

Abstract
Background
Previous reports have suggested that levels of advanced glycation end product-modified LDL (AGE-LDL) increase in patients with diabetes due to elevated plasma glucose. However, understanding of the mechanisms by which AGE-LDL may accelerate atherogenesis remains incomplete.
Methods and Results
Microarray and reverse transcription real-time PCR (RT-PCR) analyses revealed that AGE-LDL significantly increased levels of CC chemokine receptor 2 (CCR2) mRNA in human macrophages compared with LDL, an effect accompanied by increased levels of CCR2 protein. Flow cytometry also showed that AGE-LDL increases CCR2 expression on the cell surface following stimulation (48 hours) (P<0.05). This effect appeared to depend on the receptor for AGE (RAGE), since an anti-RAGE antibody significantly blocked increased CCR2 mRNA. Functional studies demonstrated that exposure of THP-1 monocytoid cells to AGE-LDL increases chemotaxis mediated by monocyte chemoattractant protein-1 (MCP-1) up to 3-fold compared to LDL treatment (P<0.05).
Conclusions
These data show that AGE-LDL can increase CCR2 expression in macrophages and stimulate the chemotactic response elicited by MCP-1. This novel mechanism may contribute to accelerated atherogenesis in diabetic patients.
Keywords: diabetes mellitus, cholesterol, lipoprotein, metabolism, atherosclerosis
Diabetes mellitus (DM) associates with increased incidence of macrovascular complications including coronary artery and peripheral vascular diseases [1-3]. Although the mechanisms for DM acceleration of atherosclerosis remain incompletely understood, the persistence of high plasma glucose levels in DM patients creates favorable conditions for non-enzymatic glycation of proteins and lipids and formation of advanced glycation end-products (AGE) [4]. AGE may promote vascular dysfunction in diabetes by various mechanisms including activation of the endothelium, thus resulting in increased monocyte chemoattractant protein-1 (MCP-1) expression [5] and induction of smooth muscle proliferation [6].
The non-enzymatic glycation of LDL can occur in vivo when blood glucose chemically modifies lysine residues of LDL apolipoprotein-B [7]. The plasma levels of AGE-modified LDL (AGE-LDL) increase in DM patients due to elevated concentrations of plasma glucose [7-9]. In vitro studies demonstrated that AGE-LDL from diabetic subjects adversely affects cultured cells relevant to atherosclerosis, resulting in cholesteryl ester accumulation in monocyte-derived macrophages and procoagulant effects on endothelial cells [9, 10]. Indeed, LDL glycation and oxidation [11, 12], alone or in combination, may contribute to the increased atherogenic risk in DM patients [13]. The toxicity of AGE-LDL [14] and its role in the pathogenesis of atherosclerosis may relate to its prolonged presence in the circulation [15], which results from impaired cellular uptake [16, 17].
Vascular inflammation plays a central role in atherogenesis [18, 19]. Chemokines regulate leukocyte migration and infiltration into the vascular wall, a critical initial step in lesion formation [19-21]. MCP-1, a monocyte/macrophage chemoattractant that contributes to the pathogenesis of chronic inflammation, belongs to the CC subfamily of chemokines [22]. The effects of MCP-1 depend primarily on CC chemokine receptor 2 (CCR2) [22]. Targeted inactivation of either the MCP-1 or the CCR2 gene markedly decreased lesion formation in apoE-deficient mice [23], indicating that CCR2 engagement contributes to the development of atherosclerotic lesions. In particular, atheromata from diabetic patients have accentuated accumulation of macrophages, although the mechanisms remain unknown [24, 25]. This study demonstrates that AGE-LDL increases CCR2 expression in human macrophages and stimulates MCP-1-mediated THP-1 monocytoid cell chemotaxis. These results contribute to the understanding of AGE-LDL-mediated mechanisms that may promote macrophage accumulation and atherosclerosis in diabetic patients.
Methods
Preparation of AGE-LDL
LDL (d= 1.019 to 1.063 g/ml) was separated from normal human plasma, dialyzed extensively at 4°C in the dark, and modified in vitro by glycation as described previously [17]. Briefly, we incubated LDL at 37°C for 7 days under argon gas in the presence of 25 mmol/L glucose, and then removed unincorporated sugars by repeated and extensive dialysis. We incubated control LDL under similar conditions, but without glucose. We passed the LDL preparations through sterile filters (0.22 μm) and stored them in the dark under argon gas at 4°C. Endotoxin was <40pg endotoxin/ml as determined by the chromogenic Limulus amoebocyte assay (Cape Cod, Falmouth, MA). Protein modification was evaluated by measuring pentosidine formation spectrofluorometrically (excitation at 335 nm, emission at 385 nm) [26]. Oxidation was measured using a highly sensitive sandwich ELISA using DH3, a monoclonal antibody that recognizes oxidatively modified lipoproteins (Kyowa Medex, Tokyo, Japan), and an anti-human apoprotein B monoclonal antibody (BD Biosciences) [27]. In the ELISA plate, various concentrations of standard oxidized LDL, which was prepared by incubating LDL with 5 μmol/L CuSO4 at 37°C for 3 hours, were run simultaneously to determine a standard curve. The concentrations of oxidized LDL are expressed in ng/5μg LDL protein.
Macrophage isolation and culture
We isolated monocytes by density gradient centrifugation that employed Lymphocyte Separation Medium (ICN Biomedicals, Aurora, OH) and subsequent adherence to cell culture dishes from leukopacs of healthy donors. Monocytes were cultured for 10 days in RPMI 1640 containing 5% human serum (Atlanta Biologicals, Lawrenceville, GA) to obtain macrophages [28]. THP-1 cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum.
Microarray analysis
Macrophages were deprived of serum in RPMI 1640 medium for 12 hours and then stimulated by adding fresh medium containing either 100μg/mL AGE-LDL or 100μg/mL LDL. Total RNA was isolated with an RNeasy Mini Kit (Qiagen) and tested for quality on agarose gels. We used total RNA (10μg) for microarray analysis on Affymetrix hg U133 Plus 2.0 chips (Affymetrix). The arrays were scanned and the data were captured using the Affymetrix GeneChip Laboratory Information Management System. Criteria for differential regulation by AGE-LDL treatment were set as >2.0-fold increase or decrease at a probability value of <0.05 (n=3).
Reverse transcription-quantitative PCR
Total RNA from human macrophages (5μg) was reverse transcribed by Superscript II (Invitrogen) following the manufacturer's instructions. Quantitative PCR was performed in a MyiQ Single-Color Real-Time PCR system (Bio-Rad, Hercules, CA) (primer sequences given in Table 1). The levels of the different mRNAs were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels and presented as fold-difference of AGE-LDL- treated cells vs. LDL-treated cells. An anti-human Receptor for AGE (RAGE) mouse monoclonal antibody and its IgG2b isotype control (R&D systems) served to test the involvement of RAGE by blocking receptor-ligand interactions.
Table 1. PCR primers.
| Forward | Reverse | |
|---|---|---|
| CCR2 | TCCATTCTCTCAGGCTTGC | TGAGCATCAAGGACATCTG |
| GAPDH | TGAAGGTCGGAGTCAACGGATTTGGTCGTA | ATCTCGCTCCTGGAAGATGGTGATGGGATT |
CC chemokine receptor 2 (CCR2); glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
Western blot analysis
Cell extracts (20-30 μg total protein/lane) were separated by standard SDS-PAGE under reducing conditions and blotted to polyvinylidene difluoride membranes (PerkinElmer Life Sciences, Boston, MA). The blots were blocked with 5% defatted dry milk in Tris-buffered saline (TBS)/0.05% Tween 20, and incubated overnight with primary antibodies (1:1,000) in blocking buffer. After washing the membranes three times with TBS/0.05% Tween 20, we incubated them with peroxidase-conjugated goat anti-mouse or goat anti-rabbit antibodies (Jackson Immunoresearch) for 1h, washed them again, and developed them with a chemiluminiscence reagent (Perkin Elmer Life Sciences). We used the following primary antibodies: anti-CCR2 from Santa Cruz, anti-human GAPDH from Biodesign, and anti-AGE from ICN. We conducted densitometric analysis using the National Institutes of Health Image J processing system.
Analysis of cell surface CCR2
We washed adherent monocyte-derived cells X 2 with sterile PBS and added 5mL 5 mM EDTA/PBS per 100mm dish and incubated at 37°C for 5 minutes. We gently pipetted the cells off the dish and collected them without scraping. This method did not injure macrophages as determined by trypan blue exclusion and phase contrast micrographic inspection. The amount of CCR2 protein on the cell surface was estimated by flow cytometry as described [29]. CCR2 surface expression was expressed as specific mean fluorescence intensity (MFI), obtained by subtracting the MFI of control cells from that of cells labeled with anti-CCR2 antibody.
Chemotaxis assay
The chemotactic activity of THP-1 monocytoid cells in response to 2 nmol/L MCP-1 was measured in a 96-well 5mm Cell Migration Plate Assembly (Chemicon) as described elsewhere [30]. The result was expressed as the number of cells migrating in response to MCP-1.
Statistical analysis
Results are mean ± SEM. Differences between groups were determined using ANOVA with Bonferroni post hoc test. Two groups were compared using Student's t test. A value of p<0.05 was regarded as a significant difference.
Results
AGE-LDL increases CCR2 mRNA expression in human macrophages
To test the hypothesis that AGE-LDL elicits programs of cell activation involved in atherogenesis, we prepared AGE-LDL by incubating LDL with glucose at 37°C for 7 days. Longer incubation times caused detectable apoB100 fragmentation (not shown). Seven days of incubation yielded a significant increase in the formation of pentosidine (Fig.1a), a marker of protein modification [26]. The apoB100 moiety of AGE-LDL remained unfragmented (Fig. 1B, right panel) but reacted with an anti-AGE antibody that did not recognize native LDL (Fig. 1B, left panel). To evaluate the possibility of oxidation, we also measured oxidized LDL levels using a highly sensitive sandwich ELISA method (Table 2). Oxidized LDL levels in AGE-LDL were similar to those in control LDL incubated for 7 days under argon gas. In contrast, levels of oxidized LDL in the LDL incubated for 7 days without argon gas were much higher than in AGE-LDL or control LDL. These results show that argon gas prevented LDL oxidation and indicate that the actions of glycated LDL on macrophages reported here do not likely result from oxidative modification.
Figure 1.
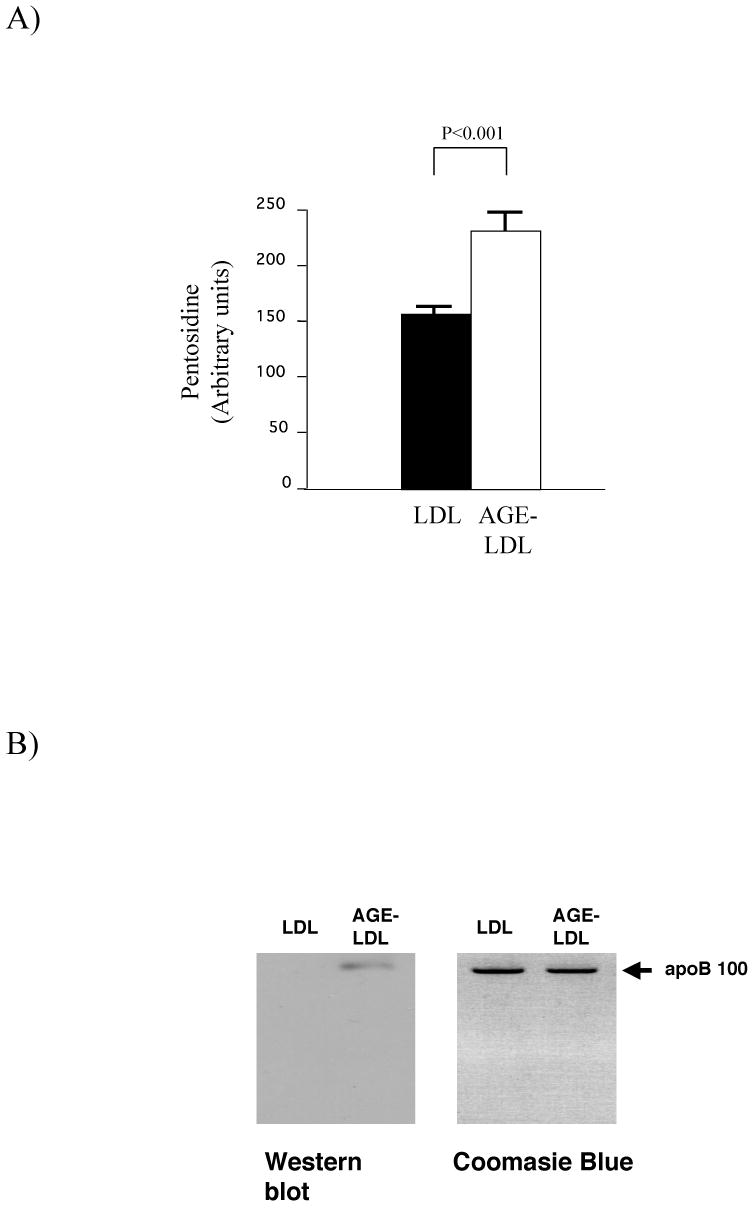
Characterization of AGE-LDL. LDL was incubated with or without glucose as described in the Methods section and: A) pentosidine formation was detected by fluorescence at 335/385 nm; B) Samples were electrophoresed through SDS-polyacrylamide gels and stained with Coomasie blue (right panel) or analyzed by Western blot using an anti-AGE antibody (left panel).
Table 2. The levels of oxidized LDL.
| Fresh LDL | LDL | AGE-LDL | LDL without argon |
|---|---|---|---|
| 0.69 | 0.75 | 0.79 | 1.56 |
The concentrations of oxidized LDL are expressed in ng/5μg LDL protein.
After treating human macrophages with AGE-LDL or LDL for 6h (100μg/ml), we isolated total RNA and subjected it to DNA microarray analysis. AGE-LDL treatment increased CCR2 mRNA levels 3.23-fold compared with LDL treatment. RT-PCR analysis verified that AGE-LDL increased the expression of CCR2 (Fig. 2a) and demonstrated that the effect depended on RAGE, because it declined significantly after treatment of macrophages with an anti-RAGE antibody (Fig. 2b) but not with isotype control IgG (data not shown).
Figure 2.

AGE-LDL increased CCR2 mRNA level in human macrophages. A) The CCR2 mRNA level was quantified by RT-PCR following 6 hours of treatment of macrophages with either LDL or AGE-LDL. B) Cells were preincubated with 50 μg/mL of either anti-RAGE or isotype control antibodies for 1h before stimulation. Data are normalized to the respective expression levels in LDL-treated cells, and shown as mean ± SEM; n=6. The experiments were performed three times for each donor.
AGE-LDL increases CCR2 protein and cell surface expression
Levels of CCR2 increased markedly (∼ 3-fold) in macrophages following AGE-LDL treatment (48 hours), as determined by Western blotting (Fig. 3a) and densitometry (Fig. 3b). AGE-LDL induced augmented expression of cell surface CCR2 (5-fold), as revealed by flow cytometry (Fig. 4).
Figure 3.

AGE-LDL increased CCR2 protein levels in human macrophages. A) Macrophages were treated with either LDL or AGE-LDL for 24 or 48 hours. Western blot analyses were performed to detect CCR2 (upper), using GAPDH (lower) as a loading control. Similar results were observed in cells from two additional donors. The experiments were done three times for each donor. B) Densitometric analysis of the immunoblots in A). The intensity of the bands corresponding to CCR2 was normalized to the intensity of GAPDH and expressed as mean ± SEM (n = 3).
Figure 4.
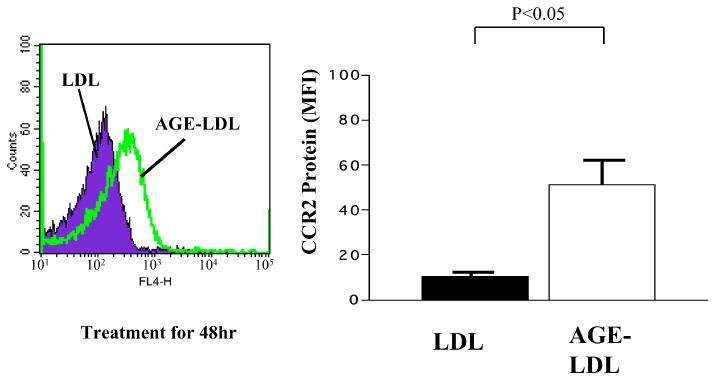
AGE-LDL increased cell surface expression of CCR2. Macrophages were incubated in the presence of either LDL or AGE-LDL for 48 hours, and CCR2 surface expression was determined by flow cytometry with anti-CCR2 IgG and expressed as specific mean fluorescence intensity (MFI). Data are shown as mean ± SEM (n=3). The experiments were done twice for each donor.
AGE-LDL increased MCP-1-mediated chemotaxis of THP-1 monocytes
Similarly to its effect in monocyte-derived macrophages, AGE-LDL augmented the expression of CCR2 in THP-1 monocytoid cells (not shown). Therefore, to examine the functional implications of AGE-LDL-induced CCR2 expression, further experiments determined the chemotactic response of THP-1 monocytoid cells to MCP-1, a specific CCR2 ligand. AGE-LDL treatment increased the chemotactic activity of THP-1 monocytoid cells 2.6-fold compared to LDL-treated cells (Fig. 5).
Figure 5.

AGE-LDL stimulated MCP-1-mediated THP-1 cell chemotaxis.
THP-1 monocytoid cells were incubated with either AGE-LDL or LDL for 48 hours before chemotaxis determination. Data are shown as mean ± SEM (n=3). The experiments were done twice for each donor.
Discussion
AGE-LDL accumulates in diabetic patients [7-9], but pathophysiological effects of AGE-LDL remain incompletely characterized. AGE-LDL can stimulate the proliferation and differentiation of vascular smooth muscle cells [31] and also increase MCP-1 expression in endothelial cells [32], potentially promoting monocyte recruitment [22]. The present study demonstrates that AGE-LDL also increases expression of the MCP-1 receptor CCR2 in macrophages and promotes chemotaxis of monocytes directed by MCP-1. Our results underscore the effect of AGE-LDL on different cell types involved in atherosclerosis.
Since an anti-RAGE antibody can suppress this cellular response to AGE-LDL, the present results demonstrate that RAGE participates in AGE-LDL-induced CCR2 expression. Because RAGE expression increases in infiltrating macrophages in the vasculature of diabetic subjects, this finding may have particular disease relevance [33]. Importantly, the anti-RAGE IgG concentration (50 μg/mL) in our antibody blocking experiments completely suppressed AGE-BSA-induced gene expression [34], but only inhibited AGE-LDL-induced CCR2 expression by 60-70% (Fig. 2b), suggesting that other receptors or AGE-binding proteins, e.g., scavenger receptors type I and type II, oligosaccharide transferase-48 (AGE-R1), 80K-H phosphoprotein (AGE-R2), and galectin-3 (AGE-R3), may also participate in CCR2 regulation by AGE-LDL [35].
In apoE-deficient mice ingesting a high-fat diet, deletion of CCR2, the only established functional receptor for MCP-1 on hematopoietic cells, afforded significant protection against macrophage accumulation and atherosclerotic lesion formation [23]. Similar studies in mice fed a regular chow diet showed that CCR2-deficient mice resisted development of atherosclerosis better than wild-type mice [36]. Such studies provide strong evidence that activation of CCR2, presumably by MCP-1, contributes to foam cell formation, one of the earliest manifestations of atherosclerosis. Thus, excessive recruitment of monocytes into the arterial intima resulting from enhanced MCP-1-mediated chemotaxis may provide a mechanism for concerted action of high serum AGE-LDL levels with LDL and other AGEs that promotes atherosclerosis in DM patients.
We used argon gas to prevent the oxidation of AGE-LDL and control LDL during the preparation, and verified the inhibition of oxidation by sandwich ELISA assay. Our preliminary data showed that oxidized LDL reduced MCP-1-mediated chemotaxis compared with LDL, a result compatible with previous reports showing that oxidized LDL caused a rapid reduction of CCR2 expression in monocytes, rendering these cells non-responsive to MCP-1 [25, 37]. Our findings suggest differing cellular responses to AGE-LDL and oxidized LDL. Oxidized LDL, found mainly in atherosclerotic lesions, may block emigration from the intima and retain monocytes at sites of inflammation, and hence promote intimal macrophage accumulation. In conclusion, AGE-LDL augments both mRNA and protein levels of CCR2 and increases MCP-1-mediated chemotaxis. As atheromata in patients with DM have accentuated accumulation of mononuclear phagocytes as well as RAGE, our findings suggest a new mechanism by which AGE-LDL can promote atherogenesis in DM patients.
Acknowledgments
This work was supported by grants from the Donald W. Reynolds Foundation and the National, Heart, Lung, and Blood Institutes (HL-34636) to P. Libby, and the National Defense Medical College (H14) to K. Isoda. We thank Elissa Simon-Morrisey, Michelle Rodrigue, and Nomeda Vaisviliene for their skillful technical assistance in this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fu MX, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. The advanced glycation end product, Nepsilon-(carboxymethyl) lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem. 1996;271:9982–6. doi: 10.1074/jbc.271.17.9982. [DOI] [PubMed] [Google Scholar]
- 2.Onorato JM, Thorpe SR, Baynes JW. Immunohistochemical and ELISA assays for biomarkers of oxidative stress in aging and disease. Ann N Y Acad Sci. 1998;854:277–90. doi: 10.1111/j.1749-6632.1998.tb09909.x. [DOI] [PubMed] [Google Scholar]
- 3.Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. Quantification of malondialdehyde and 4-hydroxynonenal adducts to lysine residuces in native and oxidized human low-density lipoproteins. Biochem J. 1997;322:317–25. doi: 10.1042/bj3220317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bierhaus A, Hofman MA, Ziegler R, Nawroth PP. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc Res. 1998;37:586–600. doi: 10.1016/s0008-6363(97)00233-2. [DOI] [PubMed] [Google Scholar]
- 5.Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu C, Kislinger T, Stern DM, Schmidt AM, De Caterina R. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation. 2002;105:816–22. doi: 10.1161/hc0702.104183. [DOI] [PubMed] [Google Scholar]
- 6.Sakata N, Meng J, Takebayashi S. Effects of Advanced Glycation End-Products on the Proliferation and Fibronectin Production of Smooth Muscle Cells. J Atheroscler Thromb. 2000;7:169–76. doi: 10.5551/jat1994.7.169. [DOI] [PubMed] [Google Scholar]
- 7.Tames FJ, Mackness MI, Arrol S, Laing I, Durrington PN. Non-enzymatic glycation of apolipoprotein B in the sera of diabetic and non-diabetic subjects. Atherosclerosis. 1992;93:237–44. doi: 10.1016/0021-9150(92)90260-n. [DOI] [PubMed] [Google Scholar]
- 8.Jack CM, Sheridan B, Kennedy L, Stout RW. Non-enzymatic glycation of low-density lipoprotein: results of an affinity chromatography method. Diabetologia. 1988;31:126–7. doi: 10.1007/BF00395561. [DOI] [PubMed] [Google Scholar]
- 9.Klein RL, Laimins M, Lopes-Virella MF. Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from type I diabetic patients and nondiabetic subjects. Diabetes. 1995;44:1093–8. doi: 10.2337/diab.44.9.1093. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Ren S, Sun D, Shen GX. Influence of glycation on LDL-induced generation of fibrinolytic regulators in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:1140–8. doi: 10.1161/01.atv.18.7.1140. [DOI] [PubMed] [Google Scholar]
- 11.Penn MS, Chisolm GM. Oxidized lipoproteins, altered cell function and atherosclerosis. Atherosclerosis. 1994;108(Suppl):S21–9. doi: 10.1016/0021-9150(94)90150-3. [DOI] [PubMed] [Google Scholar]
- 12.Lyons TJ, Jenkins AJ. Lipoprotein glycation and its metabolic consequences. Curr Opin Lipidol. 1997;8:174–80. doi: 10.1097/00041433-199706000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Howard BV. Lipoprotein metabolism in diabetes. Curr Opin Lipidol. 1994;5:216–20. doi: 10.1097/00041433-199405030-00009. [DOI] [PubMed] [Google Scholar]
- 14.Lyons TJ, Li W, Wells-Knecht MC, Jokl R. Toxicity of mildly modified low-density lipoproteins to cultured retinal capillary endothelial cells and pericytes. Diabetes. 1994;43:1090–5. doi: 10.2337/diab.43.9.1090. [DOI] [PubMed] [Google Scholar]
- 15.Steinbrecher UP, Witztum JL. Glucosylation of low-density lipoproteins to an extent comparable to that seen in diabetes slows their catabolism. Diabetes. 1984;33:130–4. doi: 10.2337/diab.33.2.130. [DOI] [PubMed] [Google Scholar]
- 16.Lorenzi M, Cagliero E, Markey B, Henriksen T, Witztum JL, Sampietro T. Interaction of human endothelial cells with elevated glucose concentrations and native and glycosylated low density lipoproteins. Diabetologia. 1984;26:218–22. doi: 10.1007/BF00252411. [DOI] [PubMed] [Google Scholar]
- 17.Zimmermann R, Panzenbock U, Wintersperger A, Levak-Frank S, Graier W, Glatter O, Fritz G, Kostner GM, Zechner R. Lipoprotein lipase mediates the uptake of glycated LDL in fibroblasts, endothelial cells, and macrophages. Diabetes. 2001;50:1643–53. doi: 10.2337/diabetes.50.7.1643. [DOI] [PubMed] [Google Scholar]
- 18.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 19.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol: modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–24. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 21.Wilcox JN, Nelken NA, Coughlin SR, Gordon D, Schall TJ. Local expression of inflammatory cytokines in human atherosclerotic plaques. J Atheroscler Thromb. 1994;1 1:S10–13. doi: 10.5551/jat1994.1.supplemment1_s10. [DOI] [PubMed] [Google Scholar]
- 22.Menten P, Wuyts A, Damme JV. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002;13:455–81. doi: 10.1016/s1359-6101(02)00045-x. [DOI] [PubMed] [Google Scholar]
- 23.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherogenesis. Nature. 1998;394:894–7. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 24.Burke AP, Kolodgie FD, Zieske A, Fowler DR, Weber DK, Varghese PJ, Farb A, Virmani R. Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler Thromb Vasc Biol. 2004;24:1266–1271. doi: 10.1161/01.ATV.0000131783.74034.97. [DOI] [PubMed] [Google Scholar]
- 25.Virmani R, Burke AP, Kolodgie F. Morphological characteristics of coronary atherosclerosis in diabetes mellitus. Can J Cardiol. 2006;22:81B–84B. doi: 10.1016/s0828-282x(06)70991-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho HM, Choi SH, Hwang KC, Oh SY, Kim HG, Yoon DH, Choi MA, Lim SY, Song H, Jang Y, Kim TW. The Src/PLC/PKC/MEK/ERK signaling pathway is involved in aortic smooth muscle cell proliferation induced by glycated LDL. Mol Cells. 2005;19:60–66. [PubMed] [Google Scholar]
- 27.Itabe H, Ueda M. Measurement of plasma oxidized low-density lipoprotein and its clinical implications. J Atheroscler Thromb. 2007;14:1–11. doi: 10.5551/jat.14.1. [DOI] [PubMed] [Google Scholar]
- 28.Schönbeck U, Mach F, Sukhova GK, Atkinson E, Levesque E, Herman M, Graber P, Basset P, Libby P. Expression of stromelysin-3 in atherosclerotic lesions: regulation via CD40-CD40 ligand signaling in vitro and in vivo. J Exp Med. 1999;189:843–53. doi: 10.1084/jem.189.5.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han KH, Chang MK, Boullier A, Green SR, Li A, Glass CK, Quehenberger O. Oxidized LDL reduces monocyte CCR2 expression through pathways involving peroxisome proliferator-activated receptor-g. J Clin Invest. 2000;106:793–802. doi: 10.1172/JCI10052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gildea JJ, Harding MA, Gulding KM, Theodrescu D. Thransmembrane motility assay of transiently transfected cells by fluorescent cell counting and luciferase measurement. Biotechniques. 2000;29:81–6. doi: 10.2144/00291st02. [DOI] [PubMed] [Google Scholar]
- 31.Velarde V, Jenkins AJ, Christopher J, Lyons TJ, Jaffa AA. Activation of MAPK by modified low-density lipoproteins in vascular smooth muscle cells. J Appl Physiol. 2001;91:1412–20. doi: 10.1152/jappl.2001.91.3.1412. [DOI] [PubMed] [Google Scholar]
- 32.Napoli C, Lerman LO, de Nigris F, Loscalzo J, Ignarro LJ. Glycoxidized low-density lipoprotein downregulates endothelial nitric oxide synthase in human coronary cells. J Am Coll Cardiol. 2002;40:1515–22. doi: 10.1016/s0735-1097(02)02306-9. [DOI] [PubMed] [Google Scholar]
- 33.Yamagishi S, Takeuchi M, Inagaki Y, Nakamura K, Imaizumi T. Role of advanced glycation end products (AGEs) and their receptor (RAGE) in the pathogenesis of diabetic microangiopathy. Int J Clin Pharmacol Res. 2003;23:129–34. [PubMed] [Google Scholar]
- 34.Isoda K, Folco EJ, Shimuzu K, Libby P. AGE-BSA decreases ABCG1 expression and reduces macrophages cholesterol efflux to HDL. Atherosclerosis. 2007;192:298–304. doi: 10.1016/j.atherosclerosis.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 35.Stitt AW, Bucala R, Vlassara H. Atherogenesis and advanced glycation: promotion, progression, and prevention. Ann NY ACad Sci. 1997;811:115–27. doi: 10.1111/j.1749-6632.1997.tb51994.x. [DOI] [PubMed] [Google Scholar]
- 36.Dawson TC, Kruziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–11. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 37.Han KH, Tangirala RK, Green SR, Quehenberger O. Chemokine receptor CCR2 expression and monocyte chemoattractant protein-1 mediated chemotaxis in human monocytes: a regulatory role for plasma low density lipoprotein. Arterioscler Thromb Vasc Biol. 1998;18:1983–91. doi: 10.1161/01.atv.18.12.1983. [DOI] [PubMed] [Google Scholar]