

Abstract
Obtaining sufficient amount of purified G-protein coupled receptors (GPCRs) is almost always one of the major challenges for their structural studies. CB2271-326, a human cannabinoid receptor 2 (CB2) fragment comprising part of the third extracellular loop (EL3), the seventh transmembrane domain (TM7) and C-terminal juxtamembrane region of the receptor, was over-expressed as a fusion protein into inclusion body (IB) of Escherichia coli. The fusion protein was purified by histidine-selected nickel affinity chromatography under denaturing conditions. Then, the fusion protein IBs were solubilized in detergent (Brij58) and the expression fusion leader sequence (TrpLE) was specifically cleaved with tobacco etch virus (TEV) protease. The target fragment, CB2271-326, was subsequently purified by reverse-phase HPLC and confirmed by SDS-PAGE and mass spectrometry. This hydrophobic fragment can refold in mild detergents digitonin and Brij58. Circular dichroism (CD) spectroscopy of CB2271-326 in digitonin and Brij58 micelles showed that the fragment adopts a more than 75% α-helical structure, with the remainder having β- strand structure. Fluorescence spectroscopy and quenching studies suggested that the C-terminal region lies near the surface of the digitonin micelles and the TM7 region is folded relatively close to the center of the micelles. This study may provide an alternative strategy for the production and structure/functional studies of GPCRs such as CB2 receptor protein produced in the form of IBs.
Keywords: Cannabinoid receptor 2, transmembrane domain, inclusion body, affinity chromatography, HPLC, circular dichroism, fluorescence
Since the discovery of the CB2 receptor in 1993 [1], there has been a growing interest to clarify the importance of this GPCR membrane protein in human physiology, and to investigate it as a possible target for current and future drug development. The CB2 receptor seems to have a significant role in mediating the immunological functions of cannabinoid receptor ligands. Thus, the CB2 receptor is a promising target for drug development that aims at management of neurological pain, inflammation, osteoporosis, and growth of malignant gliomas and tumors of immune origin, as well as immunological disorders such as multiple sclerosis [2]. However, structure-function studies and structure-based ligand design have been impeded by the absence of an experimental three-dimensional (3D) structure of the CB2 receptor. Seven transmembrane α-helical domains and the related high hydrophobicity are the major contributing factors that hinder the 3D structural study of CB2 and other GPCRs. Because of its very limited quantities obtainable from natural sources, attempts have been made to heterologously over-express CB2 [3-7]. In general, the Escherichia coli expression system is the most attractive strategy due to its advantages of simplicity of use, low cost, easy scale-up, and homogeneous protein production. Though few attempts were promising to express full length CB2 receptor in E. coli [3-5], it is still problematic to obtain the entire CB2 tertiary structure due to the difficulties to get enough yield of CB2 protein, to purify it, to entice it to fold properly, and to analyze it by NMR. Based on the above fact, we hypothesized that an alternative strategy, production of large CB2 fragments including up to two TM segments connected by a loop through over-expression in bacteria as fusion proteins, would be tractable. Structural and even functional analysis of bacterially synthesized protein fragments has become a powerful tool in the study of GPCRs and other membrane proteins [8-11].
Our lab has previously over-expressed, purified and structurally characterized CB2 fragments, including CB2180-233, CB265-101, and CB227-101, which correspond to TM1-IL1-TM2 (from the first transmembrane domain to the second intracellular loop and the second transmembrane domain), TM2 and TM5-IL3 [12-15] respectively. In these publications, the CB2 fragments were expressed as fusion proteins with a modified TrpΔLE1413 (TrpLE) leader sequence and a 9-histidine tag at the N-terminus. The TrpLE leader sequence facilitates the fusion protein to be highly expressed as IBs, which have the advantages of lower toxicity and higher yield than the strategy of insertion of the protein into the bacterial membrane. The formation of IBs also has an advantage of protecting the recombinant protein from degradation by cytosolic bacterial proteases. Also, the insolubility of IB proteins allows for removal of soluble and membrane proteins by simple centrifugation. The resulting fusion proteins were further purified by immobilized metal-ion affinity chromatography (IMAC) under denaturing conditions (6 M guanidine hydrochloride or 8 M urea). The leader sequence and His-tag were cleaved by either cyanogen bromide (CNBr) under extreme acidic conditions or proteases (e.g. Factor Xa) under mild detergent conditions (e.g. Triton X-100). Finally, the CB2 fragments were purified by RP-HPLC. In the present manuscript, we reported that another CB2 fragment (CB2271-326), including part of the extracellular loop 3, TM7, helix 8 and part of the C-terminus based on the predicted 3D structural model of CB2 [16], can be produced according to the established methods generally described above. Some important modifications, as will be detailed below, were made to optimize the protein expression level, IB solubilization, fusion protein cleavage, product purification and refolding. The obtained pure CB2 fragment was characterized by circular dichroism and fluorescence spectroscopy. It has been known from the reported mutation and function studies that Ser285, Ser292 and Tyr299 in TM7 are important for CB2 ligand binding [17, 18]. The residue Tyr299 in TM7, as well as the conserved cysteins Cys313 and Cys320 in the C-terminal juxamembrane region were shown to be critical for coupling to adenylate cyclase [17]. The solution NMR structure of CB2271-326 fragment and the possible specific interaction of the fragment with G-protein will be examined in our future work. The current study provides useful information for structure/function studies of the fragment as well as the whole CB2 receptor.
Materials and methods
Plasmid construction
A human CB2 cDNA vector pRG/II-hs-MBP-CB2-HF (gift from Dr. Reinhard Grisshammer) is used as a template to construct various CB2 fragment DNA sequences into plasmid pMMHb (gift from Dr. Peter Kim). pMMHb is a high copy plasmid and is used to express CB2 fragments as IBs in large quantities. The CB2271-326 is expressed as a fusion protein consisting of a N-terminal TrpΔLE1413 polypeptide (TrpLE, 13,669.7 Da), followed by a nine-histidine tag, a Tobacco Etch Virus (TEV) protease recognition site, and the CB2 fragment (TrpLE-9His-TEV-CB2271-326). The TEV protease was chosen in this study because of its high cleavage specificity and its activity over a broad range of temperature, pH, and different detergents [19, 20]. We have frequently observed nonspecific cleavage of the fusion membrane proteins with Factor Xa and enterokinase in the presence of various detergents. To solve this issue, a TEV protease recognition site was integrated between TrpLE encode sequence and CB2 fragment encode sequence on the expression plasmid pMMHb for improving cleavage and specificity efficiency. Recombinant DNA techniques were adopted for the expression plasmid construction, which was similarly detailed elsewhere [12].
Protein expression in E. coli
A parallel comparison study was performed to evaluate the expression level of the CB2 fusion protein in E. coli strain BL21(DE3)pLysS (purchased from Novagen) and a BL21(DE3) mutant strain C43(DE3) (purchased from Avidis Inc., France ). The protein expression level from C43(DE3) was nearly twice higher than BL21(DE3)pLysS. Therefore, C43(DE3) was employed as the host cell for the over-expression of the CB2 fragment in this study. Briefly, a single colony of a freshly transformed C43(DE3) was picked up from a LB-agar plate and a 10 ml of LB medium was inoculated. Both the plate and LB medium contained 100 μg/ml ampicillin. The solution culture was conducted at room temperature, 250 rpm for overnight. 1 L of LB medium was inoculated with the cell pellet from the 10 ml overnight culture and incubated at 37°C, 250 rpm. When the OD600 reached 0.8~1.0, the protein expression was induced with 1 mM isopropyl-β-D-thiogalactoside (IPTG) and incubated for another 4 hours at 37°C or overnight at room temperature. The cells were harvested by centrifugation (7000 g, 4°C). The cell pellets were washed in 50 mM Tris-HCl (pH 8.0) and centrifuged again. The typical yields were approximately 5 grams wet cells per liter culture. Harvested cells were then stored at -80°C for later use.
Purification, solubilization and cleavage of TrpLE-9His-TEV-CB2271-326
TrpLE-9His-TEV-CB2271-326 fusion protein was purified by His-selected nickel affinity chromatography. For a typical preparation, 20 grams of frozen C43(DE3) wet cell pellet was thawed at room temperature for 15 min and resuspended in 50 mM Tris buffer (10 ml/per gram cells), pH 8.0. PMSF was added to 1 mM and lysozyme was added to 0.2 mg/ml. After being incubated at room temperature for 30 min, the cell lysis suspension was ice-cooled for 10 min and was sonicated briefly on ice. 5 mM of MgCl2 and benzonase were added to break bacterial DNA and RNA. IBs were collected by centrifugation at 20,000 g for 30 min at 4°C. The pellet was dissolved in sample buffer (6 M GnHCl, 50 mM Tris, 0.5 M NaCl, 5 mM imidazole, pH 8.0) followed by dilution with 10 times its volumes using cold deionized water. Again, the IBs were collected by centrifugation. The IB pellet was then dissolved in the sample buffer facilitated with mild sonication on ice and incubated 4 hours to overnight to fully dissolve the pellet. The solubilized suspension was centrifuged at 20,000 g for 30 min. The supernatant was loaded on a 20 ml of nickel charged His-bind resin (Novagen) column equilibrated with binding buffer (8 M urea, 0.1 M sodium phosphate, 0.01 M Tris, pH 8.0) containing 5 mM imidazole. The column was then washed in 10 column volumes of the same buffer followed by 5 column volumes of binding buffer containing 30 mM imidazole. The target fusion protein was eluted with 3 column volumes of binding buffer containing 500 mM imidazole. The resulting fusion protein eluate was dialyzed exhaustively against deionized water and then lyophilized.
In order to remove the fusion partners, a two-step solubilization strategy was employed and proved to be very effective in this study. Briefly, the lyophilized IB was suspended in 2 × TEV cleavage buffer (100 mM Tris-Cl, pH 8.0) containing Brij58 (Sigma-Aldrich). The complete solubility of the IB was achieved by adjusting pH value to ~2.5 with 0.5 M HCl and mild sonication (micro-tip at 20% power output) on a Virsonic Cell Disruptor (Model 16-850, The Virtis Co., New York) , then quickly the pH was adjusted back to pH 6-6.5. EDTA, DTT, TEV protease and sterile deionized water were added at this moment to reach the final 0.5 mM EDTA, 1 mM DTT and IB/TEV protease ratio of 1 (mg/mg). After brief centrifugation, the supernatant was incubated at room temperature for 20 hours with shaking.
RP-HPLC purification of CB2271-326
After 20 hours of TEV protease cleavage, the reaction was stopped by adding formic acid to 0.1% and the suspension was centrifuged at 15,000 g for 15 min at 4°C. The supernatant was filtered (0.22 μm) and the resulting solution containing the target fragment was separated by a reverse phase C4 column (Vydac 214TP510, 5 μm, 10 mm × 250 mm) on an AKTAbasic chromatography system (GE Healthcare Corp.). The column was eluted with a 3-step gradient of solvent B (isopropanol/acetonitrile/TFA [60:40:0.1]) and the separation of the target fragment from TrpLE and other components in a gradient of solvent B 40%-50% over 100 min at a flow rate of 4 ml/min. The elution was monitored by UV absorbance at 220 nm and 280 nm. The peak fractions were collected and analyzed by MALDI-TOF-MS (Voyage) and SDS-PAGE. The fractions containing CB2271-326 from multiple runs were pooled and dried by rotary evaporation.
SDS-PAGE
SDS-PAGE were carried out on 4-12% or 12% NuPAGE Novex Bis-Tris gels (Invitrogen) using NuPAGE MES SDS buffer system (50 mM MES, 50 mM Tris base, 0.1% SDS, 1 mM EDTA, pH 7.3). The samples were prepared with 4 X NuPAGE LDS Sample Buffer (106 mM Tris-HCl, 141 mM Tris base, 2% LDS, 10% glycerol, 0.51 mM EDTA, 0.22 mM SERVA Blue G250, 0.175 mM Phenol Red, pH 8.5), 10 X Reducing Agent (500 mM DTT) and incubated at 70°C for 10 min. A brief centrifugation was conducted before the samples were loaded on the gels. Protein bands were visualized by staining with Coomassie brilliant blue R-250.
Circular Dichroism
The CD spectra were the average of eight scans at room temperature recorded by a JASCO J-810 Spectropolarimeter using 1-mm path length cylindrical quartz cuvette. Spectra were recorded from 185 nm to 260 nm in a bandwidth of 0.5 nm, a data pitch of 0.2 nm and a 1-sec average time per point, with a scan speed of 100 nm/min. Background spectra of samples containing detergent without protein were acquired in an identical manner and subtracted from the spectra of protein+detergent. The resulting spectra were used for the analysis of CD spectra.
The solution samples of CB2271-326 in detergents were prepared using a modified method of [9]: An aliquot of detergent stock solution in propanol/methanol (50/50, v/v) and an aliquot of CB2271-326 stock solution in propanol (containing 18.2% formic acid) were mixed in a test tube and the solvent was evaporated by Speed-Vacuum. The dried peptide-detergent pellet was dissolved in an appropriate volume buffer (10 mM Tris-HCl, pH 7.0, 50 mM NaCl) to give a final protein concentration of ~15 μM and final detergent concentrations of 20 mM Brij58, 4 mM digitonin, or 20 mM SDS. Solutions were passed through a 0.22-μm syringe filter before being used for spectroscopy.
The CB2271-326 stock solution in propanol was prepared as following. The CB2271-326 peptide dried sample was dissolved in propanol with 18.2% formic acid, mild sonication is needed. After brief centrifugation (13,000 rpm, 5 min), the supernatant was carefully transferred to a new tube and the protein concentration was determined by UV spectrometry using extinction coefficient ε280=8295 M-1cm-1 calculated by the below formula [21]for peptide in solvent propanol:
This calculated concentration was used to convert the CD spectra from CD mdeg to mean residue ellipticity [θ]. The resulting spectra were analyzed for the proportions of various secondary structures using the computer program ContinLL supported by an interactive web site DICHROWEB [22, 23].
Fluorescence spectroscopy
Fluorescence spectra were recorded in a Cary Eclipse Fluorescence Spectrophotometer (Varian) controlled by a PC computer equipped with Cary Eclipse software. Tryptophan and tyrosine quenching were measured at 23 °C. The excitation wavelength was 295 nm for tryptophan fluorescence spectroscopy and 275 nm for the combination of tryptophan and tyrosine. In peptide folding and denaturing experiment, the fluorescence emission scans were recorded at 305-450 nm for tryptophan and 285-450 nm for tryptophan + tyrosine at 600 nm/min. The excitation slit and emission slit were at 5 nm. The sample preparation was similar as described above for CD spectroscopy, except that the peptide-detergent solutions contained 4 mM detergents and the buffer is 10 mM NaPO4, pH 7.0, 50 mM NaCl for excitation at 295 nm or 10 mM Tris-HCl, pH 7.0, 50 mM NaCl for excitation at 275 nm.
In fluorescence quenching titrations, the NaI concentrations ranged from 0 to 200 mM, and NaCl concentration ranged from 200 to 0 mM which design to diminish the effect of ionic strength. 0.1 mM sodium thiosulfate was added in all samples to avoid the formation of triiodide (I− 3) which may give artifacts. All fluorescence spectroscopic parameters were same as above except the emission scans was 305-400 nm for tryptophan fluorescence and 285-400nm for tryptophan+tyrosine. All quenching experiments were carried out in triplicate.
Results and Discussion
Production of the TrpLE-9His-TEV-CB2271-326 fusion protein
The recombinant expression of hydrophobic peptides or transmembrane proteins in IBs as a fusion protein coupled with a leading partner (such as GST, KSI or TrpLE) has been shown to be an effective strategy to achieve high yield [9, 12, 24-27]. We previously showed high level expression of fusion proteins when TrpLE was fused to the N terminal of other CB2 fragments [12, 14, 15]. In the current study, the human CB2271-326 fragment was fused with TrpLE and the fusion protein was exclusively achieved in the form of IBs in E. coli cells. The strategy was modified and optimized for higher yield, specific cleavage and effective peptide purification. Typically, ~10 mg of fusion protein TrpLE-9His-TEV-CB2271-326 can be recovered from one liter of LB medium culture in C43(DE3) strain by IMAC, exceeding the yield expressed in BL21(DE3) strain by twofold. The expression level difference observed in the current study suggests that, even though the fusion protein was expressed in its insoluble form, it may still toxic to the host E. coli cells, especially at high yield. The C43(DE3) strain is more tolerable to the toxicity of the IB than its parental strain BL21(DE3). This phenomenon is similar to the expression of bovine oxoglutarate-malate transport protein and bovine phosphate carrier reported by Miroex and Walker [28].
As shown by SDS-PAGE (Figure 1) and analysis, there was no detectable expression of the CB2271-326 fusion protein without IPTG induction (lane 1). The fusion protein was expressed only after the IPTG induction (lane 2). The IB isolated from cell lysate by IMAC was mainly the fusion protein. Bradford assay (Bio-Rad Protein Assay Kit II, catalog number 500-0002) showed the IMAC purified fusion protein IB pellet contained roughly 50% (w/w) protein. We assumed that the other 50% of the materials were possibly DNA and/or lipids which hydrophobically interacted with the fusion protein and eluted together from the nickel column.
Figure 1.
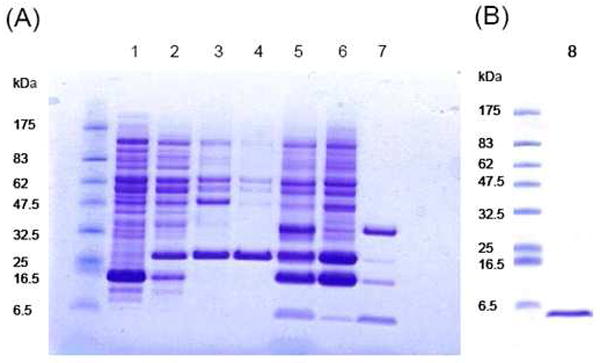
SDS-PAGE gels. (A) 1, cell lysate (not induced); 2, cell lysate (induced); 3, inclusion body; 4, fusion protein purified from nickel column; 5, suspension of TEV protease cleavage; 6, precipitate of cleavage; 7, supernatant of cleavage. Bands from top to bottom are TEV protease, uncleaved fusion protein, TrpLE tag and CB2271-326. (B) 8, purified CB2271-326 from RP-HPLC (right) and protein marker: P7708 (New England Biolabs) (left).
After cleavage, the target fragment was purified by RP-HPLC on an AKTAbasic Sytem (GE HealthCare Corp.). The cleaved suspension was centrifuged at 15000 g for 30 min and the supernatant was filtered through 0.22 μm filters. The resulting solution was injected onto HPLC column (VyDAC 214TP series C4 column with particle size 5 μm). The analyses of the eluted peaks by MALDI-TOF-MS revealed Peak 1 to be mainly DTT and Brij58, which were eluted at the wash step. Peaks 2, 3, and 4 were TEV protease, fusion tag TrpLE and CB2271-326; these three components were separated at a shallow gradient and eluted at 40%, 42% and 42.8% of solvent B, respectively (Figure 2). The majority of the fusion tag TrpLE and non-cleaved fusion protein were found in the precipitate of the centrifugation (data not shown). Figure 3 shows the Peak 4 had a mass of 6237.44 Da, which is congruent with the calculated mass of CB2271-326, 6237.36 Da, differing by only 13 ppm. This fragment contains an extra Gly residue at its N-terminal, which is from the TEV recognition site (Glu-Asn-Leu-Tyr-Phe-Gln/Gly). The mass spectrum also showed the formation of small amounts of oxidized as well as dimerized CB2271-326 (6449.28 Da and 12472.86 Da, correspondingly). The CB2271-326 fragment may easily form dimer or polymer due to its primary sequence containing four cysteine residues. The SDS-PAGE and mass spectrometry results did not show any obvious impurities. The yield of the HPLC-purified peptide fragment was determined by the Bradford method (Bio-Rad) showing 1.5~2 mg per liter of E. coli culture (Table 1).
Figure 2.
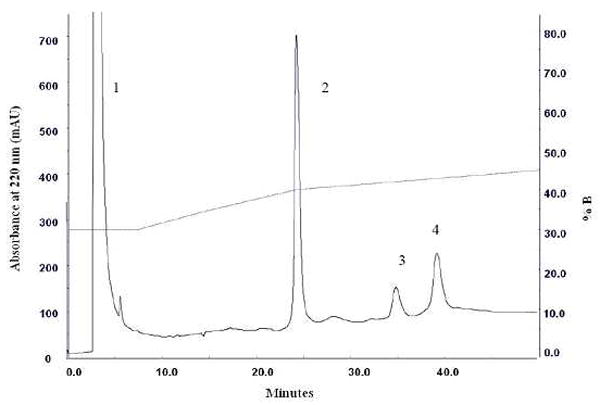
C4 RP-HPLC (VyDAC 214TP) purification of CB2271-326 from the supernatant of TEV protease cleaved TrpLE-9His-TEV- CB2271-326 fusion protein. Peaks: 1, DTT and Brij58; 2, TEV protease; 3, TrpLE tag; 4, CB2271-326.
Figure 3.
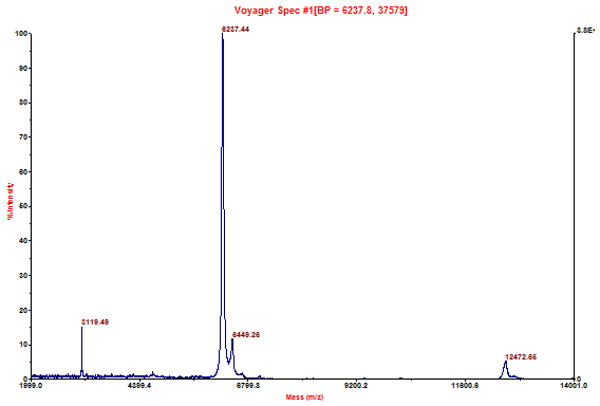
MALDI-TOF-MS (Voyager, ABI) spectrometry analysis of HPLC purified CB2271-326.
CB2271-326 sequence:
GTTLSDQVKKAFAFCSMLCLINSMVNPVIYALRSGEIRSSAHHCLAHWKKCVRGLGS (the N-terminal Gly residue is from the TEV protease recognition site).
Table 1.
Purification summary of the recombinant CB2271-326 fragment
| Step | Total protein* (mg) | Percentage of total protein | Purity (%) |
|---|---|---|---|
| Whole cell lysate | 458.6 | 100 | — |
| Fusion protein | 9.52-12.4 | 2.4 | 44-57 |
| Target protein fragment | 1.5-2 | 0.38 | >95 |
From 1 liter of cell culture (4~6 grams of wet cell).
Refolding and secondary structure of CB2271-326 in different detergents
In order to study if CB2271-326 can be properly folded, CD spectra of the peptide in different detergents (20 mM Brij58, 4 mM digitonin and 20 mM SDS) and organic solvent (TFE) were obtained on a JASCO-800 (Figure 4). The spectra of the target fragment in all four environments showed two minima at near 208 nm and 222 nm, typical for α-helical secondary structure. The raw CD data were converted to mean residue molar ellipticity [θ] and the secondary structure content of the peptide was analyzed by using the program ContinLL on DICHROWEB, an interactive web site allowing the deconvolution of data from circular dichroism spectroscopy experiments [22, 23]. The results showed that CB2271-326 adopts high α-helical structure in the all of the environments. The average α-helical contents of CB2271-326 in different solvent environments were calculated in percentage as TFE>digitonin>Brij58>SDS (Table 2). TFE has long been used as an excellent solvent to stabilize α-helices in hydrophobic peptides, and it also is known to induce α-helical structure even in parts of proteins that normally do not adopt this structure [9, 29, 30]. This could account for the higher proportion of α-helix in this solvent in comparing with the other three detergents. SDS is a strong detergent and often used to solubilize GPCR membrane proteins. The CD analysis showed CB2271-326 can retain a significant amount of α-helical structure but it may not be in the native tertiary structure in SDS. In the current CD study, the α-helix content in this harsh detergent was slightly less than in the mild detergents Brij58 and digitonin, which may be the result of either the denaturing of a small amount of α-helical stretches of CB2271-326 that are not stable, or the non-native folding of CB2271-326 in SDS. Brij58 is an efficient detergent to keep CB2271-326 solubilized as described earlier in this context. Digitonin is the most viable choice because it has been shown to keep many GPCRs in their native states [31] and, more importantly, Kiefer et al. [25] showed that OR5 olfactory receptor is not only properly folded but also exhibits functional ligand binding under reconstitution in digitonin. Therefore, the structure in the mild detergents digitonin and Brij58 likely represents the native secondary structure of CB2271-326. However, digitonin, compared to Brij58, may keep CB2271-326 in its more natural state. Furthermore, CB2271-326 fragment monomer clarified on the lane 8 in Figure 1(B) was achieved by solubilizing CB2271-326 fragment in digitonin, whereas dimer or even polymer always appeared when Brij58 was used for this purpose though with 50 mM DTT added (data not shown). Therefore, digitonin might be a better detergent for the in vitro refolding of GPCR fragments from IBs. The shortcoming of using digitonin, however, is that digitonin sometimes may precipitate in resuspension solutions during the sample preparation for CD or fluorescence studies as described in the method section.
Figure 4.
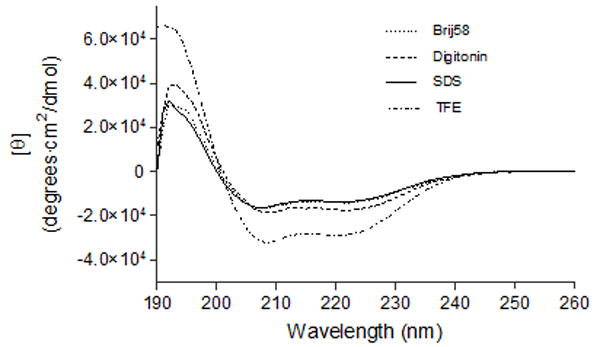
CD spectra of CB2271-326. All samples contained ~15 μM CB2271-326. Samples were solubilized either in Brij58, digitonin, or SDS containing 10 mM Tris-HCl, pH 7.0, 50 mM NaCl, or in TFE.
Table 2.
CB2271-326 secondary structure calculation from CD data and peptide sequence*
| Solvent | α-helix | β-strand | β-turn | Unordered | Total |
|---|---|---|---|---|---|
| Brij58 | 0.768 | 0.091 | 0.141 | 0 | 1 |
| Digitonin | 0.786 | 0.075 | 0.138 | 0 | 1 |
| SDS | 0.731 | 0.091 | 0.178 | 0 | 1 |
| TFE | 0.825 | 0.025 | 0.059 | 0.092 | 1 |
Fluorescence spectroscopy of CB2271-326 in folding and unfolding conditions
The intrinsic fluorescence of proteins arises primarily from the side chains of tyrosine and tryptophan residues. Tryptophans within a hydrophobic core display emission maxima at 330 nm or less, whereas those localized in more hydrophilic surroundings have an emission peak around 350 nm [32, 33]. Chemical denaturation of folded proteins can cause a dramatic shift in both the amount and the peak wavelength of their intrinsic tryptophan fluorescence as these parameters are subject to the microenvironments of each tryptophan [33].
CB2271-326 has a single tryptophan (Trp317) in its C-terminal region close to the cytoplasmic helix 8 domain of CB2 receptor. In our current CB2271-326 fluorescence experiment, the tryptophan (Trp317) in detergent micelle environment of 4 mM digitonin or Brij58 has maximum fluorescence intensity at 328 nm, but the maximum fluorescence intensity was red-shifted to about 353 nm upon denaturation in 8 M of urea (Figure 5). The red-shift in emission maximum is attributed to the micelle-folded protein fragment being denatured in the “polar” environment of urea. The tryptophan fluorescence intensity of the peptide in digitonin is 31% higher than in Brij58 and urea after solvent baseline was subtracted. The higher fluorescence intensity of CB2271-326 in digitonin than in Brij58 might be the result of the following two reasons. One possibility of the more severe quenching of tryptophan in Brij58 than in digitonin is because the peptide molecule is surrounded by more Brij58 molecules than digitonin. Brij58 has an aggregation number of 70 in one micelle while digitonin has a value of 60 (Sigma-Aldrich data). Another possibility is that CB2271-326 may form more oligomers when solubilized in Brij58 than in digitonin; this is supported in part by SDS-PAGE data (not shown). Both the oligomers and bigger detergent micelles contributed to the shielding of the tryptophan side chain and therefore caused the more severe quenching in Brij58 than that in digitonin. Regardless of mechanism, the results suggested that digitonin is a better choice to solubilize this particular set of hydrophobic peptides and membrane proteins for further structure/function study.
Figure 5.
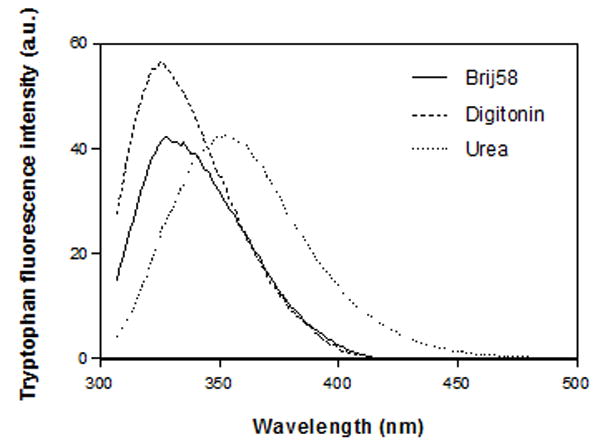
Intrinsic tryptophan fluorescence spectrum of ~15 μM CB2271-326 in 4 mM Brij58, 4 mM digitonin and 8 M urea containing 10 mM sodium phosphate buffer, pH 7.0, 50 mM NaCl. The spectra shown were baseline subtracted.
Stern-Volmer quenching
The presence of Tyr299 on TM7 and Trp317 on Helix8 offers an opportunity to characterize the helical fragment by intrinsic fluorescence. The solvent exposure and accessibility of the tryptophan and the tyrosine on peptide fragment CB2271-326 to the collisional quencher iodide was evaluated by Stern-Volmer analysis. Iodide is an ionic quencher and therefore will act only on fluorophores situated on the outer surface of proteins, and has been used for the quenching of proteins or peptides [9, 32, 34, 35]. Stern-Volmer quenching for the tryptophan and the tyrosine on CB2271-326 in iodide concentrations ranging from 0 to 200 mM was examined. When excited at 295 nm (for tryptophan alone), the fluorescence emission spectra of CB2271-326 exhibited a maximum emission wavelength at 328 nm, a typical characteristic of a tryptophan residue. These results also suggest that the tryptophan residue may be buried in a nonpolar environment, such as a detergent-based micelle. When excited at 275 nm (where both tyrosine and tryptophan residues absorb), the maximum emission wavelength was at 322 nm, which is an intermediate of the tryptophan (330 nm) and tyrosine (303 nm) residues. The steady-state fluorescence quenching data were analyzed using the Stern-Volmer equation [34]:
Where F0 and F are the fluorescence intensities in the absence and presence of quencher (Q) respectively, and the Ksv is the Stern-Volmer quenching constant, obtained from the slope of the plot F0/F versus [Q]. Figure 6 shows that the addition of sodium iodide significantly reduced the fluorescence intensity of tryptophan alone (A) and tryptophan + tyrosine (B) in a concentration-dependent manner. The fluorescence of the CB2 protein fragment follows the Stern-Volmer rule as judged by the linearity of the plot in Figure 6 (C). The quenching constant (Ksv) for tryptophan alone was 1.122 M-1, while that for tryptophan + tyrosine was 1.561 M-1. The observed Ksv enhancement for tryptophan + tyrosine is assumed to be attributed to the presence of Tyr299. Therefore, the quenching constant (Ksv) for tyrosine alone can be calculated by subtracting Ksv for tryptophan alone from the combination: 1.561-1.122=0.439 M-1. Based on the magnitudes of the quenching constants, we predict that both tryptophan and tyrosine residues are partially shielded from iodide by their local peptide structure in the detergent, but that Trp317 located in the cytoplasmic domain of CB2 is more exposed to the surface of a detergent micelle. Compared to Trp317, the relative inaccessibility of iodide to Tyr299 residue in TM7 domain is most likely the result of this fluorophore being buried in the detergent micelles and folded close to the peptide hydrophobic core.
Figure 6.
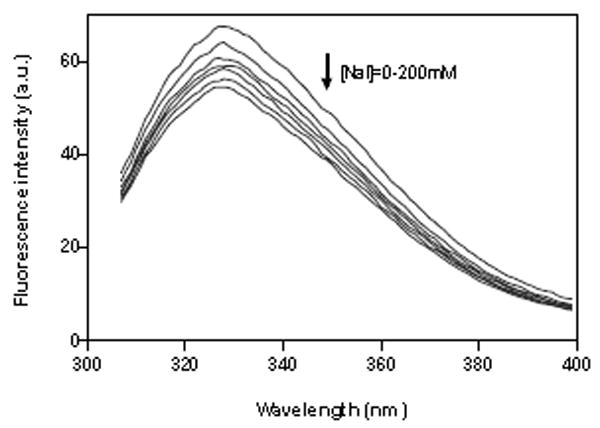
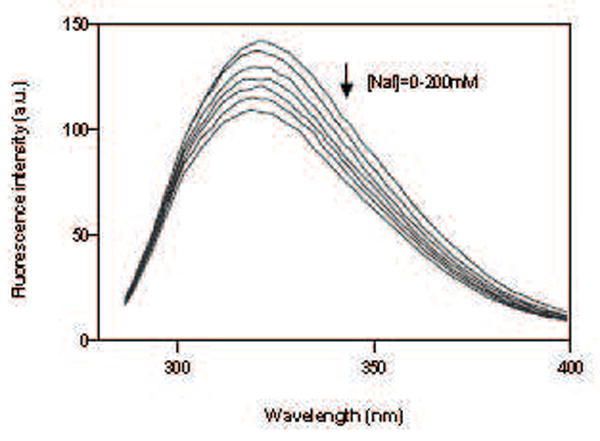
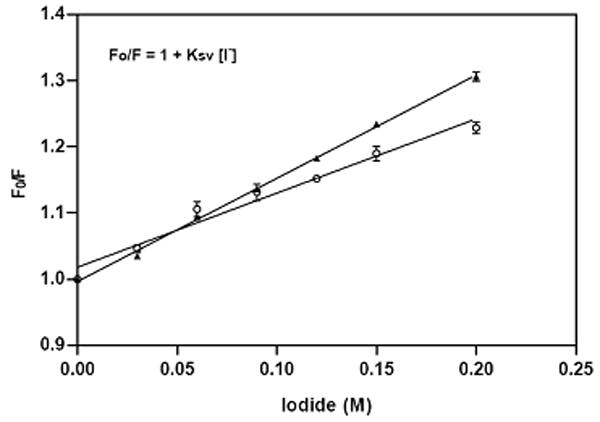
Fluorescence spectra and fluorescence quenching of ~15 μM CB2271-326 corresponds to the increasing concentrations of iodide: (A) quenching of tryptophan alone; (B) quenching of tryptophan + tyrosine; (C) Stern-Volmer plot of the effect of iodide on maximum fluorescence intensity of tryptophan (o) and tryptophan+tyrosine(▲). Each point is an average of triplicate determinations. The peptide was solubilized in 4 mM digitonin containing 10 mM sodium phosphate buffer, pH 7.0, 50 mM NaCl.
Acknowledgments
This work was supported by NIH grant R01 DA15770 to X-Q. X. We thank Dr. D. S. Waugh (NCI, NIH-Frederick) for providing pRK793 vector for expression of TEV protease. Dr. Angela Gronenbornand and Dr. Barry Gold are gratefully acknowledged for a JASCO J-810 Spectropolarimeter, a Cary Fluorescence spectrophotometer and other equipments. We thank Dr. Billy Day’s professional discussion and reading manuscript. We are also thankful to Dr. Wallace for providing online programs to estimate peptide secondary structure supported by an interactive web site DICHROWER.
Abbreviations
- CB2
cannabinoid receptor 2
- CD
circular dichroism
- CNBr
cyanogen bromide
- 3D
three-dimensional
- DTT
dithiothreitol
- E. coli
Escherichia coli
- EL
extracellular loop
- GnHCl
guanidine hydrochloride
- GPCR
G protein-coupled receptor
- IB
inclusion body
- IL
intracellular loop
- IMAC
immobilized metal-ion affinity chromatography
- IPTG
isopropyl-beta-D-thiogalactopyranoside
- Ksv
Stern-Volmer quenching constant
- LB
Luria Bertani
- LDS
lithium dodecyl sulfate
- MALDI-TOF-MS
matrix-assisted laser desorption ionization time of flight mass spectrometry
- NMR
nuclear magnetic resonance
- RP-HPLC
reverse-phase high performance liquid chromatograph
- SDS-PAGE
sodium dodecyl sulfate – polyacrylamide gel electrophoresis
- TEV
tobacco etch virus protease
- TFA
trifluoroacetic acid
- TFE
trifluoroethanol
- TM
transmembrane
- TrpLE
TrpΔLE1413 leader sequence
- UV
ultraviolet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 2.Raitio KH, Salo OM, Nevalainen T, Poso A, Jarvinen T. Targeting the cannabinoid CB2 receptor: mutations, modeling and development of CB2 selective ligands. Curr Med Chem. 2005;12:1217–1237. doi: 10.2174/0929867053764617. [DOI] [PubMed] [Google Scholar]
- 3.Calandra B, T Julie, Shire David, Grisshammer Reinhard. Expression in Escherichia coli and characterization of the human central CB1 and peripheral CB2 cannabinoid receptors. Biotechnology Letters. 1997;19:425–428. [Google Scholar]
- 4.Krepkiy D, Wong K, Gawrisch K, Yeliseev A. Bacterial expression of functional, biotinylated peripheral cannabinoid receptor CB2. Protein Expr Purif. 2006;49:60–70. doi: 10.1016/j.pep.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Yeliseev AA, Wong KK, Soubias O, Gawrisch K. Expression of human peripheral cannabinoid receptor for structural studies. Protein Sci. 2005;14:2638–2653. doi: 10.1110/ps.051550305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang R, Kim TK, Qiao ZH, Cai J, Pierce WM, Jr, Song ZH. Biochemical and mass spectrometric characterization of the human CB2 cannabinoid receptor expressed in Pichia pastoris-Importance of correct processing of the N-terminus. Protein Expr Purif. 2007 doi: 10.1016/j.pep.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 7.Zvonok N, Yaddanapudi S, Williams J, Dai S, Dong K, Rejtar T, Karger BL, Makriyannis A. Comprehensive proteomic mass spectrometric characterization of human cannabinoid CB2 receptor. J Proteome Res. 2007;6:2068–2079. doi: 10.1021/pr060671h. [DOI] [PubMed] [Google Scholar]
- 8.Karnik SS. Analysis of structure-function from expression of G protein-coupled receptor fragments. Methods Enzymol. 2002;343:248–259. doi: 10.1016/s0076-6879(02)43140-0. [DOI] [PubMed] [Google Scholar]
- 9.Kerman A, Ananthanarayanan VS. Expression and spectroscopic characterization of a large fragment of the mu-opioid receptor. Biochim Biophys Acta. 2005;1747:133–140. doi: 10.1016/j.bbapap.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Kerman A, Ananthanarayanan VS. Conformation of a double-membrane-spanning fragment of a G protein-coupled receptor: effects of hydrophobic environment and pH. Biochim Biophys Acta. 2007;1768:1199–1210. doi: 10.1016/j.bbamem.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 11.Therien AG, Glibowicka M, Deber CM. Expression and purification of two hydrophobic double-spanning membrane proteins derived from the cystic fibrosis transmembrane conductance regulator. Protein Expr Purif. 2002;25:81–86. doi: 10.1006/prep.2001.1612. [DOI] [PubMed] [Google Scholar]
- 12.Xie XQ, Zhao J, Zheng H. Expression, purification, and isotope labeling of cannabinoid CB2 receptor fragment, CB2(180-233) Protein Expr Purif. 2004;38:61–68. doi: 10.1016/j.pep.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 13.Zhao J, Zheng H, Xie XQ. NMR characterization of recombinant transmembrane protein CB2 fragment CB2(180-233) Protein Pept Lett. 2006;13:335–342. doi: 10.2174/092986606775974483. [DOI] [PubMed] [Google Scholar]
- 14.Zheng H, Zhao J, Wang S, Lin CM, Chen T, Jones DH, Ma C, Opella S, Xie XQ. Biosynthesis and purification of a hydrophobic peptide from transmembrane domains of G-protein-coupled CB2 receptor. J Pept Res. 2005;65:450–458. doi: 10.1111/j.1399-3011.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 15.Zheng H, Zhao J, Sheng W, Xie XQ. A transmembrane helix-bundle from G-protein coupled receptor CB2: biosynthesis, purification, and NMR characterization. Biopolymers. 2006;83:46–61. doi: 10.1002/bip.20526. [DOI] [PubMed] [Google Scholar]
- 16.Xie XQ, Chen JZ, Billings EM. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins. 2003;53:307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 17.Feng W, Song ZH. Functional roles of the tyrosine within the NP(X)(n)Y motif and the cysteines in the C-terminal juxtamembrane region of the CB2 cannabinoid receptor. FEBS Lett. 2001;501:166–170. doi: 10.1016/s0014-5793(01)02642-4. [DOI] [PubMed] [Google Scholar]
- 18.Rhee MH. Functional role of serine residues of transmembrane dopamin VII in signal transduction of CB2 cannabinoid receptor. J Vet Sci. 2002;3:185–191. [PubMed] [Google Scholar]
- 19.Parks TD, Leuther KK, Howard ED, Johnston SA, Dougherty WG. Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal Biochem. 1994;216:413–417. doi: 10.1006/abio.1994.1060. [DOI] [PubMed] [Google Scholar]
- 20.Mohanty AK, Simmons CR, Wiener MC. Inhibition of tobacco etch virus protease activity by detergents. Protein Expr Purif. 2003;27:109–114. doi: 10.1016/s1046-5928(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 21.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lobley A, Whitmore L, Wallace BA. DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics. 2002;18:211–212. doi: 10.1093/bioinformatics/18.1.211. [DOI] [PubMed] [Google Scholar]
- 23.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baneres JL, Mesnier D, Martin A, Joubert L, Dumuis A, Bockaert J. Molecular characterization of a purified 5-HT4 receptor: a structural basis for drug efficacy. J Biol Chem. 2005;280:20253–20260. doi: 10.1074/jbc.M412009200. [DOI] [PubMed] [Google Scholar]
- 25.Kiefer H, Krieger J, Olszewski JD, Von Heijne G, Prestwich GD, Breer H. Expression of an olfactory receptor in Escherichia coli: purification, reconstitution, and ligand binding. Biochemistry. 1996;35:16077–16084. doi: 10.1021/bi9612069. [DOI] [PubMed] [Google Scholar]
- 26.Ma C, Marassi FM, Jones DH, Straus SK, Bour S, Strebel K, Schubert U, Oblatt-Montal M, Montal M, Opella SJ. Expression, purification, and activities of full-length and truncated versions of the integral membrane protein Vpu from HIV-1. Protein Sci. 2002;11:546–557. doi: 10.1110/ps.37302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogl H, Kosemund K, Kuhlbrandt W, Collinson I. Refolding of Escherichia coli produced membrane protein inclusion bodies immobilised by nickel chelating chromatography. FEBS Lett. 1998;432:21–26. doi: 10.1016/s0014-5793(98)00825-4. [DOI] [PubMed] [Google Scholar]
- 28.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 29.Alexandrescu AT, Ng YL, Dobson CM. Characterization of a trifluoroethanol-induced partially folded state of alpha-lactalbumin. J Mol Biol. 1994;235:587–599. doi: 10.1006/jmbi.1994.1015. [DOI] [PubMed] [Google Scholar]
- 30.Kuroda Y, Hamada D, Tanaka T, Goto Y. High helicity of peptide fragments corresponding to beta-strand regions of beta-lactoglobulin observed by 2D-NMR spectroscopy. Fold Des. 1996;1:255–263. doi: 10.1016/s1359-0278(96)00039-9. [DOI] [PubMed] [Google Scholar]
- 31.Hulme EC. Receptor Biochemistry: A Practical Approach. Oxford University Press; New York: 1990. [Google Scholar]
- 32.Eftink MR, Ghiron CA. Fluorescence quenching studies with proteins. Anal Biochem. 1981;114:199–227. doi: 10.1016/0003-2697(81)90474-7. [DOI] [PubMed] [Google Scholar]
- 33.Nie Y, Hobbs JR, Vigues S, Olson WJ, Conn GL, Munger SD. Expression and purification of functional ligand-binding domains of T1R3 taste receptors. Chem Senses. 2006;31:505–513. doi: 10.1093/chemse/bjj053. [DOI] [PubMed] [Google Scholar]
- 34.Lehrer SS. Solute perturbation of protein fluorescence. The quenching of the tryptophanyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 35.Sultan NA, Swamy MJ. Fluorescence quenching and time-resolved fluorescence studies on Trichosanthes dioica seed lectin. J Photochem Photobiol B. 2005;80:93–100. doi: 10.1016/j.jphotobiol.2005.03.003. [DOI] [PubMed] [Google Scholar]