment to the TCR complex. These alterations indicate a switch from the TCR/CD3/CD3ζ/ZAP-70 receptor complex of resting T cells to the TCR/CD3/FcRγ/Syk receptor complex of effector T cells (37). Functionally SLE T cells are primed for activation and respond more rapidly to antigenic triggers than do T cells from normal individuals (30). Furthermore, they react more rapidly than healthy T cells to antigen presentation in terms of reorganization of elements known to accumulate at the IS such as F-actin and lipid rafts (6). Along this line, the significantly larger size of SLE T cells we have measured during our electrophysiological experiments indicates that in these patients T cells exist in a partially activated state as previously suggested (37). Similar to our findings all these alterations were found in freshly isolated peripheral blood T cells from SLE patients independent of their disease activity, thus suggesting a constant activation state of SLE T cells (6, 31).
However, although SLE T cells circulate as activated (or partially activated) T cells they do not display the K channel make-up characteristic of activated T cells. SLE T cells express ca. 300 Kv1.3 and ca. 30 KCa3.1 channels/cell. Similar values are reported in the literature for resting naïve, TCM and TEM cells of the CD4 and CD8 lineages (13). Although we were measuring a mixed population, we never encountered SLE T cells with either high Kv1.3 channel number (≥1,500/cell), indicative of the activated TEM phenotype, or high KCa3.1 channel number, indicative of activated naïve and TCM cells (13). Overall, freshly isolated peripheral blood SLE T cells express a number of Kv1.3 and KCa3.1 channels equal to resting healthy T cells and, likewise, Kv1.3 channels constitute the main K conductance in SLE T cells. As such, they modulate SLE T cell membrane potential. Thus alterations in Kv1.3 channel behavior might have important consequences in the Ca2+ homeostasis of SLE T cells. It is possible that alterations in dynamics of Kv1.3 localization in the IS contribute to the pronounced and sustained TCR-mediated Ca2+ influx of SLE T cells. This exaggerated Ca2+ response was observed in both SLE CD4+ and CD8+ subsets, although higher in CD4+ (5). Consistently, we showed that both CD4+ and CD8+ SLE T cells display faster dynamics of Kv1.3 translocation in and out of the IS. Furthermore, we showed that this defect is not present in T cells from RA patients. This is consistent with the fact that RA T cells do not display an exaggerated Ca2+ response to antigen presentation (5, 44).
The functional consequences of this differential dynamics of Kv1.3 protein localization in the IS of the SLE T cells are unclear at present. However, it has been suggested that ion channel localization in the IS might be necessary for guaranteeing the channel proximity to signaling molecules that control the channel’s function (45). The data we have presented indicating that in healthy resting T cells the Kv1.3 channels are maintained in the IS for about 2 hr are consistent with the notion that a prolonged interaction of naïve T cell with APC lasting 2 hr or more is required for cell division and IL-2 production/release from the cell (43, 46, 47). Although it has been shown that tyrosine phosphorylation activation mechanisms and the initial Ca2+ influx occur early upon T cell contact with the antigen (within 2–15 min), other signaling systems such as those involving Ca2+ or serine/threonine phosphorylation have been suggested to be critical during the later stages of activation. Since Kv1.3 channels are known regulators of Ca2+ homeostasis in human T cells and their activity is modulated by serine/threonine kinases it is very likely that they constitute key components of the late activation signaling complex. The prolonged time Kv1.3 channels reside in the IS may indeed be necessary for the channels to come in close proximity with signaling molecules also recruited at the IS thus facilitating the regulation of their activity and consequently the control and termination of the Ca2+ response. It has been shown that various elements that accumulate at the IS such as cholesterol and lipid rafts as well as Lck, PKC and PKA can modulate Kv1.3 channel function (28, 48–53). Furthermore these kinases move into the IS at different times after antigen presentation, with PKA and PKCθ still present at the IS well after a mature synapse is formed (54–56). Our results suggest a model in which a proper time-dependent localization of Kv1.3 in the IS is necessary for its regulation. In normal resting T cells the Kv1.3 channel remains in the IS for the time necessary for its regulation. This process responsible for bringing Kv1.3 channels into close physical proximity with signaling molecules would have particular biological relevance in the setting of SLE where there is a documented decrease in the expression and activity of multiple kinases (3). Unfortunately, since Kv1.3 channels in SLE T cells leave the IS prematurely they might not be properly regulated and an abnormal Ca2+ response might develop. On the other hand, a prolonged localization of Kv1.3 channels is instead not necessary in normal activated T cells because they also express high levels of KCa3.1 channels that could control Ca2+ homeostasis (26). We have recently shown that KCa3.1 channels are recruited in the IS of activated T cells where they reside for 30 min (17).
The data presented herein raise the possibility that Kv1.3 channels might be involved in the pathophysiology of SLE. Given the availability of pharmacological agents altering these channels, these data may lead to the discovery of new therapeutic targets for this disease.
Acknowledgments
We thank Mr. M. K. Ragupathy for his help with image analysis and the Center for Biostatistical Services at the University of Cincinnati for advice regarding data analysis. We also thank Dr C. Chougnet for critical reviewing of the manuscript.
Footnotes
This work was supported by NIH grant #CA95286 to LC and AHA-Ohio Valley Affiliate pre-doctoral fellowship #0615213B to SAN.
Abbreviations used in this paper: BF, brightfield; DIC, Differential Interference Contrast; IS, immunological synapse; MFR, mean fluorescent ratio; PHA, phytohemagglutinin; SEB, staphylococcal enterotoxin B; SLE, systemic lupus erythematosus; SLEDAI, SLE disease activity index; TCM, central memory T cells; TEM, effector memory T cells.
Publisher's Disclaimer: Disclaimer: “This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.”
References
- 1.Kammer GM. Altered regulation of IL-2 production in systemic lupus erythematosus: an evolving paradigm. J Clin Invest. 2005;115:836–840. doi: 10.1172/JCI24791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laxminarayana D, I, Khan U, Kammer G. Transcript mutations of the alpha regulatory subunit of protein kinase A and up-regulation of the RNA-editing gene transcript in lupus T lymphocytes. Lancet. 2002;360:842–849. doi: 10.1016/s0140-6736(02)09966-x. [DOI] [PubMed] [Google Scholar]
- 3.Kammer GM, Perl A, Richardson BC, Tsokos GC. Abnormal T cell signal transduction in systemic lupus erythematosus. Arthritis Rheum. 2002;46:1139–1154. doi: 10.1002/art.10192. [DOI] [PubMed] [Google Scholar]
- 4.Liossis SN, Vassilopoulos D, Kovacs B, Tsokos GC. Immune cell biochemical abnormalities in systemic lupus erythematosus. Clin Exp Rheumatol. 1997;15:677–684. [PubMed] [Google Scholar]
- 5.Vassilopoulos D, Kovacs B, Tsokos GC. TCR/CD3 complex-mediated signal transduction pathway in T cells and T cell lines from patients with systemic lupus erythematosus. J Immunol. 1995;155:2269–2281. [PubMed] [Google Scholar]
- 6.Krishnan S, Nambiar MP, Warke VG, Fisher CU, Mitchell J, Delaney N, Tsokos GC. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J Immunol. 2004;172:7821–7831. doi: 10.4049/jimmunol.172.12.7821. [DOI] [PubMed] [Google Scholar]
- 7.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 8.Cahalan MD, Wulff H, Chandy KG. Molecular properties and physiological roles of ion channels in the immune system. J Clin Immunol. 2001;21:235–252. doi: 10.1023/a:1010958907271. [DOI] [PubMed] [Google Scholar]
- 9.Gallo EM, Cante-Barrett K, Crabtree GR. Lymphocyte calcium signaling from membrane to nucleus. 2006;7:25–32. doi: 10.1038/ni1295. [DOI] [PubMed] [Google Scholar]
- 10.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 11.Wulff H, Calabresi PA, Allie R, Yun S, Pennington M, Beeton C, Chandy KG. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J Clin Invest. 2003;111:1703–1713. doi: 10.1172/JCI16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beeton C, Barbaria J, Giraud P, Devaux J, Benoliel AM, Gola M, Sabatier JM, Bernard D, Crest M, Beraud E. Selective blocking of voltage-gated K+ channels improves experimental autoimmune encephalomyelitis and inhibits T cell activation. J Immunol. 2001;166:936–944. doi: 10.4049/jimmunol.166.2.936. [DOI] [PubMed] [Google Scholar]
- 13.Chandy GK, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panyi G, Varga Z, Gaspar R. Ion channels and lymphocyte activation. Immunol Lett. 2004;92:55–66. doi: 10.1016/j.imlet.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Panyi G, Bagdany M, Bodnar A, Vamosi G, Szentesi G, Jenei A, Matyus L, Varga S, Waldmann TA, Gaspar R, Damjanovich S. Colocalization and nonrandom distribution of Kv1.3 potassium channels and CD3 molecules in the plasma membrane of human T lymphocytes. Proc Natl Acad Sci U S A. 2003;100:2592–2597. doi: 10.1073/pnas.0438057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panyi G, Vamosi G, Bacso Z, Bagdany M, Bodnar A, Varga Z, Gaspar R, Matyus L, Damjanovich S. Kv1.3 potassium channels are localized in the immunological synapse formed between cytotoxic and target cells. Proc Natl Acad Sci U S A. 2004;101:1285–1290. doi: 10.1073/pnas.0307421100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolaou SA, Neumeier L, Peng Y, Devor D, Conforti L. The Ca2+-activated K channel KCa3.1 compartmentalizes in the immunological synapse of human T lymphocytes. Am J Physiol Cell Physiol. 2006;003760 doi: 10.1152/ajpcell.00376.2006. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dustin ML. The immunological synapse. Arthritis Res. 2002;4(Suppl 3):S119–125. doi: 10.1186/ar559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 20.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 21.Kalunian K. Definition, classification, activity, and damage indices. In: Wallace D, Hahn B, editors. Dubois' Lupus Erythematosus. Williams & Wilkins; Baltimore, MD: 1996. pp. 19–30. [Google Scholar]
- 22.Robbins JR, Lee SM, Filipovich AH, Szigligeti P, Neumeier L, Petrovic M, Conforti L. Hypoxia modulates early events in T cell receptor-mediated activation in human T lymphocytes via Kv1.3 channels. J Physiol. 2005;564:131–143. doi: 10.1113/jphysiol.2004.081893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xavier R, Rabizadeh S, Ishiguro K, Andre N, Ortiz JB, Wachtel H, Morris DG, Lopez-Ilasaca M, Shaw AC, Swat W, Seed B. Discs large (Dlg1) complexes in lymphocyte activation. J Cell Biol. 2004;166:173–178. doi: 10.1083/jcb.200309044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tavano R, Gri G, Molon B, Marinari B, Rudd CE, Tuosto L, Viola A. CD28 and lipid rafts coordinate recruitment of Lck to the immunological synapse of human T lymphocytes. J Immunol. 2004;173:5392–5397. doi: 10.4049/jimmunol.173.9.5392. [DOI] [PubMed] [Google Scholar]
- 25.Round JL, Tomassian T, Zhang M, Patel V, Schoenberger SP, Miceli MC. Dlgh1 coordinates actin polymerization, synaptic T cell receptor and lipid raft aggregation, and effector function in T cells. J Exp Med. 2005;201:419–430. doi: 10.1084/jem.20041428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, Cahalan MD, Chandy KG. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem. 2000;275:37137–37149. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- 27.Szabo I, Nilius B, Zhang X, Busch AE, Gulbins E, Suessbrich H, Lang F. Inhibitory effects of oxidants on n-type K+ channels in T lymphocytes and Xenopus oocytes. Pflugers Arch. 1997;433:626–632. doi: 10.1007/s004240050323. [DOI] [PubMed] [Google Scholar]
- 28.Szigligeti P, Neumeier L, Duke E, Chougnet C, Takimoto K, Lee SM, Filipovich AH, Conforti L. Signalling during hypoxia in human T lymphocytes - critical role of the src protein tyrosine kinase p56Lck in the O2 sensitivity of Kv1.3 channels. J Physiol. 2006;573:357–370. doi: 10.1113/jphysiol.2006.109967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grissmer S, Nguyen AN, Cahalan MD. Calcium-activated potassium channels in resting and activated human T lymphocytes. Expression levels, calcium dependence, ion selectivity, and pharmacology. J Gen Physiol. 1993;102:601–630. doi: 10.1085/jgp.102.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jury EC, Kabouridis PS, Flores-Borja F, Mageed RA, Isenberg DA. Altered lipid raft-associated signaling and ganglioside expression in T lymphocytes from patients with systemic lupus erythematosus. J Clin Invest. 2004;113:1176–1187. doi: 10.1172/JCI20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pavon EJ, Munoz P, Navarro MD, Raya-Alvarez E, Callejas-Rubio JL, Navarro-Pelayo F, Ortego-Centeno N, Sancho J, Zubiaur M. Increased association of CD38 with lipid rafts in T cells from patients with systemic lupus erythematosus and in activated normal T cells. Mol Immunol. 2006;43:1029–1039. doi: 10.1016/j.molimm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Jury EC, Kabouridis PS. T-lymphocyte signalling in systemic lupus erythematosus: a lipid raft perspective. Lupus. 2004;13:413–422. doi: 10.1191/0961203304lu1045rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- 34.Kalman K, Pennington MW, Lanigan MD, Nguyen A, Rauer H, Mahnir V, Paschetto K, Kem WR, Grissmer S, Gutman GA, Christian EP, Cahalan MD, Norton RS, Chandy KG. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J Biol Chem. 1998;273:32697–32707. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- 35.Dykstra M, Cherukuri A, Sohn HW, Tzeng SJ, Pierce SK. Location is everything: lipid rafts and immune cell signaling. Annu Rev Immunol. 2003;21:457–481. doi: 10.1146/annurev.immunol.21.120601.141021. [DOI] [PubMed] [Google Scholar]
- 36.Wulff H, Knaus HG, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- 37.Krishnan S, Farber DL, Tsokos GC. T Cell Rewiring in Differentiation and Disease. J Immunol. 2003;171:3325–3331. doi: 10.4049/jimmunol.171.7.3325. [DOI] [PubMed] [Google Scholar]
- 38.Fanger CM, Rauer H, Neben AL, Miller MJ, Wulff H, Rosa JC, Ganellin CR, Chandy KG, Cahalan MD. Calcium-activated potassium channels sustain calcium signaling in T lymphocytes. Selective blockers and manipulated channel expression levels. J Biol Chem. 2001;276:12249–12256. doi: 10.1074/jbc.M011342200. [DOI] [PubMed] [Google Scholar]
- 39.Beeton C, Wulff H, Singh S, Botsko S, Crossley G, Gutman GA, Cahalan MD, Pennington M, Chandy KG. A Novel Fluorescent Toxin to Detect and Investigate Kv1.3 Channel Up-regulation in Chronically Activated T Lymphocytes. J Biol Chem. 2003;278:9928–9937. doi: 10.1074/jbc.M212868200. [DOI] [PubMed] [Google Scholar]
- 40.Horwitz DA, SW, Gray JD. T lymphocytes, natural killer cells, cytokines, and immune regulation. In: Wallace DaHBH., editor. Dubois' Lupus Erythematosus. Wiliams & Wilkins; Baltimore: 1997. pp. 155–194. [Google Scholar]
- 41.Han BK, White AM, Dao KH, Karp DR, Wakelan EK, Davis LS. Increased prevalence of activated CD70+CD4+ T cells in the periphery of patients with systemic lupus erythematosus. Lupus. 2005;14:598–606. doi: 10.1191/0961203305lu2171oa. [DOI] [PubMed] [Google Scholar]
- 42.Gascoigne NR, Zal T. Molecular interactions at the T cell-antigen-presenting cell interface. Curr Opin Immunol. 2004;16:114–119. doi: 10.1016/j.coi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 43.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 44.Carruthers DM, Naylor WG, Allen ME, Kitas GD, Bacon PA, Young SP. Characterization of altered calcium signalling in T lymphocytes from patients with rheumatoid arthritis (RA) Clin Exp Immunol. 1996;105:291–296. doi: 10.1046/j.1365-2249.1996.d01-768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panyi G, Vamosi G, Bodnar A, Gaspar R, Damjanovich S. Looking through ion channels: recharged concepts in T-cell signaling. Trends Immunol. 2004;25:565–569. doi: 10.1016/j.it.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 46.Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 47.Weiss A, Shields R, Newton M, Manger B, Imboden J. Ligand-receptor interactions required for commitment to the activation of the interleukin 2 gene. J Immunol. 1987;138:2169–2176. [PubMed] [Google Scholar]
- 48.Cayabyab FS, Khanna R, Jones OT, Schlichter LC. Suppression of the rat microglia Kv1.3 current by src-family tyrosine kinases and oxygen/glucose deprivation. Eur J Neurosci. 2000;12:1949–1960. doi: 10.1046/j.1460-9568.2000.00083.x. [DOI] [PubMed] [Google Scholar]
- 49.Chung I, Schlichter LC. Native Kv1.3 channels are upregulated by protein kinase C. J Membr Biol. 1997;156:73–85. doi: 10.1007/s002329900189. [DOI] [PubMed] [Google Scholar]
- 50.Bock J, Szabo I, Gamper N, Adams C, Gulbins E. Ceramide inhibits the potassium channel Kv1.3 by the formation of membrane platforms. Biochem Biophys Res Commun. 2003;305:890–897. doi: 10.1016/s0006-291x(03)00763-0. [DOI] [PubMed] [Google Scholar]
- 51.Hajdu P, Varga Z, Pieri C, Panyi G, Gaspar R., Jr Cholesterol modifies the gating of Kv1.3 in human T lymphocytes. Pflugers Arch. 2003;445:674–682. doi: 10.1007/s00424-002-0974-y. [DOI] [PubMed] [Google Scholar]
- 52.Chung I, Schlichter LC. Regulation of native Kv1.3 channels by cAMP-dependent protein phosphorylation. Am J Physiol. 1997;273:C622–633. doi: 10.1152/ajpcell.1997.273.2.C622. [DOI] [PubMed] [Google Scholar]
- 53.Payet MD, Dupuis G. Dual regulation of the n type K+ channel in Jurkat T lymphocytes by protein kinases A and C. J Biol Chem. 1992;267:18270–18273. [PubMed] [Google Scholar]
- 54.Altman A, Villalba M. Protein kinase C-theta (PKCtheta): it's all about location, location, location. Immunol Rev. 2003;192:53–63. doi: 10.1034/j.1600-065x.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- 55.Torgersen KM, Vang T, Abrahamsen H, Yaqub S, Tasken K. Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell Signal. 2002;14:1–9. doi: 10.1016/s0898-6568(01)00214-5. [DOI] [PubMed] [Google Scholar]
- 56.Zhou W, Vergara L, Konig R. T cell receptor induced intracellular redistribution of type I protein kinase A. Immunology. 2004;113:453–459. doi: 10.1111/j.1365-2567.2004.01992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]