

Abstract
The mechanism/s leading to diabetic neuropathy are complex. Transforming growth factor-β1 (TGF-β1) has been associated with diabetic nephropathy and retinopathy but not neuropathy. In this study, changes in TGF-β isoforms were examined in-vivo and in-vitro. Two groups of animals, streptozotocin diabetic with neuropathy and non-diabetic controls were examined at 4 weeks (n=10/group) and 12 weeks (n=8/group). In diabetic DRG using quantitative real-time PCR (QRT-PCR), TGF-β1 and TGF-β2 mRNA, but not TGF-β3, was increased at 4 and 12 weeks. In sciatic nerve TGF-β3 mRNA was primarily increased. Immunohistochemistry (DRG) and immunoblotting (sciatic nerve) showed similar differential protein expression. In sciatic nerve TGF-β formed homo- and heterodimers, of which β2/β3, β1/β1, and β1/β3 were significantly increased, while that of the TGF-β2/β2 homodimer was decreased, in diabetic compared to non-diabetic rats. In-vitro, pretreatment of embryonic DRG with TGF-β neutralizing antibody prevents the increase in total TGF-β protein observed with high glucose using immunoblotting. In high glucose conditions, combination with TGF-β2 > β1 increases the percent of cleaved caspase-3 compared to high glucose alone and TGF-β neutralizing antibody inhibits this increase. Furthermore, consistent with the findings in diabetic DRG and nerve, TGF-β isoforms applied directly in vitro reduce neurite outgrowth, and this effect is partially reversed by TGF-β neutralizing antibody. These findings implicate upregulation of TGF-β in experimental diabetic peripheral neuropathy and indicate a novel mechanism of cellular injury related to elevated glucose levels. In combination, these findings indicate a potential new target for treatment of diabetic peripheral neuropathy.
Keywords: cell death, cytokines, diabetes, DRG, neuropathy, sciatic nerve, TGF-β
Introduction
Diabetes mellitus is associated with multi-organ complications of which the commonest is neuropathy that occurs in up to 60% of diabetic subjects (Dyck et al., 1993). Despite the significant pathology associated with nerve degeneration, the etiology of neuropathy is still unclear. Several pathways have been suggested to be associated with glucose neurotoxicity (Tomlinson and Gardiner 2008) and diabetic neuropathy, for example oxidative stress (Cameron et al., 1993; Coppey et al., 2001; Obrosova et al., 2005; Pop-Busui et al., 2006; Russell et al., 2002), altered polyol metabolism (Cameron and Cotter 1997), mitochondrial dysfunction (Huang et al., 2003a; Montal, 1998; Russell et al., 2002), activation of certain cysteine proteases (caspases) (Russell et al., 1999; Srinivasan et al., 2000) (Cheng and Zochodne 2003; Schmeichel et al., 2003), and regulation of growth factors and their intermediate signaling pathways (Sayers et al., 2003; Tomlinson et al., 1996). However, the role of cytokines such as transforming growth factor β (TGF-β) in development of diabetic neuropathy is not clearly defined.
Three isoforms of TGF-β are reported in mammals (TGF-β1, -β2, & -β3). TGF-β comprises a family of cytokine growth factors that have several functions in the areas of cellular development, differentiation, extra cellular matrix formation and apoptosis (Lin and Moustakas 1994; Schuster and Krieglstein 2002). It has been generally accepted that functions of TGF-β family members may vary depending on cellular status and cell types. TGF-β isoforms have been implicated in a broad diversity of biological activities, including cell growth, cell death, cell differentiation, inflammation, and immunological reactions, by modifying the expression of specific sets of target genes (Massague 1998; Massague et al., 2000). TGF-β has been shown to be both pro- and anti- apoptotic, influenced by both context and location. Increases or decreases in the production of TGF-β have been linked to numerous disease states, including atherosclerosis and fibrotic disease of the kidney, liver and lung. TGF-β is reported to be increased by elevated glucose and is a known powerful stimulus of extra cellular matrix production (Chen et al., 2003; Oh et al., 1998). High levels of glucose significantly increase levels of TGF-β1 and lead to fibrosis of these organs (Park et al., 1997; Sharma et al., 1997) and induction of diabetic nephropathy (Pantsulaia 2006).
In neuronal tissues, it is clear that TGF-βs play a neurotrophic role in some situations (Chalazonitis et al., 1992; Krieglstein et al., 1995; Martinou et al., 1990; Poulsen et al., 1994), while they elicit cell-death-inducing effects in other situations (Krieglstein et al., 2000; Schuster and Krieglstein 2002). However, the exact role of various isoforms of TGF-β and its functions are not understood in the peripheral nervous system or in diabetic neuropathy.
The current study examines the expression of differential expression of TGF-β isoforms in DRG and sciatic nerve of diabetic animals and the association of TGF-β with cellular injury in the peripheral nervous system.
Experimental procedures
Materials
Streptozotocin (STZ), Sigma-Aldrich, St. Louis, MO. Biomedia Gel/Mount: Electron Microscopy Services (EMS), Ft. Washington, PA. Trypsin, B-27, neurobasal medium, MEM: Gibco-BRL, Gaithersberg, MD. Nerve Growth Factor (NGF): Harlan Bioproducts for Science, Indianapolis, IN. Pen/Strep/Neo, l-glutamine, selenium, hydrocortisone, β-estradiol, transferrin, 4′,6-diamidino-2-phenylindole (DAPI), adeno cAMP, forskolin, bovine pituitary extract, fetal bovine serum (FBS), 5-fluoro-2-deoxyuridine (FUDR): Sigma-Aldrich, St. Louis, MO. Rabbit polyclonal cleaved caspase-3 antibody: Pharmingen, San Diego, CA. 3,3′-diaminobenzidine (DAB), Invitrogen Corporation, California, USA. Alexafluor 596 goat-anti-rabbit antibody: Molecular Probes, Eugene, OR. Protein assay reagents: Bio-Rad, South San Francisco, CA. Chemiluminescence’s reagents: Amersham, Piscataway, NJ. Nitrocellulose: Schleicher and Schuell, Keene, NH. Horseradish peroxidase-conjugated goat-anti-rabbit, goat-anti-mouse, TGF-β1, TGF-β2 antibodies: Santa Cruz, Santa Cruz, CA. β-Actin antibodies: Chemicon, Temecula, CA. TGF-β1, & -β3 antibody (Sigma-Aldrich, St. Louis, MO). FITC-conjugated goat anti-rabbit IgG secondary antibodies (Molecular Probes). Apoptag fluorescein In Situ Detection Kit: Serologicals, Norcross, GA. Purified TGF-β1, -β2, & -β3 protein & pan-specific TGF-β neutralizing antibody: R&D Systems, Minneapolis, MN. TaqMan universal PCR master mix and Primers of TGF-β, Applied Biosystems, CA, USA. Anti-human rabbit PGP 9.5 was purchased from Accurate Chemical & Scientific Co., NY, USA.
Generation of STZ Induced Diabetic Rats
Male Sprague Dawley (SD) rats (Harlan, Indianapolis, IN) were maintained on regular rat chow diet and light cycle. The animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee and conform to U.S. National Institutes of Health guidelines. All efforts were made to minimize stress to the animals, and multiple technologies were used to maximize the amount of data obtained from each animal and to minimize the total number of animals required. Animals were housed under approved conditions with a 12-h light/dark cycle with free access to food and water. Diabetes was induced in 12 week old SD rats with a 45 mg/kg of STZ injection as previously described (Russell et al., 1999). Diabetes was verified after 2 days by evaluating blood glucose levels with the use of a glucose monitor and glucose-oxidase reagent strip. Rats having blood glucose levels of 300 mg/dl (16.7 mM) or greater were considered to be diabetic. After 4 and 12 weeks of STZ, diabetic animals were euthanized, and the DRG and sciatic nerves were collected.
Nerve Conduction Studies (NCS)
Before performing the electrophysiological examination, rats were anesthetized with ketamine 60 mg/kg and acepromazine 0.75 mg/kg by intraperitoneal injection. The hind limbs were kept injection free to avoid nerve or muscle injury.
NCS were performed in the tail and left hind limb using platinum electrodes adjacent to the nerve to obtain near nerve recordings. The G1 (active) and G2 (indifferent) recording electrodes were separated by a distance of 10 mm. Tail nerve conduction studies were measured over a 9 cm distance from the base of the tail. Proximal nerve conduction velocities were measured over the proximal 5 cm from the base of the tail, and distal nerve conductions over the terminal 4 cm. Orthodromic tail motor conduction velocities (MCV) and distal latencies were obtained by recording at the tip of the tail and stimulating with the cathode 4 and 9 cm proximal to the G1 recording electrode. Othodromic tail sensory conduction velocities (SCV) were recorded using the same measuring distances but with the G1 electrode place proximally on the tail. In the hind limb MCV and distal latencies were recorded from the dorsal foot with stimulation at the malleolus, and proximally in the sciatic notch. All sensory responses were averaged until the waveform was stable. Tail and limb temperatures were maintained at 34° C using a near nerve temperature probe, and stimulus intensities were controlled as previously described (Russell et al., 2001).
Behavioral Assessment for Mechanical Stimulation (Von-Frey test)
Mechanical withdrawal threshold was measured using an electronic Von-Frey device (Somedic Inc.) as previously described (Hong et al., 2004; Whiteside et al., 2004). Rats were placed in an individual chamber on a metal mesh floor and allowed to habituate for 30 min. Initially, probe tips in the range of 0.1 to 1.0 mm were tested. Trials were repeated four times for each probe tip with increasing strength until the animal first showed a response to the monofilament stimuli. The smallest tip where the animal showed a response was used for subsequent testing (0.3 mm). The 0.3 mm probe was attached to the probe tip holder and a force was applied on the plantar surface of the left hindlimb for 2–4 seconds. At the beginning of the experiment, the equipment was calibrated and standardized using the threshold for a 20 gram weight. The 20 gram weight was placed on the tip of the 0.3 mm probe for standardization and the peak stimuli based on this force was measured. For subsequent testing, the probe strength at threshold on the footpad was recorded automatically for each rat using a computer.
Skin Biopsy
Fifty µm sections were cut using a cryostat (Leica CM 1850) as described previously (Hirai et al., 2000). Briefly, endogenous peroxidase activity was blocked by incubation for 0.5 hr with 1% H2O2 in 100% methanol. Sections were incubated in primary, anti-human rabbit PGP 9.5 antibody (Accurate Chemical & Scientific Co., NY, USA) in 1/20,000 dilution in 0.1 M PBS with 0.3% Triton X-100 and 1% normal bovine serum for 1 hr at room temperature. Non-immune serum was used as a control. After sections were washed three times in 0.1 M PBS with 0.3% Triton X-100, they were incubated in goat biotinylated anti-rabbit IgG (1/ 500; Vector) overnight at room temperature. Following additional washes, sections were placed in an avidin / biotin complex solution (Vector ABC Kit). Nerve fibers were demonstrated using 3,3’-diaminobenzidine (DAB) chromogen (Invitrogen, CA, USA). Sections were mildly counterstained with eosin to better localize the basement membrane and epidermal layer. Images were captured digitally using a Zeiss microscope and software with the help of a Nikon digital camera. The average intraepidermal nerve fibers density (IENFD) and length were measured using standardized measurement protocols and compared to controls (Lauria et al., 2005).
Quantitative RT-PCR
DRG neurons and sciatic nerve tissues were collected from both non-diabetic control and diabetic rats 4 and 12 weeks after STZ treatment. We performed quantitative real-time PCR (QRT-PCR) to assess the amount of endogenous mRNA for all TGF-β (β1, β2, β3) genes. RNA was isolated from rat DRG and sciatic nerve tissues using TRIZOL® (Invitrogen), treated with DNaseI (New England Biolabs), and then reverse-transcribed with the High-Capacity cDNA Archive Kit® (Applied Biosystems). QRT-PCR was carried out by an Applied Biosystems 7500 PCR system with TaqMan universal PCR master mix. Primers for TGF-β were supplied by Applied Biosystems and were: Rn00572010_m1 (TGF-β1), Rn00579674_m1 (TGF-β2), Rn00565937_m1 (TGF-β3). Reactions were run with an initial ramp time of 2 minutes at 50° C, then 10 minutes at 95° C, and 40 subsequent cycles of 15 seconds at 95° C and 1 minute at 60° C. As a negative control for the RT reaction, reverse transcriptase was omitted in the reaction mix. For negative controls for the PCR reaction, either the primer sets or the cDNA were omitted from the reactions. Relative concentrations of TGF-β RNA were calculated with comparison to a standard curve. Data analysis was performed using Sequence Detection System software, version 1.9.1 (Applied Biosystems). In each experiment the relative measurement of QRT-PCR was normalized against β-actin and expressed as a ratio. The mean of the ratio was obtained from three separate experiments. Since several reference genes may themselves vary in diabetic conditions, we determined the stability of endogenous reference control genes. Analysis was performed using the GeNorm 3.4 visual basic application for Microsoft Excel validated previously (Vandesompele et al., 2002). Total RNA was also measured spectrophotometrically and all these data were analyzed for stability. Based on this data, β-actin was found to be the most suitable reference gene for DRG.
Immunohistochemistry for Different Isoforms of TGF-β
Both DRG and sciatic nerve tissues were collected from non-diabetic control and diabetic rats and embedded in OCT solution. Sections were washed with phosphate-buffered saline (PBS) before incubation with 10% normal goat serum (NGS) in PBS containing 0.3% Triton-X 100 and 0.5% dried milk for 1 hour. Immunostaining was performed using isoform-specific anti-TGF-β antibodies: TGF-β1 (T0688) 1:100, TGF-β2 (s90) 1:100, TGF-β3 (T4692) 1:100, and TGF-β Neutralizing antibody at a dilution of 1:50. Each slide received the same concentration and volume of primary and secondary antibody for an identical period of time. Non-diabetic controls and diabetic slides were batch processed to assume uniformity of staining. The reaction was visualized using FITC-conjugated goat anti-rabbit and rabbit ant-goat IgG secondary antibodies at dilution of 1:100–1:200 in 10% NGS/PBS/Triton-X 100. Images of DRG and sciatic nerve were collected on a Zeiss Axiovision microscope using a X63 objective and each image was captured digitally using Zeiss software. Each DRG image was collected at 1300 × 1030 pixels, gain 3, and 450 ms exposure and each sciatic nerve image was collected at 1300 × 1030 pixels, gain 3, and 1125 ms exposure. For DRG, 7 sections with a total of 50 DRG were counted for each condition. For sciatic nerve, 7 sections were counted. Within each section, the number of axons in which 50% of the axon showed staining for the specific antibody were counted and recorded as a percentage of the total number of axons in the section.
Cell Culture
Cultured DRG neurons
DRG cell cultures were prepared as previously described (Russell et al., 1999). Dissociated DRG from 15 day old embryonic (E15) SD rats were plated on rat tail collagen coated glass cover slips and pure neuronal cultures were grown in 24-well cell culture dishes as previously described (Russell et al., 2002). Dissociated DRG were prepared by incubating whole DRG in 0.25% trypsin for 40 minutes at 37°C, centrifuging at 800 × g for 5 minutes, then removing trypsin. Explant cultures were grown by placing whole DRG onto collagen-coated 6-well culture dishes. Cultures were grown for 3 days in Neurobasal Medium containing 25 mM glucose (optimal basal glucose for neurons) (Russell et al., 1999; Russell et al., 2002; Srinivasan et al., 2000), with 0.5% B27 without antioxidants, 10 ng/ml Nerve Growth Factor, 0.5% Pen/Strep/Neo, and 0.7 mM l-glutamine, and 40 µM FUDR. Media was changed every 48 hours, and immediately before drug treatment. During experimental drug treatment, all cells were treated in 25 mM glucose, unless exposed to high glucose concentrations (20 mM added glucose, 45 mM total glucose), as indicated. Embryonic rat DRG were used in this study because other studies have shown that adult rat DRG are less susceptible to glucose induced-injury apparently because unlike embryonic rat neurons they are surrounded by a sheath of satellite cells that may protect the adult neurons from injury (Berent-Spillson and Russell 2007; Huang et al., 2003b; Huang et al., 2005), although the duration of treatment in previous studies (1 day) may have been inadequate to induce cellular injury.
TGF-β Treatment
Before beginning the experimental drug treatment, dissociated DRG cell cultures were grown for 2 days at 37°C. Before addition of drugs, cells were rinsed with MEM. TGF-β1, -β2 & -β3 were each used at 10 ng/ml, and the pan-specific TGF-β neutralizing antibody at 10 µg/ml (determined as the optimal concentration on prior dose response experiments). This antibody was selected for its ability to neutralize the biological activity of TGF-β1, -β2 & -β3. In high glucose treated cultures, glucose 20 mM was added, bringing the total concentration of glucose in glucose-treated cells to 45 mM. Cells were treated for 6, 24, or 48 hours before fixation in 4% paraformaldehyde.
TGF-β Neutralization in DRG culture
After growing the embryonic DRG cell cultures for 3 days as described above, cells were incubated as follows: 1) control (without extra glucose), 2) high glucose, 3) high glucose with TGF-β neutralizing antibody, 4) TGF-β internal control. The TGF-β neutralizing antibody neutralizes all TGF-β1, -β2 & -β3 isoforms. The dose of TGF-β neutralizing antibody was 10 µg/ml and treated for 48 hr. The cells were collected and TGF-β protein expression in all conditions was measured by western immunoblotting. Because of the small amount of tissue, a pan-TGF-β antibody was used.
TGF-β Western Immunoblotting
This was performed as previously described (Delaney et al., 2001; Vincent et al., 2002). Purified DRG cultures and sciatic nerve tissues were obtained, cultures were pooled, and protein extracted in RIPA buffer and protease inhibitor (Roche Diagnostics, Indianapolis, IN). Protein (25 µg per sample) was separated by SDS-PAGE and transferred to nitrocellulose. Immunostaining was performed using isoform-specific anti- TGF-β antibodies: TGF-β1 (T0688) 1:500, TGF-β2 (s90) 1:1000, TGF-β3 (T4692) 1:1000, and anti-TGF-β (T9429) 1:1000. TGF-β primary antibody (1:500) was incubated for 2 hr followed by horseradish peroxidase-conjugated secondary (1:3000) over night. We used actin-β as internal control at a 1:1000 dilution, followed by horseradish peroxidase-conjugated goat-anti rabbit antibody at a 1:3000 concentration.
Measurement of Neuronal Injury
Immunohistochemistry for Cleaved Caspase-3
At each experimental time point, cultures were fixed for 30 minutes with 4% paraformaldehyde in PBS, and then stained for active caspase-3 as previously described (Russell et al., 1999; Russell et al., 2002). Cultures were incubated in 0.5 µg/ml rabbit polyclonal cleaved caspase-3 antibody for 2 hours at 37°C. Cells were then incubated in 5 µg/ml Alexafluor 596 goat-anti-rabbit antibody at room temperature for 1 hour. Specificity and concentration of the anti active antibody was determined by immunoabsorption against cleaved peptide. Samples were then co-stained with 1 µg/ml DAPI in PBS for five minutes at room temperature. Cells were finally mounted on slides with Biomedia Gel/Mount, and analyzed at 40x magnification on a Zeiss Axiophot microscope by a blinded observer in a random fashion. For each slide, 10 random fields of 25 to 100 cells per field were counted, and the percent caspase-3 positive neurons were determined. The percentages for each field were averaged for each condition. Each experiment was repeated a minimum of 3 times.
TdT-Mediated dUTP-Biotin Nick End Labeling (TUNEL) Staining
Cultured DRG neurons were fixed in 4% paraformaldehyde for 20 minutes and then TUNEL stained using the Apoptag fluorescein In Situ Detection Kit to detect nuclear fragmentation, as previously described (Delaney et al., 2001; Russell et al., 1999). Slides were analyzed at 40x on a Zeiss Axiophot microscope by a blinded observer in a random fashion; for each slide, 10 random fields of 25–100 cells each were counted, and the percent TUNEL positive neurons determined for each field. The percentages for each field were averaged for each condition. Each experiment was repeated a minimum of 3 times.
Measurement of Neurite Outgrowth
Whole DRG explant cultures were grown for 12 hours with endogenous Schwann cells, and then exposed to experimental conditioned media. Measurements were obtained every 24 hours as previously described (Russell et al., 1994) by a trained observer blinded to the experimental condition. Briefly, the longest neurite from each of 24 explants per condition was measured every 24 hours for the specified number of hours, and subtracted from baseline growth at the start of the experiment. Results were standardized against control conditions. Each experiment was repeated a minimum of 3 times.
Statistics
Assumptions about the Gaussian distribution of data and rules for transformation of non-normative data were made as previously described (Russell et al., 1999; Russell et al., 2002). Comparison of dependent variables was performed on transformed data using factorial analysis of variance (ANOVA), and individual comparisons were made using students T-test, assuming unequal variances. Levels of significance were determined from a 2-tailed T-test.
Results
Animal characteristics and quantitative measurements
Data was obtained in 10 non-diabetic controls and 10 diabetic animals after 4 weeks of diabetes, and a separate group of 8 non-diabetic control and 8 diabetic rats after 12 weeks of diabetes. The mean weights, glucose, glycosylated hemoglobin, and insulin were significantly different at 4 and 12 weeks between diabetic and non-diabetic animals (Table 1). Proximal nerve MCV and SCV in meters/second were significantly reduced in the tail at both 4 and 12 weeks (sensory>motor) consistent with development of a peripheral sensorimotor neuropathy. The sciatic MCV were reduced in diabetic animals at 4 weeks. However, because of variability of NCS between animals, sciatic MCVs were only significantly different at 12 weeks (Table 1). In 4 week diabetic animals, paw mechanical withdrawal threshold was decreased (hyperalgesia) as compared to age matched non-diabetic control rats (Fig. 1). Measurement of skin biopsy sections at 4 weeks, immunostained with antibody to PGP 9.5 (Fig. 2), indicates that there is a significant decrease in both IENFD as compared to non-diabetic controls. These data results for IENFD in controls and diabetics are similar to previously published reports in rats and humans (Bianchi et al., 2004; Hirai et al., 2000; Leonelli et al., 2007).
Table 1.
General characteristics of non-diabetic and diabetic animals at 4 and 8 weeks after STZ injection
| 4 weeks |
12 weeks |
|||||
|---|---|---|---|---|---|---|
| Non-Diabetic | Diabetic | Significance | Non-Diabetic | Diabetic | Significance | |
| Weight (grams) | 507.64 ± 24.60 | 255.88 ± 16.50 | P<0.001 | 459.30 ± 8.32 | 316.67 ± 12.08 | P<0.001 |
| Final Glucose (mg/dl) | 144.00 ± 4.10 | 480.00 ± 28.60 | P<0.001 | 128.14 ± 8.09 | 424.75 ± 26.76 | P<0.001 |
| %Glycolylated Hb | 8.10 ± 0.38 | 15.63 ± 0.29 | P<0.001 | 6.73 ± 0.34 | 16.44 ± 0.73 | P<0.001 |
| Insulin (mU/ml) | 6.89 ± 1.22 | 0.50 ± 0.85 | P<0.001 | 8.1 ± 2.8 | 1.25 ± 0.8 | P<0.001 |
| Tail proximal MCV (m/s) | 35.9 ± 1.9 | 29.8 ± 2.5 | P<0.05 | 35.5 ± 3.1 | 23.8 ± 0.5 | P<0.01 |
| Tail proximal SCV (m/s) | 45.8 ± 1.0 | 38.5 ± 2.1 | P<0.01 | 43.3 ± 2.1 | 32.5 ± 0.9 | P<0.01 |
| Sciatic MCV (m/s) | 50.7 ± 7.9 | 40.6 ± 7.0 | NS | 66.4 ± 3.6 | 51.6 + 2.9 | P<0.05 |
MCV = motor conduction velocity
SCV = sensory conduction velocity
Fig. 1. Behavioral assessment of tactile allodynia in 4 week non-diabetic control and STZ-induced diabetic rats using electronic Von-Frey testing.

Paw mechanical withdrawal threshold (g) was significantly decreased in diabetic rats. Data is expressed as mean ± SEM. **P<0.001 as compared with non-diabetic control.
Fig. 2. Paw skin biopsy for nerve fibers.

Images show sections immunostained with anti-PGP 9.5 antibody in 50 µm cryosections from the hind limb. The arrow indicates intraepidermal nerve fibers; arrow heads indicate dermal nerve fibers (scale bar 30 µm). There is a marked reduction in IENFD in the diabetic rats (B) as compared with the non-diabetic control (A). Lower panel (C) indicates the measurement of IENFD both in non-diabetic and diabetic animals. **P<0.001 as compared to non-diabetic control animals.
Expression of different TGF-β mRNA in DRG and sciatic nerve
In order to determine the expression of specific isoforms of TGF-β under diabetic conditions, we collected both DRG and sciatic nerves from diabetic and non-diabetic control rats. QRT-PCR data of DRG and sciatic nerve tissue samples from non-diabetic control (n=10) and diabetic rats (n=10) indicate that at 4 weeks there is an increase in TGF-β1 expression (23.7% increase, P<0.05) and TGF-β2 (53.2% increase, P<0.05), but no increase in TGF-β3 in diabetic DRG as compared with non-diabetic control rats (Fig. 3A). At 12 weeks, the changes in expression ratio to β-actin for DRG from diabetic compared to non-diabetic animals were as follows: TGF-β1 (110.1% increase, P<0.001), TGF-β2 (45.8% increase, p<0.05), TGF-β3 (no significant difference) (Fig. 3B). In contrast, in sciatic nerve, only the TGF-β3 isoform (64.1% increase, P<0.05) was increased after 4 weeks in STZ rats (Fig. 3C). Similar results were also obtained at 12 weeks (Fig. 3D) in non-diabetic control (n=8) and diabetic (n=8) animals.
Fig. 3. Expression of different isoforms of TGF-β in 4 & 12 week non-diabetic control and STZ-induced diabetic rats in DRG (A – 4 weeks, B – 12 weeks) and sciatic nerve (C – 4 weeks, D – 12 weeks).

QRT-PCR was performed using specific primers as described in experimental procedures. TGF-β1 and TGF-β2, but not TGF-β3 was increased in DRG. In sciatic nerve, only TGF-β3 is increased. Data is expressed as mean ± SEM. *P<0.05 & **P<0.001 as compared with non-diabetic control.
TGF-β detection in both DRG and sciatic nerve of diabetic rats
Because we wanted to determine that TGF-β protein was present in neurons and Schwann cells rather than simply being expressed in connective tissue, immunohistochemistry was performed in DRG and sciatic nerve obtained from non-diabetic and diabetic animals. In DRG neurons and sciatic nerve tissues stained for TGF-β1, -β2 & -β3, immunoreactivity for TGF-β2 was significantly increased in DRG (P<0.001, Fig. 4), and TGF-β3 in sciatic nerve (42.45 ± 1.59; P<0.001). There was no antibody staining in the non-diabetic control tissue and when the tissue sections were incubated with TGF-β neutralizing antibody, no antibody staining was observed.
Fig. 4. Immunohistochemistry of 4 weeks non-diabetic control and diabetic rat DRG tissue.

(A) Light microscopic images: a, c, e; immunofluoresence (FITC staining) images: b, d, f. TGF-β2 was significantly increased in diabetic DRG (d) compared to non-diabetic control (b) There was no immunofluorescence using pretreatment with pan- TGF-β neutralizing antibody on the section as an internal control (f). (B) Intensity measurement: TGF-β2 staining was significantly increased in diabetic DRG (**P<0.001).
Differential accumulation of homo- and hetro-dimeric isoforms of TGF-β dimer in sciatic nerve of diabetic and non-diabetic rats
To further investigate protein expression of TGF-β in sciatic nerve, we performed quantitative Western blot analysis using protein samples obtained from 4 week diabetic and non-diabetic control tissues. After electrophoresis, the gels were subjected to immunoblotting with antibodies against each isoform of TGF-β and to actin. The relative level of TGF-β in each sample was measured by quantification of the intensity of the TGF-β band and normalized to the actin level in the corresponding sample (Fig. 5). We observed four distinct isoforms of TGF-β dimer, corresponding to molecular masses of 96 (TGF-β2/β2 dimer), 60 (TGF-β2/β3 dimer), 48 (TGF-β1/β1 dimer), and 37 (TGF-β1/β3 dimer) kDa, respectively. Of the four identified TGF-β dimer isoforms, the protein level of three isoforms (60, 48, and 37 kDa) of TGF-β dimer were significantly increased 2.4-, 2.1-, and 3.8-fold, respectively, in diabetic compared to non-diabetic rats. In contrast, the protein level of the 96 kDa isoform of TGF-β2/β2 homodimer was significantly decreased 5-fold in diabetic compared to non-diabetic rats.
Fig. 5. Western blot analysis of TGF-β isoforms in 4 week non-diabetic and diabetic rat sciatic nerve.
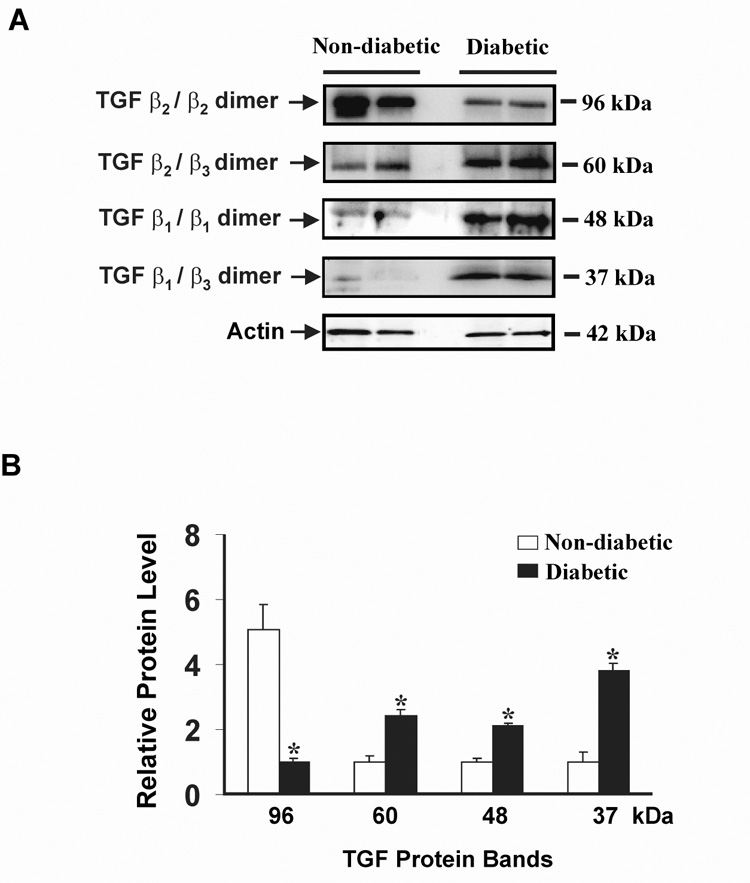
(A) Lane 1 & 2 represents non-diabetic protein bands; lane 3 & 4 represents protein bands from diabetic rats. In this blot actin-β was used as an internal control. (B) The density of the relative protein levels were measured and expressed as a graph. Clear bars indicate the relative protein level in non-diabetic controls, black bars indicate the relative protein level in diabetic rats. Data is expressed as mean ± SEM. *P<0.05 as compared with non-diabetic rats.
Effect of TGF-β on DRG neuronal injury
Experiments in tissue culture using western immunoblot analysis indicate that there is an increase in TGF-β protein levels in DRG neurons exposed to high glucose when compared to control neurons. In the presence of TGF-β neutralizing antibody and high glucose, TGF-β protein levels were normalized to control levels as compared to neurons exposed to high glucose in the absence of neutralizing antibody (P<0.05, Fig. 6).
Fig. 6. Pretreatment with TGF-β neutralizing antibody normalizes the increase in total TGF-β antibody levels with high glucose.

Western immunoblot analysis of TGF-β was performed in pooled cultured embryonic DRG neurons with high glucose ± neutralizing antibody: (A) Control DRG neurons (lane 1), high glucose treated neurons (lane 2), high glucose plus TGF-β neutralizing antibody (Lane 3), TGF-β control protein (lane 4). In this blot actin-β was used as internal control. (B) The density of the relative protein levels were measured and expressed as a graph. Data is expressed as mean ± SEM. *P<0.05 as compared with control, #P<0.05 as compared with high glucose,
In dose-ranging experiments we evaluated the dose-effect of TGF-β after 24 hours on caspase-3 cleavage in cultured DRG neurons. Combined TGF-β protein increased the percent of cleaved caspase-3 positive neurons in a dose dependent manner in both control and high glucose conditions (Fig. 7). These dose ranges were selected for the further experiments to determine the effect of different isoforms of TGF-β on caspase-3 activity. After 48 hours, TGF-β increased cell injury in neurons as compared to non TGF-β treated controls as measured by propidium iodide exclusion, or chromatin condensation as detected by DAPI nuclear staining (data not shown).
Fig. 7. Increased glucose levels and purified combined TGF-β induces DRG neuronal injury.

Purified DRG neurons were treated for 24 hours with high glucose, and/or purified TGF-β protein and stained and counted for caspase-3 cleavage. There is an increase in the percent of caspase-3 positive neurons in DRG neurons exposed to high glucose or TGF-β in a dose dependent manner and there is an additive effect with glucose and higher doses of TGF-β. Data is expressed as mean ± SEM. ***, ###P<0.01 as compared with control, +++P<0.01 as compared with high glucose treatment.
Pure DRG neuronal cultures were treated with 10 ng/ml each of purified TGF-β1, -β2 & -β3 proteins for 24 hours indicating that caspase-3 is increased in the high glucose condition as compared with control (Fig. 8A). Cells in the high glucose condition treated with TGF-β1, -β2 & -β3 showed a significant increase in the percent of caspase-3 positive neurons (P<0.001), but not with TGF-β3. TGF-β neutralizing antibody reduced glucose-induced neuronal injury as indicated by the percent caspase-3 positive cells compared to glucose treated neurons (P<0.001). The percent of TUNEL positive neurons were increased in high glucose conditions as compared with control neurons, and TGF-β neutralizing antibody reduced neuronal injury (Fig. 8B).
Fig. 8. TGF-β1, -β2, & -β3 increases glucose-induced neuronal injury.

(A) A higher percentage of cultured DRG neurons expressed cleaved caspase-3 after incubation in high glucose + TGF-β1, -β2 & -β3 for 24 hours than treated or non-treated controls. Addition of glucose to TGF-β1, -β2 & -β3 had an additive effect on the percent of caspase-3 positive neurons. TGF-β Neutralizing antibody prevented glucose-induced caspase-3 cleavage. Data expressed as mean ± SEM. #P<0.05 as compared with non-treated control cells, *P<0.05, **P<0.01 in glucose-treated neurons as compared with the corresponding control condition.
(B) DRG neurons that were exposed to high glucose for 48 hours had a higher rate of neuronal injury than non-treated control cells as indicated by TUNEL staining of nuclei for DNA degradation. TGF-β Neutralizing antibody prevented glucose-induced neuronal injury. Data is expressed as mean ± SEM. **P<0.001 in glucose-treated neurons as compared to control.
Effect of TGF-β on DRG neurite outgrowth
In addition to increasing cellular injury, TGF-β treatment also affects neurite growth in cultured DRG explants. Whole DRG explants were grown in 10 ng/ml TGF-β1, -β2 & -β3 for 48 hours, and mean neurite length determined at 24 hour intervals. Neurons treated with each of the isoforms exhibited slower neurite growth and shorter mean neurite length than did non-treated controls (Fig. 9). At 48 h neurite growth in controls was significantly different from glucose treated and TGF-β1, -β2 & -β3 treated cultures (P<0.001). Neurite outgrowth was reduced in the following order TGF-β3> TGF-β2> TGF-β1 (Fig. 9A) and each isoform was significantly different from the other (P<0.001). There was no significant difference between glucose treated and TGF-β1 or -β2 and between glucose and glucose + TGF-β1 or -β2 suggesting maximal inhibition of neurite outgrowth with TGF-β generated by high glucose alone. There was a slight reduction in neurite growth at 48 h in glucose + TGF-β3 treated cultures at 48 h (0.74 ± 0.06 mm) compared to glucose treated cultures (0.88 ± 0.05 mm, P=0.08). With TGF-β neutralizing antibody (Fig. 9B) there was a partial reversal of glucose-induced inhibition of neurite growth (P<0.05 compared to glucose treated cultures). There was no significant difference in neurite growth between control and cultures treated with TGF-β neutralizing antibody alone.
Fig. 9. TGF-β1, -β2, & -β3 reduce neurite outgrowth.

(A) DRG explants cultured with 10 ng/ml of purified TGF-β1, -β2, or -β3 for 48 hours had a reduced mean neurite length compared to control explants. Data expressed as mean ± SEM. (B) high glucose reduces neurite outgrowth but this effect is partially reversed by the use of the TGF-β neutralizing antibody. TGF-β neutralizing antibody alone does not affect neurite growth.
Discussion
Cytokines are generally defined as any polypeptide that affects the functions of other cells and are trophic regulatory proteins that can be broadly classified into four major groups: growth factors, interleukins, interferon’s, and tumor necrosis factors. Cytokines and growth factors have been strongly implicated in the generation of pathological pain states at both peripheral and central nervous system sites (Lewin and Mendell 1993; Watkins et al., 1994) and in the development and progression of diabetic neuropathy (Skundric and Lisak 2003), although the role of cytokine transforming factors such as TGF-β. is unknown. Furthermore, TGF-β mRNA in tissue can be used as a diagnostic or prognostic marker for human disease (for review, (Blobe et al., 2000).
The present study indicates that there is increased expression of TGF-β. at both gene and protein level in DRG and sciatic nerve in the diabetic peripheral nervous system and furthermore that TGF-β. in vitro is able to induce cellular injury both in embryonic DRG and neurites, a structure that mimics changes in the axon in vivo (Berent-Spillson et al., 2004; Russell et al., 2000). In the present study, Using QRT-PCR, TGF-β2 mRNA was primarily increased in DRG tissue of diabetic rats. In early diabetes, TGF-β1 mRNA was only minimally increased in DRG and there was essentially no immunoreactivity in DRG, however mRNA increased after 12 weeks of diabetes. TGF-β3 mRNA and immunoreactivity was essentially unchanged in DRG. In contrast, in sciatic nerve only TGF-β3 mRNA, but not TGF-β1 and -β2 mRNA were increased. Immunoblot analysis revealed the presence of various TGF-β dimers and their relative protein levels were differentially regulated in sciatic nerve of diabetic animals. In sciatic nerve from animals with diabetic neuropathy, there was an increase primarily in TGF-β3 dimers consistent with the mRNA data. However, there was also a lesser increase in TGF-β1 dimers, that may account for the subsequent increase in TGF-β3 mRNA. Consistent with our results, TGF-β has previously been shown to capable of dimerization (Border and Noble 1994; Venkataraman et al., 1995). Furthermore, the biologically active forms of the TGF-β family are disulfide-linked dimers (Border and Noble 1994; Venkataraman et al., 1995). Overall the mRNA and protein data are consistent with previous observations that there is consistent co-expression of TGF-β2 in neurons and -β3 in Schwann cells, and there are low levels of TGF-β1 in the unlesioned nervous system (Bottner et al., 2000). Previous published findings also show that that the TGF-β3 mRNA signal in neurons is weak, but neurons are strongly labeled following treatment with NGF or fibroblast growth factor-2 in the presence of Schwann cells (Bottner et al., 2000; Unsicker et al., 1996). This further indicates that different isoforms of TGF-β will express differently at different sites within the peripheral nervous system and may have alternative effects on signaling or cell biology in different disease states.
TGF-β has been clearly implicated in the pathogenesis of one microvascular complication, diabetic nephropathy, and has been shown to promote renal cell hypertrophy and stimulate formation of extra cellular matrix in the kidney (Ohashi et al., 2004); Gore-Hyer et al., 2002; Kim et al., 2001; Park et al., 1997; Sharma et al., 1997). It also has been shown that overeexpression of TGF-β in diabetic rat glomeruli promotes extra cellular matrix accumulation that is responsible for glomerular injury, and treatment with anti-TGF-β antibody ameliorates diabetic nephropathy (Hill et al., 2001; Ma et al., 2004). In previous tissue culture and animal studies, cellular matrix production was stimulated by high glucose levels, was associated with increased TGF-β expression, and the matrix stimulatory effects of high glucose were prevented by anti-TGF-β therapy (Gore-Hyer et al., 2002; Kim et al., 2001; Ohashi et al., 2004; Park et al., 1997; Sharma et al., 1997). Despite a strong association of TGF-β with renal disease no association with neuropathy has so far been established. In the present study, all the animals with both early diabetes (4 weeks) and more chronic diabetes (12 weeks) had evidence of a sensorimotor axonal neuropathy based on the nerve conduction data, sensory behavioral testing, or IENFD. Glucose and glycosylated hemoglobin data were similar in 4 and 12 week animals. The timing of increases in TGF-β mRNA and protein show an association with decreased nerve conduction velocities, paw withdrawal threshold, IENFD and fiber length in the STZ diabetic model. IENFD is a good marker of early neuropathy both in rodents and humans (Bianchi et al., 2004; Hirai et al., 2000; Leonelli et al., 2007; Polydefkis et al., 2004). Other models of nerve injury, for example sciatic cryoneurolysis and chronic constriction injury, show a similar increase in cytokine production including TGF-β in the spinal cord and dorsal and ventral roots (DeLeo et al., 1994; Willenbring et al., 1994). This supports our hypothesis that specific cytokines may be mediators of neuronal injury and neuropathic pain.
The mechanism/s of cytokine induction of neuropathic pain is unclear. However, it is known that cytokines have a pain facilitatory effect following nerve injury by activating central pathways via: (1) axonal or non-axonal transport to the dorsal root ganglion, spinal cord, and brain and/or; (2) induction of central cytokines by either peripheral cytokines or glial/neuronal activation (Rutkowski and DeLeo 2002). In nerve injury, cytokines may also indirectly activate mediators of pain transmission, such as glutamate and nitric oxide, or affect neurotrophin signaling in the peripheral nerve (Rutkowski and DeLeo 2002; Watkins et al., 1994). These are similar to pathways activated in diabetic neuropathy or neuropathic pain (Russell et al., 2006b; Russell et al., 2006a; Russell and Kaminsky 2005).
Direct application of various isoforms of exogenous TGF-β in vitro is in agreement with the in vivo data. TGF-β was able to increase cell toxicity in cultured embryonic neurons and this effect was increased with added glucose. Interestingly, consistent with the DRG neuronal mRNA and protein data in vivo, TGF-β1 and in particular TGF-β2 produced the greatest direct neuronal injury, whereas there was no increase in neuronal injury with TGF-β3. These data are in accordance with the proapoptotic role of endogenous TGF-β during ontogenetic neuron death in vivo, most recently demonstrated by Krieglstein et al. (Krieglstein et al., 2000). There is further evidence of TGF-β injury in non-neuronal tissue. For example, using transgenic mice generated with a fusion gene (Alb/TGF-beta 1), hepatic overexpression in TGF-β1 results in hepatic fibrosis and apoptotic death of hepatocytes (Sanderson et al., 1995). In the current study, prior treatment with TGF-β neutralizing antibody was able to reduce the level of TGF-β protein in embryonic DRG treated with high glucose conditions and measured by western immunoblotting. This is consistent with observation in non-neuronal tissue that (1) inhibition of TGF-β signaling by an ALK5 serine/threonine kinase receptor inhibitor (GW6604) protects from dimethylnitrosamine-induced liver fibrosis (de Gouville et al., 2005), and (2) recombinant human keratinocyte growth factor which lowers TGF-β1 levels, also reduces radiation-induced fibrosis (Chen et al., 2004).
Although some studies indicate that TGF-β can both prevent and induce cell injury under different conditions depending on the specific paradigm (Bottner et al., 2000; Krieglstein et al., 2000), many other studies show that TGF-β is able to induce apoptosis and that Akt and downstream signaling intermediates block TGF-β induction of cell death (Samatar et al., 2002; Schuster and Krieglstein 2002). In the current study, increased glucose and TGF-β1 or TGF-β2 in combination produced greater neuronal injury in embryonic DRG than TGF-β used alone. There are several possible pathways by which TGF-β can induce cell and axonal injury including induction of ROS (Jardine et al., 2002; Yao et al., 2007); by activation of programmed cell death pathways including activation of caspases, Smads, release of cytochrome c; and by down regulating antiapoptotic Bcl proteins for example Bcl2 and Bcl-XL, while up regulating proapoptotic proteins (Bax and Bad) (Barna et al., 2002; Derynck and Zhang 2003; Parkinson et al., 2001; Schuster and Krieglstein 2002; Ten Dijke et al., 2002).
TGF-β also reduces neurite growth from embryonic DRG neurons, where TGF-β neutralizing antibody partially reversed glucose-induced inhibition of neurite growth. Consistent with the finding that TGF-β3 is the primary isoform increased in diabetic sciatic nerve, TGF-β3 produced the greatest reduction in neurite growth. These findings are consistent with previous observations that TGF-β blocks the myelination of axons (Guenard et al., 1995) and mediates the death of mature SCs resulting in axonal injury (Parkinson et al., 2001).
Our results show that TGF-β is differentially upregulated in diabetic DRG and sciatic nerve in vivo. In vitro, using high glucose conditions, TGF-β neutralizing antibody prevents an increase in total TGF-β protein and specific TGF-β isoforms directly injure embryonic DRG neurons and neurites. These findings suggest a novel mechanism of injury in diabetes. However, further animal studies will be needed to determine if inhibition of TGF-β in vivo prevents diabetic neuropathy.
Acknowledgments
This work was supported in part by NIH NS42056, The Juvenile Diabetes Research Foundation Center for the Study of Complications in Diabetes (JDRF), Office of Research Development (Medical Research Service), Department of Veterans Affairs, American Diabetes Association (ADA) (JWR), and NIH 1T32 DC05341 (ABS). We also thank Thomas Morrow, PhD for technical advice with the mechanical Von-Frey sensory testing, and Aurora Anderson, MS for assistance with cell culture experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section topic: Cellular and Molecular Neuroscience, S. Gilman, Editor-in-Chief, Michigan, USA
Reference list
- Barna G, Sebestyen A, Chinopoulos CC, Nagy K, Mihalik R, Paku S, Kopper L. TGF beta 1 kills lymphoma cells using mitochondrial apoptotic pathway with the help of caspase-8. Anticancer Res. 2002;22:3867–3872. [PubMed] [Google Scholar]
- Berent-Spillson A, Robinson A, Golovoy D, Slusher B, Rojas C, Russell JW. Protection against glucose-induced neuronal death by NAAG and GCP II inhibition is regulated by mGluR3. J. Neurochem. 2004;89:90–99. doi: 10.1111/j.1471-4159.2003.02321.x. [DOI] [PubMed] [Google Scholar]
- Berent-Spillson A, Russell JW. Metabotropic glutamate receptor 3 protects neurons from glucose-induced oxidative injury by increasing intracellular glutathione concentration. J. Neurochem. 2007;101:342–354. doi: 10.1111/j.1471-4159.2006.04373.x. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Buyukakilli B, Brines M, Savino C, Cavaletti G, Oggioni N, Lauria G, Borgna M, Lombardi R, Cimen B, Comelekoglu U, Kanik A, Tataroglu C, Cerami A, Ghezzi P. Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proc. Natl. Acad. Sci. U. S. A. %20. 2004;101:823–828. doi: 10.1073/pnas.0307823100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N. Engl. J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- Bottner M, Krieglstein K, Unsicker K. The transforming growth factor-betas: structure, signaling, and roles in nervous system development and functions. J. Neurochem. 2000;75:2227–2240. doi: 10.1046/j.1471-4159.2000.0752227.x. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA. Metabolic and vascular factors in the pathogenesis of diabetic neuropathy. Diabetes. 1997;46 Suppl 2:S31–S37. doi: 10.2337/diab.46.2.s31. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA, Maxfield EK. Anti-oxidant treatment prevents the development of peripheral nerve dysfunction in streptozotocin-diabetic rats. Diabetologia. 1993;36:299–304. doi: 10.1007/BF00400231. [DOI] [PubMed] [Google Scholar]
- Chalazonitis A, Kalberg J, Twardzik DR, Morrison RS, Kessler JA. Transforming growth factor beta has neurotrophic actions on sensory neurons in vitro and is synergistic with nerve growth factor. Dev. Biol. 1992;152:121–132. doi: 10.1016/0012-1606(92)90162-a. [DOI] [PubMed] [Google Scholar]
- Chen L, Brizel DM, Rabbani ZN, Samulski TV, Farrell CL, Larrier N, Anscher MS, Vujaskovic Z. The protective effect of recombinant human keratinocyte growth factor on radiation-induced pulmonary toxicity in rats. Int. J Radiat. Oncol. Biol. Phys. 2004;60:1520–1529. doi: 10.1016/j.ijrobp.2004.07.729. [DOI] [PubMed] [Google Scholar]
- Chen S, Jim B, Ziyadeh FN. Diabetic nephropathy and transforming growth factor-beta: transforming our view of glomerulosclerosis and fibrosis build-up. Semin. Nephrol. 2003;23:532–543. doi: 10.1053/s0270-9295(03)00132-3. [DOI] [PubMed] [Google Scholar]
- Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes. 2003;52:2363–2371. doi: 10.2337/diabetes.52.9.2363. [DOI] [PubMed] [Google Scholar]
- Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- de Gouville AC, Boullay V, Krysa G, Pilot J, Brusq JM, Loriolle F, Gauthier JM, Papworth SA, Laroze A, Gellibert F, Huet S. Inhibition of TGF-beta signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br. J Pharmacol. 2005;145:166–177. doi: 10.1038/sj.bjp.0706172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney CL, Russell JW, Cheng H-L, Feldman EL. Insulin-like growth factor-I and over-expression of Bcl-xL prevent glucose-mediated apoptosis in Schwann cells. J. Neuropathol. Exp. Neurol. 2001;60:147–160. doi: 10.1093/jnen/60.2.147. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Coombs DW, Willenbring S, Colburn RW, Fromm C, Wagner R, Twitchell BB. Characterization of a neuropathic pain model: sciatic cryoneurolysis in the rat. Pain. 1994;56:9–16. doi: 10.1016/0304-3959(94)90145-7. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Kratz KM, Karnes JL, Litchy WJ, Klein R, Pach JM, Wilson DM, O'Brien PC, Melton LJ., III The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: The Rochester Diabetic Neuropathy Study. Neurology. 1993;43:817–824. doi: 10.1212/wnl.43.4.817. [DOI] [PubMed] [Google Scholar]
- Gore-Hyer E, Shegogue D, Markiewicz M, Lo S, Hazen-Martin D, Greene EL, Grotendorst G, Trojanowska M. TGF-beta and CTGF have overlapping and distinct fibrogenic effects on human renal cells. Am. J. Physiol Renal Physiol. 2002;283:F707–F716. doi: 10.1152/ajprenal.00007.2002. [DOI] [PubMed] [Google Scholar]
- Guenard V, Gwynn LA, Wood PM. Transforming growth factor-beta blocks myelination but not ensheathment of axons by Schwann cells in vitro. J. Neurosci. 1995;15:419–428. doi: 10.1523/JNEUROSCI.15-01-00419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill C, Flyvbjerg A, Rasch R, Bak M, Logan A. Transforming growth factor-beta2 antibody attenuates fibrosis in the experimental diabetic rat kidney. J Endocrinol. 2001;170:647–651. doi: 10.1677/joe.0.1700647. [DOI] [PubMed] [Google Scholar]
- Hirai A, Yasuda H, Joko M, Maeda T, Kikkawa R. Evaluation of diabetic neuropathy through the quantitation of cutaneous nerves. J Neurol. Sci. 2000;172:55–62. doi: 10.1016/s0022-510x(99)00290-7. [DOI] [PubMed] [Google Scholar]
- Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and - resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol. Chem. 2004;279:29341–29350. doi: 10.1074/jbc.M404167200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Chance B, Wang X, Kime R, Nioka S, Chance EM. Modeling of oxygen diffusion and metabolism from capillary to muscle. Adv. Exp. Med. Biol. 2003a;540:325–330. doi: 10.1007/978-1-4757-6125-2_46. 325–330. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Price SA, Chilton L, Calcutt NA, Tomlinson DR, Verkhratsky A, Fernyhough P. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes. 2003b;52:2129–2136. doi: 10.2337/diabetes.52.8.2129. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Verkhratsky A, Fernyhough P. Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Mol. Cell Neurosci. 2005;28:42–54. doi: 10.1016/j.mcn.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Jardine H, MacNee W, Donaldson K, Rahman I. Molecular mechanism of transforming growth factor (TGF)-beta1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP-1/ARE and Fra-1. J. Biol. Chem. 2002;277:21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim BC, Song CY, Hong HK, Moon KC, Lee HS. Advanced glycosylation end products stimulate collagen mRNA synthesis in mesangial cells mediated by protein kinase C and transforming growth factor-beta. J. Lab Clin. Med. 2001;138:59–68. doi: 10.1067/mlc.2001.115494. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Richter S, Farkas L, Schuster N, Dunker N, Oppenheim RW, Unsicker K. Reduction of endogenous transforming growth factors beta prevents ontogenetic neuron death. Nat. Neurosci. 2000;3:1085–1090. doi: 10.1038/80598. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Suter-Crazzolara C, Fischer WH, Unsicker K. TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 1995;14:736–742. doi: 10.1002/j.1460-2075.1995.tb07052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria G, Cornblath DR, Johansson O, McArthur JC, Mellgren SI, Nolano M, Rosenberg N, Sommer C. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur. J Neurol. 2005;12:747–758. doi: 10.1111/j.1468-1331.2005.01260.x. [DOI] [PubMed] [Google Scholar]
- Leonelli E, Bianchi R, Cavaletti G, Caruso D, Crippa D, Garcia-Segura LM, Lauria G, Magnaghi V, Roglio I, Melcangi RC. Progesterone and its derivatives are neuroprotective agents in experimental diabetic neuropathy: a multimodal analysis. Neuroscience. 2007;144:1293–1304. doi: 10.1016/j.neuroscience.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Nerve growth factor and nociception. Trends Neurosci. 1993;16:353–359. doi: 10.1016/0166-2236(93)90092-z. [DOI] [PubMed] [Google Scholar]
- Lin HY, Moustakas A. TGF-β receptors: Structure and function. Cell. Mol. Biol. 1994;40:337–349. [PubMed] [Google Scholar]
- Ma LJ, Jha S, Ling H, Pozzi A, Ledbetter S, Fogo AB. Divergent effects of low versus high dose anti-TGF-beta antibody in puromycin aminonucleoside nephropathy in rats. Kidney Int. 2004;65:106–115. doi: 10.1111/j.1523-1755.2004.00381.x. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Le Van TA, Valette A, Weber MJ. Transforming growth factor beta 1 is a potent survival factor for rat embryo motoneurons in culture. Brain Res. Dev. Brain Res. 1990;52:175–181. doi: 10.1016/0165-3806(90)90233-o. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. 753–791. [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Montal M. Mitochondria, glutamate neurotoxicity and the death cascade. Biochim Biophys Acta. 1998;1366:113–126. doi: 10.1016/s0005-2728(98)00124-8. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JH, Ha H, Yu MR, Lee HB. Sequential effects of high glucose on mesangial cell transforming growth factor-beta 1 and fibronectin synthesis. Kidney Int. 1998;54:1872–1878. doi: 10.1046/j.1523-1755.1998.00193.x. [DOI] [PubMed] [Google Scholar]
- Ohashi S, Abe H, Takahashi T, Yamamoto Y, Takeuchi M, Arai H, Nagata K, Kita T, Okamoto H, Yamamoto H, Doi T. Advanced glycation end products increase collagen-specific chaperone protein in mouse diabetic nephropathy. J. Biol. Chem. 2004;279:19816–19823. doi: 10.1074/jbc.M310428200. [DOI] [PubMed] [Google Scholar]
- Pantsulaia T. Role of TGF-beta in pathogenesis of diabetic nephropathy. Georgian. Med. News. 2006:13–18. [PubMed] [Google Scholar]
- Park IS, Kiyomoto H, Abboud SL, Abboud HE. Expression of transforming growth factor-beta and type IV collagen in early streptozotocin-induced diabetes. Diabetes. 1997;46:473–480. doi: 10.2337/diab.46.3.473. [DOI] [PubMed] [Google Scholar]
- Parkinson DB, Dong Z, Bunting H, Whitfield J, Meier C, Marie H, Mirsky R, Jessen KR. Transforming growth factor beta (TGFbeta) mediates Schwann cell death in vitro and in vivo: examination of c-Jun activation, interactions with survival signals, and the relationship of TGFbeta-mediated death to Schwann cell differentiation. J. Neurosci. 2001;21:8572–8585. doi: 10.1523/JNEUROSCI.21-21-08572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydefkis M, Hauer P, Sheth S, Sirdofsky M, Griffin JW, McArthur JC. The time course of epidermal nerve fibre regeneration: studies in normal controls and in people with diabetes, with and without neuropathy. Brain. 2004;127:1606–1615. doi: 10.1093/brain/awh175. [DOI] [PubMed] [Google Scholar]
- Pop-Busui R, Sima A, Stevens M. Diabetic neuropathy and oxidative stress. Diabetes Metab Res. Rev. 2006;22:257–273. doi: 10.1002/dmrr.625. [DOI] [PubMed] [Google Scholar]
- Poulsen KT, Armanini MP, Klein RD, Hynes MA, Phillips HS, Rosenthal A. TGF beta 2 and TGF beta 3 are potent survival factors for midbrain dopaminergic neurons. Neuron. 1994;13:1245–1252. doi: 10.1016/0896-6273(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Russell JW, Cheng H-L, Golovoy D. Insulin-like growth factor-I promotes myelination of peripheral sensory axons. J. Neuropathol. Exp. Neurol. 2000;59:575–584. doi: 10.1093/jnen/59.7.575. [DOI] [PubMed] [Google Scholar]
- Russell JW, Cowell RM, Feldman EL. Neuronal and Schwann Cell Death in Diabetic Neuropathy. In: Veves A, Malik R, editors. The Clinical Management of Diabetic Neuropathy. Totowa: Humana Press Inc.; 2006a. [Google Scholar]
- Russell JW, Gill JS, Sorenson EJ, Schultz DA, Windebank AJ. Suramin-induced neuropathy in an animal model. J. Neurol. Sci. 2001;192:71–80. doi: 10.1016/s0022-510x(01)00633-5. [DOI] [PubMed] [Google Scholar]
- Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB. 2002;16:1738–1748. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- Russell JW, Kaminsky AJ. Oxidative injury in diabetic neuropathy. In: Opara E, editor. Nutrition and Diabetes: Pathophysiology and Management. Boca Raton: Taylor & Francis; 2005. pp. 381–397. [Google Scholar]
- Russell JW, Smith AG, Singleton JR. Impaired glucose regulation and neuropathy. In: Gilman S, editor. Neurobiology of Diseases. San Diego: Elsevier; 2006b. pp. 849–869. [Google Scholar]
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol. Dis. 1999;6:347–363. doi: 10.1006/nbdi.1999.0254. [DOI] [PubMed] [Google Scholar]
- Russell JW, Windebank AJ, Podratz JL. Role of nerve growth factor in suramin neurotoxicity studied in vitro. Ann. Neurol. 1994;36:221–228. doi: 10.1002/ana.410360215. [DOI] [PubMed] [Google Scholar]
- Rutkowski MD, DeLeo JA. The Role of Cytokines in the Initiation and Maintenance of Chronic Pain. Drug News Perspect. 2002;15:626–632. [PubMed] [Google Scholar]
- Samatar AA, Wang L, Mirza A, Koseoglu S, Liu S, Kumar CC. Transforming growth factor-beta 2 is a transcriptional target for Akt/protein kinase B via forkhead transcription factor. J. Biol. Chem. 2002;277:28118–28126. doi: 10.1074/jbc.M203686200. [DOI] [PubMed] [Google Scholar]
- Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. U. S. A. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers NM, Beswick LJ, Middlemas A, Calcutt NA, Mizisin AP, Tomlinson DR, Fernyhough P. Neurotrophin-3 prevents the proximal accumulation of neurofilament proteins in sensory neurons of streptozocin-induced diabetic rats. Diabetes. 2003;52:2372–2380. doi: 10.2337/diabetes.52.9.2372. [DOI] [PubMed] [Google Scholar]
- Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002;307:1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- Sharma K, Ziyadeh FN, Alzahabi B, McGowan TA, Kapoor S, Kurnik BR, Kurnik PB, Weisberg LS. Increased renal production of transforming growth factor-beta1 in patients with type II diabetes. Diabetes. 1997;46:854–859. doi: 10.2337/diab.46.5.854. [DOI] [PubMed] [Google Scholar]
- Skundric DS, Lisak RP. Role of neuropoietic cytokines in development and progression of diabetic polyneuropathy: from glucose metabolism to neurodegeneration. Exp. Diabesity. Res. 2003;4:303–312. doi: 10.1155/EDR.2003.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Stevens MJ, Wiley JW. Diabetic peripheral neuropathy: Evidence for apoptosis and associated mitochondrial dysfunction. Diabetes. 2000;49:1932–1938. doi: 10.2337/diabetes.49.11.1932. [DOI] [PubMed] [Google Scholar]
- Ten Dijke P, Goumans MJ, Itoh F, Itoh S. Regulation of cell proliferation by Smad proteins. J. Cell Physiol. 2002;191:1–16. doi: 10.1002/jcp.10066. [DOI] [PubMed] [Google Scholar]
- Tomlinson DR, Fernyhough P, Diemel LT. Neurotrophins and peripheral neuropathy. Philos. Trans. R. Soc. Lond B Biol. Sci. 1996;351:455–462. doi: 10.1098/rstb.1996.0042. [DOI] [PubMed] [Google Scholar]
- Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nat. Rev. Neurosci. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- Unsicker K, Meier C, Krieglstein K, Sartor BM, Flanders KC. Expression, localization, and function of transforming growth factor-beta s in embryonic chick spinal cord, hindbrain, and dorsal root ganglia. J. Neurobiol. 1996;29:262–276. doi: 10.1002/(SICI)1097-4695(199602)29:2<262::AID-NEU10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034 (epub) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman G, Sasisekharan V, Cooney CL, Langer R, Sasisekharan R. Complex flexibility of the transforming growth factor beta superfamily. Proc. Natl. Acad. Sci. U. S. A. 1995;92:5406–5410. doi: 10.1073/pnas.92.12.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Brownlee M, Russell JW. Oxidative stress and programmed cell death in diabetic neuropathy. Ann. N. Y. Acad. Sci. 2002;959:368–383. doi: 10.1111/j.1749-6632.2002.tb02108.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. Characterization of cytokine-induced hyperalgesia. Brain Res. 1994;654:15–26. doi: 10.1016/0006-8993(94)91566-0. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Harrison J, Boulet J, Mark L, Pearson M, Gottshall S, Walker K. Pharmacological characterisation of a rat model of incisional pain. Br. J Pharmacol. 2004;141:85–91. doi: 10.1038/sj.bjp.0705568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenbring S, DeLeo JA, Coombs DW. Differential behavioral outcomes in the sciatic cryoneurolysis model of neuropathic pain in rats. Pain. 1994;58:135–140. doi: 10.1016/0304-3959(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Yao K, Tan J, Gu WZ, Ye PP, Wang KJ. Reactive oxygen species mediates the apoptosis induced by transforming growth factor beta(2) in human lens epithelial cells. Biochem. Biophys. Res. Commun. 2007;354:278–283. doi: 10.1016/j.bbrc.2006.12.198. [DOI] [PubMed] [Google Scholar]