

Abstract
The pro-apoptotic BH3-only protein Bim is established to be an important mediator of signaling pathways that induce cell death. Multi-site phosphorylation of Bim by several members of the MAP kinase group is implicated as a regulatory mechanism that controls the apoptotic activity of Bim. To test the role of Bim phosphorylation in vivo, we constructed mice with a series of mutant alleles that express phosphorylation-defective Bim proteins. We show that mutation of the phosphorylation site Thr-112 causes decreased binding of Bim to the anti-apoptotic protein Bcl2 and can increase cell survival. In contrast, mutation of the phosphorylation sites Ser-55, Ser-65, and Ser-73 can cause increased apoptosis because of reduced proteasomal degradation of Bim. Together, these data indicate that phosphorylation can regulate Bim by multiple mechanisms and that the phosphorylation of Bim on different sites can contribute to the sensitivity of cellular apoptotic responses.
Introduction
Bim is a BH3-only member of the Bcl2-related protein family that is implicated as a mediator of signaling pathways that can initiate apoptosis (Strasser, 2005). Studies of Bim knockout mice have established that Bim contributes to the apoptotic response of cells to cytokine and growth factor withdrawal (Bouillet et al., 1999; Putcha et al., 2001; Whitfield et al., 2001). Bim is also required for the normal apoptosis of autoreactive T cells during thymic development (Bouillet et al., 2002) and termination of immune responses mediated by apoptosis of activated T cells (Hildeman et al., 2002; Pellegrini et al., 2003). It has been established that Bim can induce apoptosis by engaging anti-apoptotic members of the Bcl-2 family, including Bcl2, Bcl-XL, and Mcl-1 (Willis et al., 2007).
A major mechanism of functional regulation of Bim-dependent apoptosis is the regulation of Bim expression. Indeed, the Bim promoter is regulated by stress-responsive transcription factors, including members of the FOXO and AP-1 transcription factor families (Gilley et al., 2003; Stahl et al., 2002; Whitfield et al., 2001). A second regulatory mechanism implicated in the control of Bim-dependent apoptosis is the covalent modification of Bim by phosphorylation (Ley et al., 2005). Bim is phosphorylated on multiple sites by members of the MAP kinase family, including ERK, JNK, and p38 isoforms. Transfection studies using mutant Bim proteins in cultured cells have indicated that phosphorylation may increase or decrease the apoptotic activity of Bim (Ley et al., 2005). Bim phosphorylation may influence the formation of Bim protein complexes (Ewings et al., 2007; Harada et al., 2004; Lei and Davis, 2003) and may regulate Bim protein stability (Ley et al., 2003; Luciano et al., 2003). Whether phosphorylation represents a mechanism of Bim regulation in vivo is unclear.
The purpose of this study was to test the hypothesis that Bim phosphorylation contributes to the regulation of apoptosis by constructing mice with germ-line mutations in the Bim gene that substitute the major Bim phosphorylation sites with Ala residues. We report that these phosphorylation sites influence Bim-dependent apoptotic responses.
Results
Transcripts derived from the Bim gene are alternatively spliced to create several different Bim proteins (Bouillet et al., 2001b). BimEL is encoded by sequences derived from exons 2–6 (Figure 1A). Alternative splicing can delete sequences derived from exon 3 (BimL) or exons 3 & 4 (BimS) to create additional Bim isoforms. These alternatively spliced exons encode the major sites of Bim phosphorylation (Figure 1A). To study the role of Bim phosphorylation, we examined the effect of replacement of these phosphorylation sites with Ala residues. Transfection studies using a cDNA expression vector demonstrated that the mutant Bim proteins can be expressed (Figure 1B). Furthermore, co-immunoprecipitation analysis demonstrated that the mutant proteins were able to interact with the pro-survival Bcl2-family protein Mcl-1 (Figure 1B). Substitution of the major Bim phosphorylation sites with Ala residues therefore does not result in the expression of Bim proteins that completely lack functional activity. These data suggest that the physiological role of Bim phosphorylation can be tested by phenotypic analysis of mutant mice that express phosphorylation-defective Bim proteins.
Figure 1. Phosphorylation of Bim isoforms.
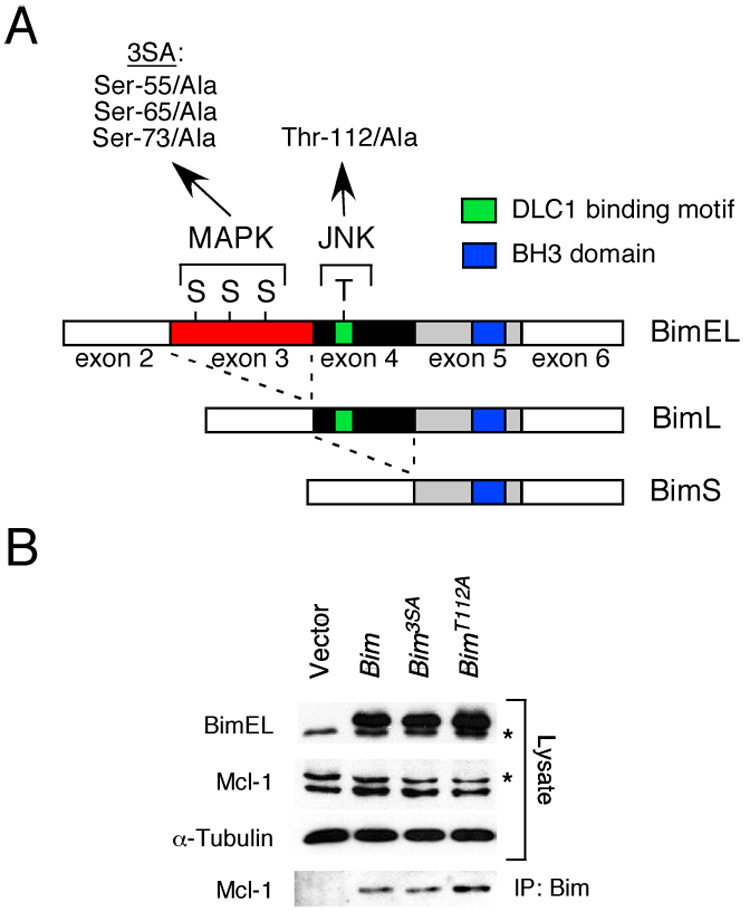
A) The structure of BimEL, which is encoded by sequences derived from exons 2–6 of the Bim gene, is illustrated schematically. Alternative splicing can delete sequences derived from exon 3 (BimL) or exons 3 & 4 (BimS) and creates two additional Bim isoforms. Three sites of MAPK phosphorylation are encoded by exon 3 (Ser-55, Ser-65, and Ser-73) and one site of JNK phosphorylation is encoded by exon 4 (Thr-112). Mutation of the three sites of Ser phosphorylation (3SA) and the site of Thr phosphorylation (Thr112A) by replacement with Ala residues is indicated. The BH3 domain and the dynein light chain 1 (DLC1) binding motif are illustrated.
B) Recombinant BimEL proteins with mutations at the three sites of MAPK phosphorylation (3SA) or the JNK phosphorylation site (Thr-112) were expressed in 293T cells. The endogenous (asterisk) and recombinant BimEL proteins were detected by immunoblot analysis. Endogenous Mcl-1 (asterisk indicates a non-specific band) and α-Tubulin were also examined. Bim/Mcl-1 complexes were examined by immunoprecipitation (IP) of recombinant Bim proteins with an antibody to the Flag epitope-tag and immunoblot analysis with an antibody to Mcl-1.
Creation of mice with defects in Bim phosphorylation
To study the role of Bim phosphorylation, we constructed mice with germ-line point mutations in the Bim gene using homologous recombination in ES cells (Figure 2). A targeting vector was designed to insert a floxed NeoR cassette within intron 4 and introduce specific mutations in exons 3 and 4. The NeoR cassette was excised with Cre recombinase to create a Bim genomic locus with a single LoxP site within intron 4. We created four mouse strains with this single LoxP site in intron 4. First, we constructed mice that lack mutations within the coding regions of the Bim gene. These mice (BimLoxP) express wild-type Bim protein. Second, we constructed mice with mutations within Bim exon 3 that replace the three MAP kinase phosphorylation sites (Ser-55/65/73) with Ala residues (Bim3SA). Translationally silent nucleotide changes were introduced to create novel restriction sites (NcoI, PstI, and XhoI) at each of the mutated phosphorylation sites. Third, mice were constructed with mutations within exon 4 that replace the JNK phosphorylation site Thr-112 with Ala (BimT112A) and create a novel restriction site (KasI). Fourth, we constructed mice (BimΔEL) with a deletion of alternatively spliced exon 3. Heterozygous matings of the mutant mice led to the creation of homozygous mice for the mutant Bim alleles (Figure 2G). The average litter size obtained from matings of homozygous mice with mutant Bim alleles was not significantly different (p > 0.05) from matings of wild-type mice.
Figure 2. Construction of mice with phosphorylation-defective Bim.
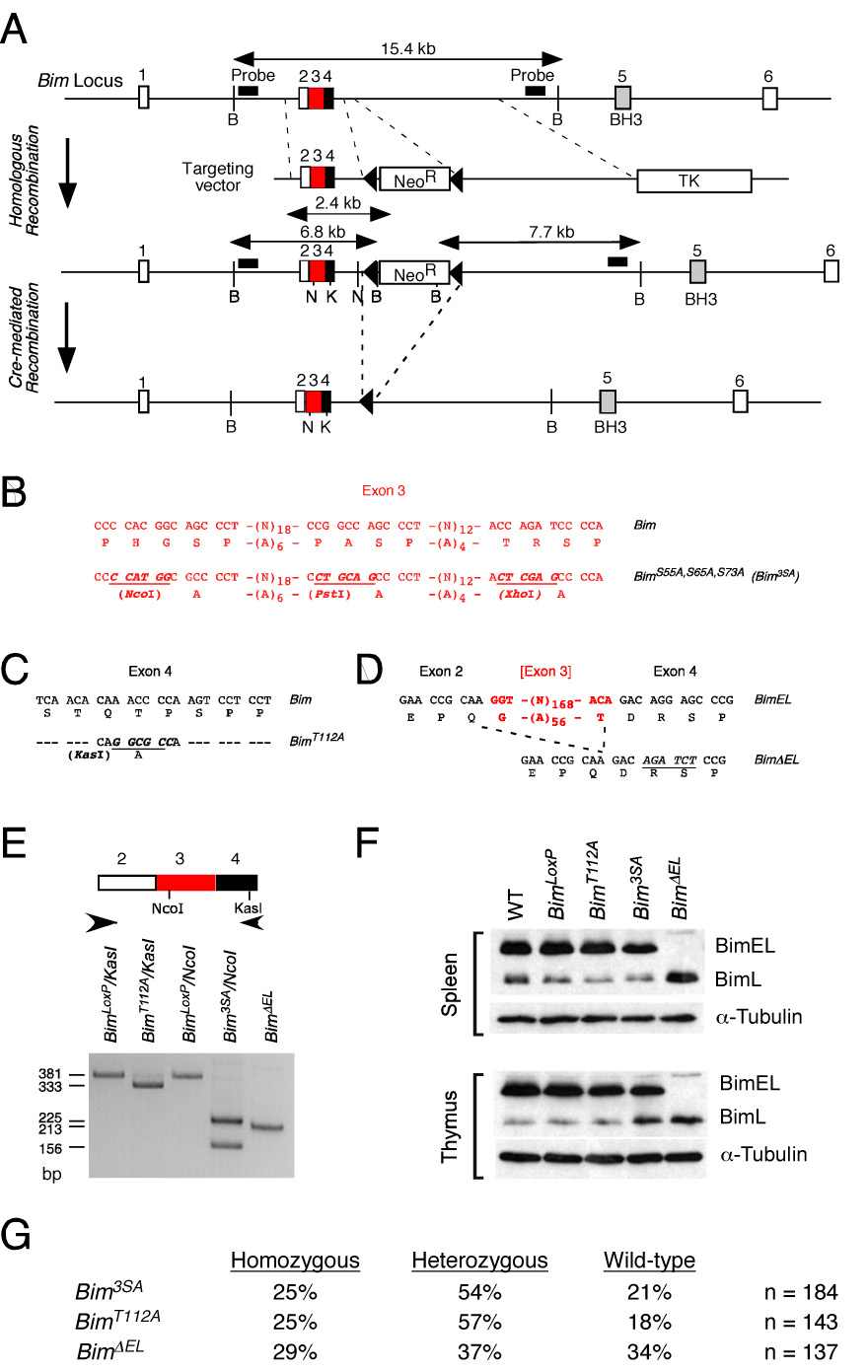
A) Strategy for construction of mice with phosphorylation-defective Bim proteins. The structure of the Bim genomic locus and the targeting vectors that were employed to create germ-line mutations in exons 3 and 4 are illustrated.
B–D) The point mutations in exon 3 (B) and exon 4 (C) are illustrated. The strategy for deletion of exon 3 is also illustrated (D).
E) PCR analysis of mouse genomic DNA isolated from BimLoxP mice (wild-type Bim) and from mice with homozygous mutations: BimT112A; Bim3SA; and BimΔEL. The presence of the T112A (KasI) and 3SA (NcoI) mutations were detected by digestion with the indicated restriction enzymes.
F) Splenocytes and thymocytes were isolated from wild-type C57BL/6J mice and mice with homozygous modifications in the Bim gene: BimLoxP, BimT112A; Bim3SA; and BimΔEL. The expression of Bim and α-Tubulin was examined by immunoblot analysis.
G) Mice obtained from heterozygous crosses of Bim mutant mice were genotyped. The number of homozygous, heterozygous, and wild-type mice are presented as the percentage of the total number (n) of mice.
We examined Bim protein expression by immunoblot analysis of extracts prepared from the thymus and spleen. The major Bim isoform detected in wild-type mice was BimEL, but smaller amounts of BimL were also found (Figure 2F). The low abundance BimS isoform was not reproducibly detected. A similar pattern of Bim expression was observed in studies of control BimLoxP mice. This finding indicates that the presence of a single LoxP site within intron 4 does not markedly alter Bim expression. A similar expression pattern of Bim proteins was observed in BimT112A mice and Bim3SA mice. In contrast, no BimEL was detected in BimΔELmice. The BimΔEL mice expressed increased amounts of BimL because of the deletion of alternatively spliced exon 3.
Together, these data establish that control BimLoxP mice express normal amounts of wild-type Bim proteins. Moreover, the mutant Bim3SA, BimT112A, and BimΔEL mice that express phosphorylation-defective Bim proteins are viable.
Bim is a target of MAP kinase phosphorylation in vivo
To test whether Bim is subject to multi-site phosphorylation in vivo, we examined the phospho-isomers of BimEL using 2D gel electrophoresis (Bunin et al., 2005; Hacker et al., 2006; Kuroda et al., 2006; Puthalakath et al., 2007). Immunoblot analysis demonstrated that BimEL in serum-starved MEF exhibited a heterogeneous distribution between a number of phospho-isoforms (Figure 3). Serum-treatment caused a large shift in the distribution of the BimEL phospho-isoforms to a more homogeneous group with higher negative charge, consistent with increased phosphorylation on multiple sites. Studies of homozygous Bim3SA MEF indicated that the replacement of the three major MAP kinase phosphorylation sites (Ser-55/65/73) with Ala strongly suppressed the effect of serum on BimEL phospho-isomers (Figure 3). In contrast, studies of homozygous BimT112A MEF demonstrated that the replacement of the Thr-112 phosphorylation site with Ala did not prevent the major effects of serum on BimEL phospho-isomers (Figure 3). These data are consistent with previous reports that Ser-55/65/73 represent major sites of phosphorylation by serum-stimulated ERK and that Thr-112 is a major site of Bim phosphorylation by stress-activated JNK (Ley et al., 2005).
Figure 3. Analysis of Bim phosphorylation in vivo.
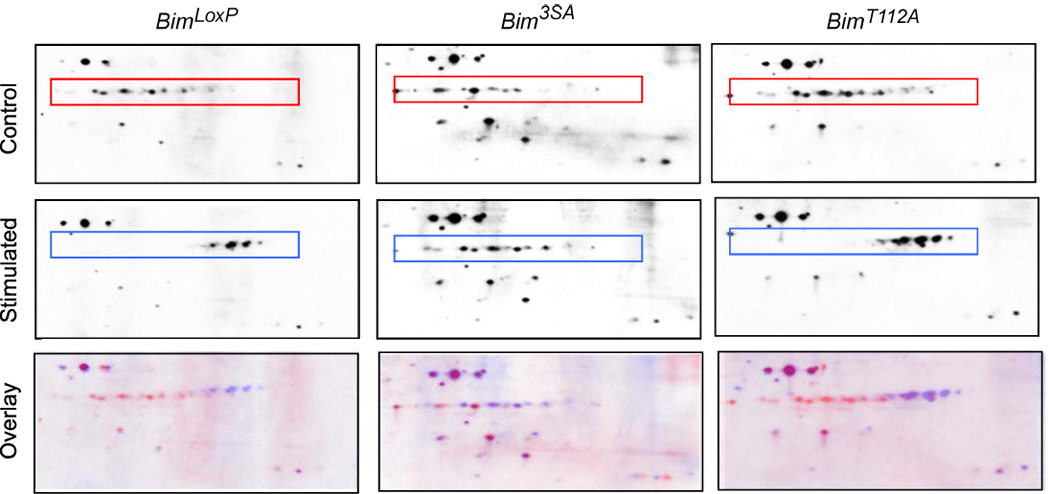
Homozygous BimLoxP, Bim3SA, and BimT112A primary MEF were serum-starved (Control) or treated with 10% fetal bovine serum (Stimulated) for 30 mins. Extracts were prepared from the cells and examined by 2D gel electrophoresis (isoelectric focusing (pI range 4 – 7) in the horizontal dimension and SDS-PAGE in the vertical dimension) and immunoblot analysis by probing with an antibody to Bim. Proteins corresponding to isoforms of BimEL are indicated with a box. The Control (red) and Stimulated (blue) images were superimposed using Adobe Photoshop CS3 software (Overlay).
To examine the phosphorylation of BimEL on specific sites we performed immunoblot analysis using phospho-specific antibodies. Phosphorylation of BimEL on the MAP kinase phosphorylation site Ser-65 can be detected using previously characterized antibodies (Putcha et al., 2003). However, an antibody to the JNK phosphorylation site Thr-112 has not been described. We therefore prepared a phospho-specific antibody to the Thr-112 phosphorylation site on BimEL. Immunoblot analysis demonstrated that this antibody detected recombinant BimEL in cells expressing activated JNK and that replacement of Thr-112 with Ala prevented the detection of BimEL (Figure 4A). Together, these data indicate that BimEL phosphorylation on Ser-65 and Thr-112 can be examined by immunoblot analysis.
Figure 4. Phosphorylation of Bim on Ser65 and Thr112 in vivo.
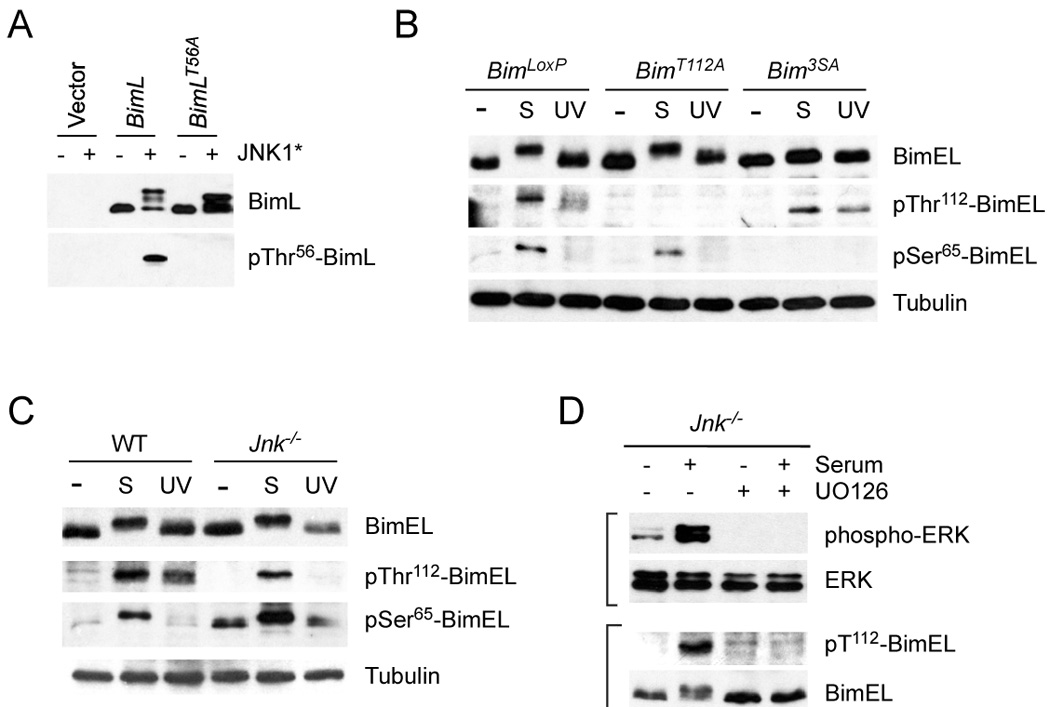
A) Characterization of an antibody to the Bim phosphorylation site Thr112 on BimEL (Thr56 on BimL). 293T cells were transfected with an empty expression vector or a vector that expresses BimL. The expression of BimL and phosphoThr56-BimL was detected by immunoblot analysis of cells extracts. The effect of co-expression of constitutively activated JNK1 (JNK1*) and the replacement of Thr56 with an Ala residue is shown.
B) Homozygous BimLoxP, BimT112A, and Bim3SA primary MEF were serum-starved or treated with serum (S) (10% fetal bovine serum; 30 mins) or UV light (60 J/m2; 60 mins). Cell extracts were examined using antibodies to Bim, phospho-Thr112-Bim, phosphoSer65-Bim, and Tubulin.
C) Wild-type and Jnk1−/− Jnk2−/− (Jnk−/−) fibroblasts were serum-starved or treated with serum (S) or UV light. Cell extracts were examined using antibodies to Bim, phospho- Thr112-Bim, phosphoSer65-Bim, and Tubulin. The basal phosphorylation of BimEL on Ser65 was slightly increased in JNK-deficient fibroblasts compared with wild-type fibroblasts.
D) Jnk1−/− Jnk2−/− (Jnk−/−) fibroblasts were serum-starved or treated with serum and/or the MEK inhibitor U0126 (30 mins). Cell extracts were examined using antibodies to phospho-ERK, ERK, phospho-Thr112-Bim, and Bim.
Treatment of BimLoxP MEF with serum caused markedly increased phosphorylation of wild-type BimEL on Ser-65 (Figure 4B). A similar amount of serum-induced phosphorylation on Ser-65 was detected in homozygous BimT112A MEF, but no Ser-65 phosphorylation was detected in homozygous Bim3SA MEF (Figure 4B). Exposure of the MEF to stress (UV radiation) caused no change in the phosphorylation of these Bim proteins on Ser-65 (Figure 4B). Together, these data are consistent with the conclusion that Ser-65 is phosphorylated by serum-stimulated ERK (Ley et al., 2005) and that mutation of the Thr-112 phosphorylation site does not affect the phosphorylation of BimEL on Ser-65 (Figure 4B).
Exposure of primary BimLoxP MEF to stress (UV radiation) caused increased phosphorylation of wild-type BimEL on Thr-112 (Figure 4B). A similar amount of UV-induced phosphorylation on Thr-112 was detected in homozygous Bim3SA MEF, but no Thr-112 phosphorylation was detected in homozygous BimT112A MEF (Figure 4B). To test whether this UV-stimulated phosphorylation was mediated by JNK, we examined BimEL phosphorylation in compound mutant Jnk1−/− Jnk2−/− fibroblasts that express no JNK. These studies demonstrated that no UV-stimulated phosphorylation of BimEL on Thr-112 was detected in JNK-deficient fibroblasts (Figure 4C). Together, these data indicate that JNK is the physiogically relevant UV-stimulated kinase that phosphorylates BimEL on Thr-112 (Figure 4C) and that mutation of the Ser-65 phosphorylation site does not affect the phosphorylation of BimEL on Thr-112 (Figure 4B).
One unexpected finding was that BimEL was also phosphorylated on Thr-112 in response to serum-stimulation (Figure 4B). It was unlikely that this phosphorylation was solely mediated by JNK because it is established that the stress-regulated JNK pathway is not strongly activated by serum-stimulation (Davis, 2000). Indeed, studies of JNK-deficient fibroblasts indicated that JNK was not required for serum-stimulated phosphorylation of BimEL on Thr-112 (Figure 4C). A different class of MAP kinase may account for this serum-stimulated phosphorylation. A role for p38 MAP kinase was possible, but UV-stimulated p38 MAP kinase activity in JNK-deficient fibroblasts did not cause BimEL phosphorylation on Thr-112 (Figure 4C). We therefore tested the contribution of ERK to serum-stimulated BimEL phosphorylation on Thr-112. Studies of JNK-deficient fibroblasts demonstrated that serum-stimulated BimEL phosphorylation on Thr-112 was strongly suppressed by an inhibitor (U0126) of the ERK signaling pathway (Figure 4D). Together, these data indicate that Thr-112 is a target of both the ERK and JNK signaling pathways and that Thr-112 phosphorylation is mediated primarily by JNK in response to UV radiation and by ERK in response to serum-stimulation (Figure 4C,D). In contrast, Ser-65 is a selective target of the ERK pathway in serum-stimulated fibroblasts (Ley et al., 2005) and Ser-65 is not phosphorylated by JNK in response to the exposure of fibroblasts to UV radiation (Figure 4B).
Interaction of Bim with members of the Bcl2 family
It is established that one mechanism that mediates Bim apoptotic activity is an interaction of Bim with other members of the Bcl2 family, including the anti-apoptotic proteins Bcl2, Bcl-XL, and Mcl-1 (Willis et al., 2007). We therefore performed co-immunoprecipitation assays to test whether mutation of Bim phosphorylation sites caused altered interaction of Bim with Bcl2 family proteins. Studies of primary MEF prepared from homozygous BimLoxP, Bim3SA, BimT112A, and BimKO mice demonstrated that the anti-apoptotic proteins Bcl2, Bcl-XL, and Mcl-1 co-immunoprecipitated with Bim (Figure 5). In contrast, the pro-apoptotic protein Bax did not co-immunoprecipitate with Bim (Figure 5), consistent with previous observations (Willis et al., 2007).
Figure 5. Interaction of Bim with Bcl2 family proteins.
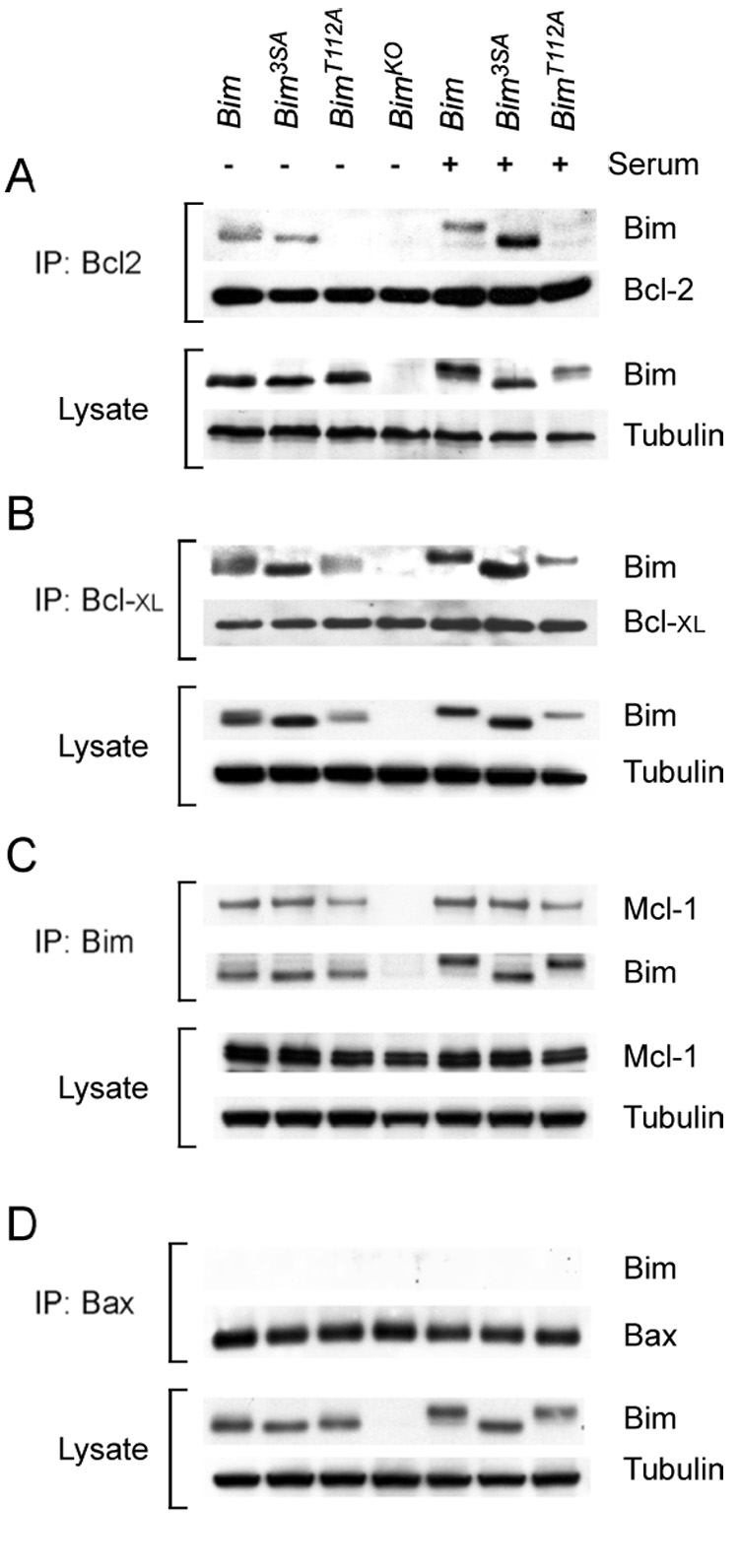
A) Homozygous BimLoxP, BimT112A, and Bim3SA primary MEF were serum-starved or treated with 10% fetal bovine serum (30 mins). The interaction of Bim with Bcl2 was examined by immunoprecipitation (IP) of cell extracts with an antibody to Bcl2 and examination of the amount of co-immunoprecipitated Bim and Bcl2 by immunoblot analysis. The amount of Bim and Tubulin in the cell lysate was probed by immunoblot analysis.
B) The interaction of Bim with Bcl- XL was examined by immunoprecipitation of cell extracts with an antibody to Bcl- XL and examination of the amount of co-immunoprecipitated Bcl- XL and Bim by immunoblot analysis. The amount of Bim and Tubulin in the cell lysate was probed by immunoblot analysis.
C) The interaction of Bim with Mcl-1 was examined by immunoprecipitation of cell extracts with an antibody to Bim and examination of the amount of co-immunoprecipitated Mcl-1 and Bim by immunoblot analysis. The amount of Mcl-1 and Tubulin in the cell lysate was probed by immunoblot analysis
D) The interaction of Bim with Bax was examined by immunoprecipitation of cell extracts with an antibody to Bax and examination of the amount of co-immunoprecipitated Bax and Bim by immunoblot analysis. The amount of Bim and Tubulin in the cell lysate was probed by immunoblot analysis
Mutation of Bim phosphorylation sites (Bim3SA and BimT112A) did not cause marked changes in the co-immunoprecipitation of Bim with Mcl-1 or Bcl-XL (Figure 5). Similarly, both wild-type Bim and Bim3SA co-immunoprecipitated with Bcl2. In contrast, BimT112A did not co-immunoprecipitate with Bcl2 (Figure 5). Since the binding of Bim to Bcl2 contributes to Bim-induced apotptosis (Willis et al., 2007), these data suggest that the BimT112A protein may cause reduced apoptosis in cells that depend on Bcl2 for survival.
Phosphorylation sites encoded by alternatively spliced exon 3 contribute to the regulation of Bim expression
Serum-starvation of primary MEF causes increased Bim expression that is associated with decreased activation of ERK MAPK (Figure 6A). In contrast, serum-treatment caused ERK activation and decreased Bim expression (Figure 6B). Previous studies have established that changes in Bim levels are mediated by altered Bim gene expression and Bim protein stability (Strasser, 2005). Indeed, the Bim protein is rapidly degraded by a ubiquitin-mediated mechanism in response to ERK MAP kinase activation (Ley et al., 2003; Luciano et al., 2003). Thus, the serum-induced decrease in Bim protein expression observed in primary MEF is attenuated if the cells are incubated with the MEK inhibitor U0126 to block ERK MAPK activation or with the proteasome inhibitor MG132 (Figure 6B).
Figure 6. The BimEL specific domain encoded by exon 2 is required for MAPK-induced Bim degradation.
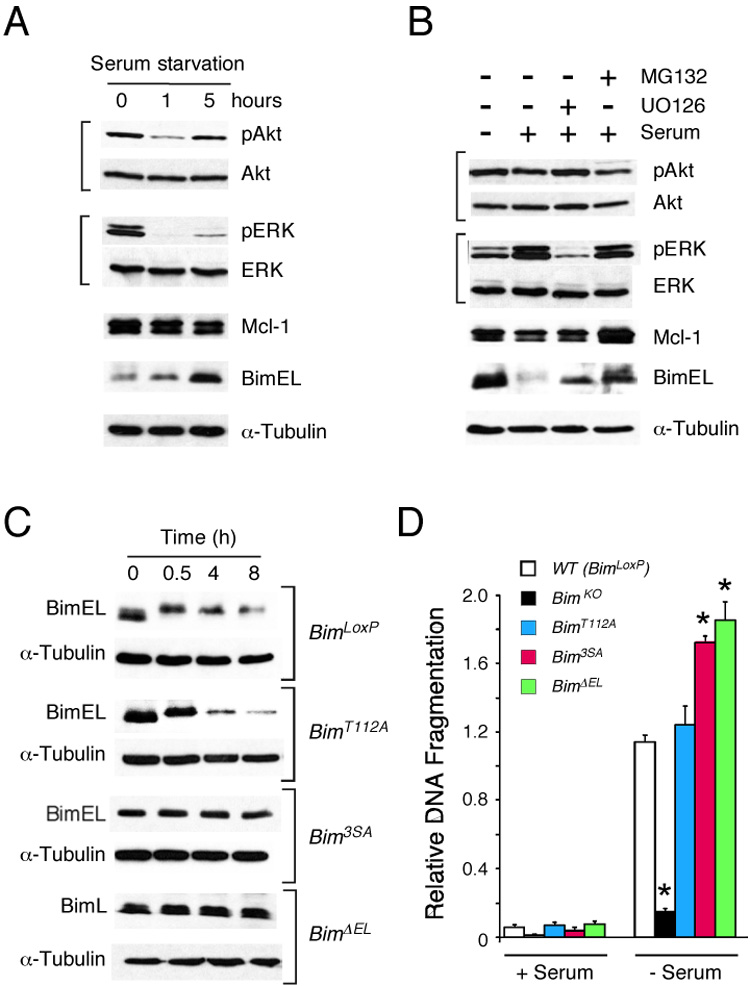
A) Serum starvation induces the expression of BimEL. Wild-type primary MEF were transferred to serum-free medium. Cell extracts were prepared at 0, 1, and 5 h following serum withdrawal. Protein expression was examined by immunoblot analysis.
B) Serum addition causes BimEL degradation. Wild-type primary MEF serum-starved (12 h) and then transferred to fresh medium supplemented with 10% FBS. Cell extracts were prepared at 0 and 8 hrs following serum treatment. The effect of pre-incubation (30 mins) with 10 µM U0126 or 20 µM MG132 was examined. Protein expression was examined by immunoblot analysis.
C) Primary MEF were prepared from embryos that express wild-type Bim (BimLoxP) and also from homozygous mutants embryos (BimT112A, Bim3SA, and BimΔEL). The cells were transferred to fresh medium supplemented with 10% fetal bovine serum plus 100 µg/ml cycloheximide. Protein expression was examined by immunoblot analysis.
D) Primary MEF were incubated in medium supplemented without and with 10% FBS (25 h.). Apoptotic DNA fragmentation was examined by ELISA. The effect of serum withdrawal to cause apoptosis of MEF that express wild-type Bim (BimLoxP) and also from homozygous mutants embryos (BimKO, BimT112A, Bim3SA, and BimΔEL) is presented as mean relative DNA fragmentation ± SD (n = 4). A statistically significant difference (P < 0.05) in DNA fragmentation compared with BimLoxP MEF is indicated with an asterisk.
To test the role of Bim phosphorylation during serum-induced Bim protein degradation, we compared the time course of Bim degradation in primary MEF prepared from embryos that express wild-type and phosphorylation-defective Bim proteins. Control studies using MEF obtained from BimLoxP mice demonstrated that serum caused a rapid reduction in the electrophoretic mobility of BimEL prior to degradation (Figure 6C). Similar serum-induced degradation of BimEL protein was detected in MEF isolated from homozygous BimT112A embryos (Figure 6C). In contrast, no serum-induced loss of BimEL protein expression was detected in MEF prepared from homozygous Bim3SA mice (Figure 6C). These data indicate that the MAP kinase phosphorylation sites encoded within exon 3 of the Bim gene are required for serum-induced Bim degradation. To test this hypothesis, we examined MEF isolated from Bim∆EL mice that lack alternatively spliced exon 3 which encodes the three sites of MAPK phosphorylation (Ser-55/65/73). These studies demonstrated that the deletion of exon 3 results in the expression of BimL as the major Bim isoform (Figure 2F) and that this Bim protein is resistant to serum-induced degradation (Figure 6C).
To examine the functional significance of phosphorylation-induced changes in Bim protein expression, we examined the apoptotic responses of primary MEF. Previous studies have demonstrated that MEF isolated from Bim knockout (BimKO) mice retain sensitivity to many stresses, indicating that Bim may play only a redundant role in stress-induced apoptosis of MEF (Naik et al., 2007). Nevertheless, BimKO MEF are resistant to serum withdrawal-induced apoptosis (Ewings et al., 2007). We therefore tested the serum withdrawal-induced apoptotic response of primary MEF isolated from mice that express either wild-type or phosphorylation-deficient Bim proteins.
MEF that express wild-type Bim (BimLoxP) exhibited apoptotic fragmentation of genomic DNA in response to serum withdrawal (Figure 6D). This apoptotic response was suppressed in Bim knockout MEF (BimKO). Studies of MEF isolated from BimT112A mice indicated that the Thr-112 phosphorylation site did not contribute to the apoptotic response (Figure 6D). In contrast, loss of the ERK MAP kinase phosphorylation sites encoded by exon 3, caused by substitution of Ser-55/65/73 with Ala residues (Bim3SA) or deletion of exon 3 (BimΔEL), increased serum withdrawal-induced apoptosis (Figure 6D). This increased apoptotic response is consistent with the hypothesis that the MAP kinase phosphorylation sites encoded by exon 3 act to inhibit Bim function. These phosphorylation sites may reduce Bim apoptotic activity by causing proteasomal degradation of Bim (Figure 6).
A phosphorylation site encoded by alternatively spliced exon 4 contributes to the regulation of Bim apoptotic activity
Studies of Bim knockout (BimKO) mice indicate that Bim plays a major role in the apoptotic response of thymocytes and T cells (Strasser, 2005). Indeed, BimKO mice exhibit defects in the deletion of autoreactive thymocytes and the response to stress (Bouillet et al., 1999; Bouillet et al., 2002). To test whether Bim phosphorylation may contribute to these Bim-dependent apoptotic processes, we examined thymocyte apoptosis in mutant mice that express phosphorylation-defective Bim proteins.
Treatment of wild-type mice with the anti-inflammatory drug dexamethasone caused apoptosis of double-positive CD4+ CD8+ (DP) thymocytes. This deletion of DP thymocytes is partially suppressed in BimKO mice (Erlacher et al., 2005). Studies of control BimLoxP mice and mice with phosphorylation-defective Bim alleles (Bim3SA, BimT112A, and BimΔEL) by flow cytometry demonstrated comparable sub-populations of CD4/8 thymocytes. Treatment with dexamethasone caused similar deletion of DP thymocytes in BimLoxP, Bim3SA, and BimΔEL mice (Figure 7A). This observation suggests that the MAP kinase phosphorylation sites encoded by exon 3 do not contribute to dexamethasone-stimulated apoptosis of DP thymocytes. In contrast, reduced deletion of DP thymocytes was observed in BimT112A mice following treatment with dexamethasone (Figure 7A). These data suggest that the phosphorylation site Thr-112 may contribute to the regulation DP thymocyte apoptosis.
Figure 7. Effect of phosphorylation-defective Bim on thymocyte apoptosis in vivo.
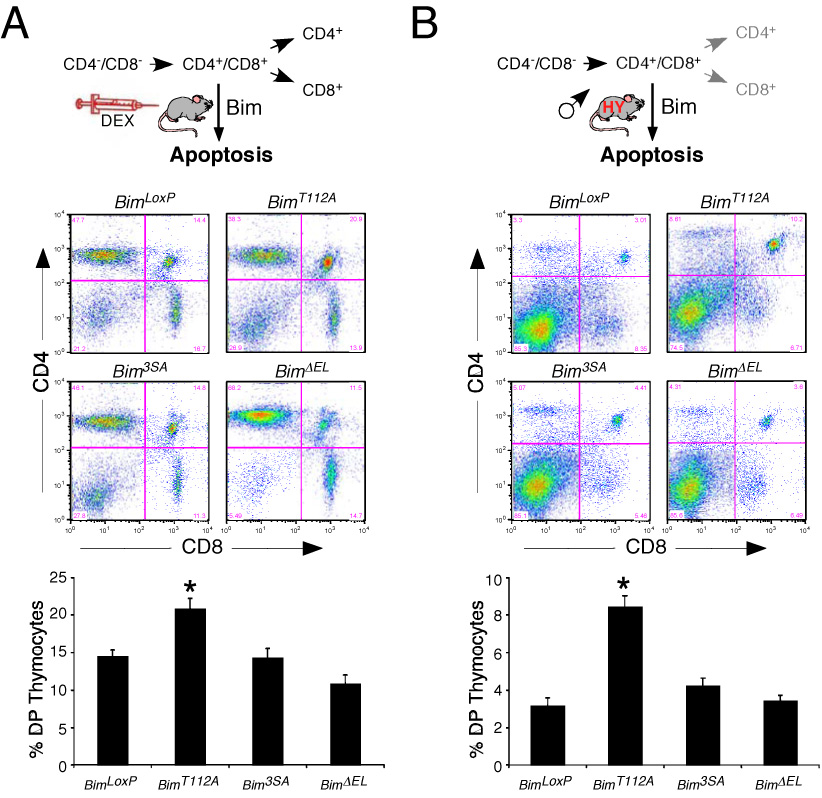
A) Mice (8 – 12 weeks old) were injected with dexamethasone (8.3 µg / g body mass) or solvent (saline). Thymocytes were isolated at 20 h post-injection and the CD4+ and CD8+ sub-populations were examined by flow cytometry. Representative FACS profiles of thymocytes from dexamethasone-treated homozygous BimLoxP, BimT112A, Bim3SA, and BimΔEL mice are illustrated. The data obtained from groups of 8 –12 mice per genotype are presented as the mean ± SD in the lower panel. A statistically significant increase (t-test; P < 0.05) in the survival of double positive (DP) CD4+CD8+ thymocytes from Bim mutant mice compared with mice that express wild-type Bim (BimLoxP) is indicated with an asterisk.
B) The thymocytes of male HY-TCR positive mice (5 – 8 weeks old) on genetic backgrounds that are homozygous for the wild-type Bim protein (BimLoxP) or a phosphorylation-defective Bim protein (BimT112A, Bim3SA, and BimDEL) were examined by flow cytometry. Representative FACS profiles of thymocytes are illustrated. The data obtained from groups of 13 –14 mice per genotype are presented as the mean ± SD in the lower panel. A statistically significant increase (P < 0.05) in the survival of double positive (DP) CD4+CD8+ thymocytes from Bim mutant mice compared with mice that express wild-type Bim (BimLoxP) is indicated with an asterisk.
BimKO mice exhibit defects in the deletion of autoreactive thymocytes (Bouillet et al., 2002). To test the role of Bim phosphorylation, we initially examined the effect of engagement of the T cell receptor with anti-CD3 to cause deletion of DP thymocytes. These studies demonstrated that the administration of anti-CD3 caused similar deletion of DP thymocytes in BimLoxP, Bim3SA, and BimΔEL mice, but BimT112A mice exhibited reduced deletion of DP thymocytes. These data suggested that the Thr-112 phosphorylation site may contribute to negative selection of thymocytes. To test this hypothesis we examined the deletion of autoreactive thymocytes in a mouse model with transgenic expression of a T cell receptor (TCR) that recognizes the HY male-specific antigen. In this model, male (but not female) mice exhibit strong negative selection of thymocytes (Kisielow et al., 1988).
We found that female HY-TCR+ and HY-TCR− mice exhibited similar CD4/8 thymocyte sub-population profiles in the context of homozygous control (BimLoxP) and phosphorylation-defective Bim alleles (Bim3SA, BimΔELand BimT112A). In contrast, studies of male HY-TCR+ mice demonstrated marked deletion of autoreactive DP thymocytes compared with male HY-TCR− mice. The extent of deletion of autoreactive thymocytes was similar in studies of male HY-TCR+ BimLoxP, Bim3SA, and BimΔELmice (Figure 7B). However, reduced deletion of DP thymocytes was observed in male HY-TCR+BimT112A mice (Figure 7B). These data indicate that mutation of the phosphorylation site Thr-112 causes reduced apoptosis of autoreactive DP thymocytes. This reduction in apoptosis correlates with the observation that the phosphorylation-defective BimT112A protein exhibits a defect in binding Bcl2 (Figure 5).
Discussion
Bim is regulated by multi-site phosphorylation
Phosphorylation has been implicated as a mechanism of regulation of Bim-dependent apoptosis. This conclusion is based on the results of transient transfection assays and the over-expression of Bim proteins in cultured cells (Ley et al., 2005). Here we report studies of mice with germ-line mutations in the Bim gene that establish that Bim phosphorylation contributes to the physiological regulation of cell death. Mechanisms that contribute to the effects of phosphorylation on Bim function include the regulation of Bim protein stability (Figure 6) and the interaction of Bim with other members of the Bcl2 family (Figure 5).
Phosphorylation regulates Bim protein stability
We report that mutation of the MAP kinase phosphorylation sites encoded within exon 3 (Ser-55/65/73) causes Bim protein stabilization by preventing proteasomal degradation in vivo (Figure 6). These data support the conclusion that MAP kinase signaling induces ubiquitination of Bim on Lys-3 & Lys-108 and subsequent destruction by the proteasome (Akiyama et al., 2003; Ley et al., 2003; Luciano et al., 2003). This phosphorylation may be mediated by members of the ERK MAP kinase family (Ley et al., 2003; Luciano et al., 2003). This is consistent with our finding that serum-induced BimEL phosphorylation and degradation is inhibited by a pharmacological inhibitor of the ERK pathway (Figure 6B). Indeed, we found that phosphorylation-induced proteasomal degradation can suppress Bim-dependent apoptosis (Figure 6). While all three phosphorylation sites are implicated in the regulation of Bim protein stability, the Ser-65 phosphorylation site may play a central role in this process (Ley et al., 2005).
Although a role for the ERK group of MAP kinases in BimEL phosphorylation on Ser-55/65/73 is established, it is possible that BimEL phosphorylation on these sites may also be mediated by other protein kinases, including JNK (Becker et al., 2004; Putcha et al., 2003) and p38 MAP kinases (Cai et al., 2006). However, our analysis of MEF indicates that UV-stimulated JNK and p38 MAP kinase does not cause the phosphorylation of BimEL on Ser-65 (Figure 4C). Nevertheless, it is possible that these stress-activated MAP kinases may contribute to BimEL phosphorylation on these sites in specific cell-types or in response to particular stimuli (Ley et al., 2005).
Our analysis of Bim3SA mice supports the conclusion that phosphorylation on Ser- 55/65/73 negatively regulates BimEL expression and function (Figure 6). However, some studies using transfected cells have indicated that these phosphorylation sites may increase BimEL apoptotic activity by an undefined mechanism. This effect of phosphorylation to increase apoptotic activity may reflect both cell-type specific and MAP kinase isoform-specific effects of BimEL phosphorylation on apoptosis (Becker et al., 2004; Cai et al., 2006; Putcha et al., 2003).
Interaction of Bim with members of the Bcl2 family
Bim can induce apoptosis by interacting with anti-apoptotic members of the Bcl2 family, including Bcl2, Bcl-XL and Mcl-1 (Willis et al., 2007). Phosphorylation-induced changes in the interaction of Bim with these Bcl2 family proteins may therefore influence the apoptotic activity of Bim. Studies of Bim3SA MEF indicated that mutation of the phosphorylation sites Ser-55/65/73 did not cause marked changes in co-immunoprecipitation of Bim with Bcl2, Bcl-XL or Mcl-1 (Figure 5). In contrast, our analysis of BimT112A MEF indicated that the loss of the Bim (Kuwana et al., 2005) phosphorylation site Thr-112 caused a selective defect in the interaction of Bim with Bcl2 (Figure 5).
Although it is established that Bim interacts with anti-apoptotic members of the Bcl2 family, it is unclear whether Bim interacts with pro-apoptotic members of the Bcl2 family (e.g. Bax). Some studies indicate that Bim may directly interact with Bax (Kuwana et al., 2005; Letai et al., 2002), while other studies have failed to detect this interaction (O'Connor et al., 1998; Willis et al., 2007). It is therefore possible that Bim may directly activate Bax (Kuwana et al., 2005; Letai et al., 2002) or may indirectly activate Bax by sequestering anti-apoptotic Bcl2 proteins (O'Connor et al., 1998; Willis et al., 2007). These actions of Bim may be cell type- or context- dependent. One previous study using over-expressed proteins indicated that serine phosphorylation of Bim may inhibit the binding of Bim to Bax (Harada et al., 2004). We were unable to test this hypothesis using the mutant mouse models we have created because we did not observe co-immunoprecipitation of endogenous Bim with Bax (Figure 5). Further studies are therefore required to examine the possible effects of Bim phosphorylation on the interaction of Bim with Bax.
The Bim phosphorylation site Thr-112 site is located within the dynein light chain 1 (DLC1) binding motif (Figure 1A). Previous studies have demonstrated that the binding of Bim to DLC1 decreases the apoptotic activity of Bim (Puthalakath et al., 1999), that Bim phosphorylation on Thr-112 disrupts the interaction of Bim with DLC1 Bim (Lei and Davis, 2003), and that exposure to stress (e.g. UV light) causes increased binding of Bim to Bcl2 (Puthalakath et al., 1999). Together, these data indicate that Bim phosphorylation on Thr-112 may increase Bim apoptotic activity by decreasing the binding of Bim to DLC1 and by increasing the binding of Bim to Bcl2. This conclusion is consistent with our finding that the phosphorylation-defective BimT112A protein exhibits reduced interaction with Bcl2 (Figure 5) and reduced apoptotic activity (Figure 7). It is also possible that the decreased interaction of Bim with DLC1 may regulate the sub-cellular localization of Bim (Puthalakath et al., 1999), although in some cell types, including T cells, Bim is found to be constitutively associated with mitochondria (Hildeman et al., 2002).
Previous studies have demonstrated strong genetic interactions between mutant alleles of the Bim and Bcl2 genes in mice (Bouillet et al., 2001a). Thus, Bcl2 knockout mice exhibit a number of degenerative disorders; this phenotype is markedly suppressed by loss of Bim expression. These data suggest that Bcl2 is a physiologically relevant target of pro-apoptotic Bim. The finding that the Bim phosphorylation site Thr-112 selectively influences the binding of Bim to Bcl2 (Figure 5) provides a possible mechanism that may account for the reduced apoptotic activity of the BimT112A protein (Figure 7). These considerations suggest that Bim phosphorylation on Thr-112 may be relevant to cell types in which Bcl2 plays a key role in maintaining survival. Indeed, Bcl2 is important for preventing Bim-dependent thymocyte apoptosis (Bouillet et al., 2001a) and BimT112A mice were found to exhibit a defect in thymocyte apoptosis (Figure 7). Studies of other cell types from BimT112A mice in which Bcl-XL or Mcl-1 may play a more prominent role in the maintenance of cell survival (e.g. MEF) did not exhibit defects in apoptosis (Figure 6). Together, these data indicate that the Thr-112 phosphorylation site on Bim may influence apoptosis in a cell-type specific manner.
Integration of JNK and ERK signaling to Bim
JNK can phosphorylate BimEL on Thr-112 in vitro and transfection studies have also implicated a role for this phosphorylation in Bim-dependent apoptosis (Lei and Davis, 2003). This role of JNK is strongly supported by our observation that UV-radiation causes Bim phosphorylation on Thr-112 in wild-type MEF, but not in JNK-deficient fibroblasts (Figure 4C). However, JNK is not the only kinase that can phosphorylate this site on Bim. Indeed, studies using JNK-deficient fibroblasts demonstrated that ERK MAP kinases can also phosphorylate Bim on Thr-112 in serum-treated cells (Figure 4D). However, the significance of this finding is unclear because although ERK-mediated phosphorylation on Thr-112 may increase Bim apoptotic activity, ERK-mediated phosphorylation on Ser-55/65/73 causes rapid proteasomal degradation of Bim. Together, these observations suggest a possible mechanism of cross-talk between JNK-induced Bim apoptotic activity and a dominant effect of ERK signaling to suppress Bim expression and consequently promote cell survival.
Conclusions
The results of this study establish that multi-site phosphorylation of Bim represents a physiological mechanism that can control the sensitivity and threshold response of Bim to activation by pro-apoptotic stimuli.
Experimental Procedures
Mice
Mice with Jnk1 and Jnk2 gene disruption have been described (Tournier et al., 2000). Mice with transgenic expression of a T cell receptor (HY-TCR) specific for male antigen HY presented by class I MHC H2-Db (Kisielow et al., 1988) and mice with targeted disruption of the Bim gene (Bouillet et al., 1999) were obtained from Taconic Farms and The Jackson Laboratories, respectively. Mice with germ-line mutations in exons 3 or 4 of the Bim gene were constructed using homologous recombination in TC1 embryonic stem (ES) cells. Targeting vectors were constructed using a mouse strain 129/Svev genomic BAC clone containing the Bim gene, a floxed NeoR cassette, and a thymidine kinase cassette (Figure 2). ES cells were electroporated and selected with 200µg/ml G418 and 2µM gangcyclovir. Correctly targeted ES cell clones were identified by Southern blot analysis and PCR analysis. The floxed NeoR cassette was excised using Cre recombinase. Positive ES cell clones were injected into C57BL/6J blastocysts to create chimeric mice that transmitted the mutated Bim alleles through the germ-line. The mice were backcrossed for ten generations to the C57BL/6J strain (Jackson Laboratories) and were housed in a facility accredited by the American Association for Laboratory Animal Care (AALAC). The animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Massachusetts Medical School.
Genotype analysis
The Bim locus was examined by Southern blot analysis of BamHI restricted genomic DNA by probing with a random-primed 32P-labeled 5′ (369 bp) and 3′ (384 bp) probes isolated by PCR using a genomic BAC clone as the template and the amplimers 5′- TGGGGACACAGCAGGTGAAGTTG plus 5′-ATCGTAACCAATGGGCAGAGG and 5′-CAGATGAGTGGACTGTCACCATAAAC plus 5′- AAACGCTGCTGTCAAAGAAAGC, respectively. The wild-type and mutated alleles of Bim were also detected by PCR amplification of genomic DNA using the amplimers 5′- TGGTTTAGTGTTTGGGGTGGCG and 5′-AGACTGCCTTGGGAAAAGCG to create a 2.4 kb PCR product that was incubated with restriction enzymes that are specific for each mutation (Figure 2B,C). The genotypes of homozygous mutant mice were confirmed by PCR using genomic DNA as template and the primers 5′- ATGGCCAAGCAACCTTCTGATGTAAGTTCTGAG and 5′- CATTGCACTGAGATAGTGGTTGAAGGCCTGGC to create a 380 bp product (or a 211 bp product in the case of BimΔEL) that was incubated with restriction enzymes that are specific for each mutation (Figure 2E). The wild-type and mutated Bim alleles were also confirmed by sequence analysis. Routine genotyping of these mice was performed by PCR with the amplimers 5′-ATCACATCATCACCAGGCGAGC, 5′- AAGGCATACTCTGTCTCCTACCGC, and 5′-CGTGGTAGAATGGTAGCAAGTGC to distinguish between wild-type (441 bp) and mutant (642 bp) Bim alleles.
Plasmids
A BimEL cDNA was isolated by PCR amplification of reverse-transcribed mRNA and was cloned with an NH2-terminal epitope tag (Flag) in the expression vector pCMV-Tag2B (Stratagene) at the BamH1 and EcoR1 restriction sites. Point mutations were created using the Quick-change procedure (Stratagene) and were confirmed by DNA sequencing. BimL expression vectors and the constitutively activated JNK1 expression vector (MKK7-JNK1) have been described previously (Lei and Davis, 2003). Retroviral vectors expressing Bcl2 (#8793), Bcl-XL (#3541), and Bax (#3694) were obtained from Add-gene.
Tissue culture
Primary fibroblasts prepared from E13.5 murine embryos and 293T cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS (Invitrogen). The 293T cells were transfected with plasmids using Lipofectamine (Invitrogen).
Immunoblot analysis
Cell extracts were prepared using Triton lysis buffer [20 mM Tris (pH 7.4), 1% Triton X-100, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 10 µg/mL of aprotinin and leupeptin]. Cell extracts (50 µg of protein) and immunoprecipitates prepared from cell extracts were resolved by SDS-PAGE or 2D gels (isoelectric focusing in the horizontal dimension (pI range 4 – 7) and SDS-PAGE in the vertical dimension) and examined by protein immunoblot analysis. Immunoblots of cell lysates were probed using enhanced chemiluminescence (NEN). Immunobots of immunoprecipitates were probed using a co-immunoprecipitation kit obtained from Genescript Inc. The antibodies employed in these studies were obtained from Cell Signaling (Akt and phosphoSer473 Akt, Bax, Bcl2, Bcl- XL, ERK and phosphoERK, phosphoSer65-Bim), Calbiochem (Bim), Rockland (Mcl-1), and Sigma (α-Tubulin). The polyclonal antibody to phosphoThr112- Bim was purified from serum obtained from rabbits immunized with the peptide DKSTQpTpPSPP (Biosource).
Apoptosis assays
DNA fragmentation assays were performed using the Cell Death Detection ELISAplus kit, following the manufacturer's recommendations (Roche).
Flow cytometry
Cells (5 × 105) were incubated with R-phycoerythrin-conjugated anti-CD4 (L3T4; PharMingen) and Allophycocyanin-conjugated anti-CD8α (Ly-2; PharMingen) antibodies (30 min at 4°C), washed with PBS plus 2% BSA, fixed in PBS plus 2% formaldehyde, and examined by flow cytometry.
Acknowledgments
We thank Dr Sunny Tam for assistance with 2D gel electrophoresis, Dr Stephen Jones for assistance with blastocyst injections, Vicky Benoit and Judy Reilly for expert technical assistance, and Kathy Gemme for administrative assistance. These studies were supported by a grant from the National Institutes of Health. Core facilities used by these studies were supported by the NIDDK Diabetes and Endocrinology Research Center (DK52530) at the University of Massachusetts. R.A.F. and R.J.D. are Investigators of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, Fukuda A, Hikita A, Seto H, Okada T, et al. Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. Embo J. 2003;22:6653–6664. doi: 10.1093/emboj/cdg635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker EB, Howell J, Kodama Y, Barker PA, Bonni A. Characterization of the c-Jun N-terminal kinase-BimEL signaling pathway in neuronal apoptosis. J Neurosci. 2004;24:8762–8770. doi: 10.1523/JNEUROSCI.2953-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev Cell. 2001a;1:645–653. doi: 10.1016/s1534-5807(01)00083-1. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P, Eyre HJ, Sutherland GR, Adams JM. Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm Genome. 2001b;12:163–168. doi: 10.1007/s003350010242. [DOI] [PubMed] [Google Scholar]
- Bunin A, Khwaja FW, Kersh GJ. Regulation of Bim by TCR signals in CD4/CD8 double-positive thymocytes. J Immunol. 2005;175:1532–1539. doi: 10.4049/jimmunol.175.3.1532. [DOI] [PubMed] [Google Scholar]
- Cai B, Chang SH, Becker EB, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J Biol Chem. 2006;281:25215–25222. doi: 10.1074/jbc.M512627200. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, Adams JM, Strasser A, Villunger A. BH3-only proteins Puma and Bim are rate-limiting for gamma-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, Cook SJ. ERK1/2- dependent phosphorylation of Bim(EL) promotes its rapid dissociation from Mcl- 1 and Bcl-x(L) Embo J. 2007 doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker G, Suttner K, Harada H, Kirschnek S. TLR-dependent Bim phosphorylation in macrophages is mediated by ERK and is connected to proteasomal degradation of the protein. Int Immunol. 2006;18:1749–1757. doi: 10.1093/intimm/dxl109. [DOI] [PubMed] [Google Scholar]
- Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci U S A. 2004;101:15313–15317. doi: 10.1073/pnas.0406837101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by proapoptotic bcl- family member bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 1988;333:742–746. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–14912. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Ley R, Ewings KE, Hadfield K, Cook SJ. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ. 2005;12:1008–1014. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- Naik E, Michalak EM, Villunger A, Adams JM, Strasser A. Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J Cell Biol. 2007;176:415–424. doi: 10.1083/jcb.200608070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci U S A. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson EM., Jr JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–296. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c- mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]