

Abstract
Toll-like receptors (TLRs) play a critical role in the induction of innate immune responses which have been implicated in neuronal death induced by global cerebral ischemia/reperfusion (GCI/R). The present study investigated the role and mechanisms-of-action of TLR4 signaling in ischemia-induced hippocampal neuronal death. Neuronal damage, activation of the TLR4 signaling pathway, expression of pro-inflammatory cytokines and activation of the PI3K/Akt signaling pathway in the hippocampal formation (HF) were assessed in wild type (WT) mice and TLR4 knockout mice (TLR4-/-) mice after GCI/R. GCI/R increased expression of TLR4 protein in the hippocampal formation (HF) and other brain structures in WT mice. Phosphorylation of the inhibitor of kappa B (p-IκB) as well as activation of nuclear factor kappa B (NFκB) increased in the HF of WT mice. In contrast, there were lower levels of p-IκB and NFκB binding activity in TLR4-/- mice subjected to GCI/R. Pro-inflammatory cytokine expression was also decreased, while phosphorylation of Akt and GSK3β were increased in the HF of TLR4-/- mice after GCI/R. These changes correlated with decreased neuronal death/apoptosis in TLR4-/- mice following GCI/R. These data suggest that activation of TLR4 signaling contributes to ischemia-induced hippocampal neuronal death. In addition, these data suggest that modulation of TLR4 signaling may attenuate ischemic injury in hippocampal neurons.
Keywords: TLR4, cerebral, ischemia, reperfusion
1. Introduction
Global cerebral ischemia/reperfusion (GCI/R) induces neuronal death, especially in the hippocampal formation (HF) (Nitatori et al., 1995). Recent evidence suggests that innate immune and inflammatory responses play a critical role in ischemia-induced neuronal damage (Lambertsen et al., 2004; Liao et al., 2001). However, the precise molecular mechanisms by which immune/inflammatory responses are involved in ischemia-induced neuronal death are unclear.
Toll-like receptors (TLRs) are a family of signal transduction molecules and play a critical role in the induction of innate and adaptive immunity (Aderem and Ulevitch, 2000). TLR-mediated signaling pathways mainly stimulate the activation of NFκB which is an important nuclear transcription factor for regulating expression of genes involved in innate and inflammatory responses (Hoshino et al., 2002; Porter and Janicke, 1999; Toshchakov et al., 2002). Recent studies have shown that activation of the TLR4-mediated NFκB pathway plays a role in ischemia/reperfusion (I/R) injury. For example, we (Hua et al., 2007; Li et al., 2005) and others (Ha et al., 2006) have reported that TLR4-mediated NFκB signaling contributes to myocardial ischemia/reperfusion injury and TLR4 deficiency protects the myocardium from ischemic injury. TLR4 is also involved in the pathogenesis of I/R injury in liver (Zhai et al., 2004), kidney (Kim et al., 2005) and lung tissues (Shimamoto et al., 2006), TLRs have been identified in the central nervous system (CNS) and are thought to play an important role in the brain's response to pathogens as well as toxic cell debris (Bottcher et al., 2003; Bsibsi et al., 2002; Maslinska et al., 2004) and inflammatory or autoimmune CNS diseases (Chakravarty and Herkenham, 2005; Kerfoot et al., 2004). Recent studies (Cao et al., 2006; Caso et al., 2007) using a permanent and longstanding focal cerebral ischemia model have shown that infarct size is reduced in TLR4-deficient mice compared with wild type (WT) mice. It is still unclear why TLR4 deficiency results in the protection of brain from ischemic injury.
In the present study, we investigated the role and mechanisms-of-action of TLR4 in ischemia-induced hippocampal neuronal death using a murine model of global cerebral ischemia/reperfusion (GCI/R). We observed that neuronal death/apoptosis and the levels of pro-inflammatory cytokine expression in the HF of TLR4 deficient mice were significantly less than in age-matched wild type mice following GCI/R. Importantly, the levels of phosphorylated Akt and GSK3β in the HF of TLR4-/- mice were significantly higher compared with WT type mice after GCI/R. Our data indicates that TLR4-mediated signaling contributes to ischemia-induced hippocampal neuronal death. Our results suggest that modulation of TLR4 may attenuate the inflammatory response and concomitantly enhance activation of the PI3K/Akt pathway, thus protecting hippocampal neurons from ischemia injury.
2. Materials and methods
2.1 Animals
C57BL/10ScCr (TLR4-/-) and C57BL/10ScSn (wild type, WT) mice (male, 25∼30g, age: 8∼12 weeks) were obtained from Jackson Laboratory and maintained in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU). The experiments outlined in this manuscript conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 1996). All aspects of the animal care and experimental protocols were approved by the ETSU Committee on Animal Care. The mice were divided into four groups: TLR4-/- mice subjected to GCI/R (TLR4-/--I/R, n=11), wild type mice subjected to GCI/R (WT-I/R, n=11), TLR4-/- sham operation (TLR4-/--S, n=8) and WT sham operation (WT-S, n=8). Seventy-two hrs after GCI/R (n = 6/group) or sham operation (n= 5/group) mice were anesthetized with Ketamine and transcardially perfused with normal saline followed by 30 ml of 4% buffered paraformaldehyde, pH 7.4. The brains were removed, postfixed, embedded in paraffin and cut into sections (7μm). In a separate experiment, six hours after GCI/R (n = 5/group) or sham operation (n = 3/group), the mice were sacrificed by ketamine overdose and the brains were removed and stored at -80°C for isolation of cellular proteins.
2.2 Induction of global cerebral ischemia/reperfusion (GCI/R)
Our laboratory established a mouse model of GCI/R, in which cerebral ischemia was induced by occlusion of the common carotid arteries (CCA) bilaterally and the left subclavian artery (LSA) together with right subclavian artery (RSA) stenosis under controlled ventilation (Hua et al., 2006). In brief, anesthesia was induced by 5.0% Isoflurane and was maintained by inhalation of 1.5% Isoflurane driven by 100% oxygen flow using the EZ-Anesthesia system (Euthanex Corp., Palmer, PA). The trachea was intubated and the lungs were mechanically ventilated at a rate of 110 breaths per min with a total delivered volume of 0.5 ml. Body temperature was regulated at 37.0°C by surface water heating. Under a surgical microscope, stenosis of the RSA was produced as previously described (Hua et al., 2006). Then the left CCA, the LSA and the right CCA were gently isolated and clamped with microsurgical clamps, respectively. Cerebral ischemia was maintained for 12 min and reperfusion started when the clamps were removed. The ischemic condition and the sufficiency of reperfusion were confirmed by regional cerebral blood flow (rCBF) detected by the PeriFlux system 5000 (Type PE5001, Jarfalla, Sweden). The chest was closed in layers and all of the incisions were sutured. The mice were placed in a cage kept at 31°C for the following 3 hours and then returned to the animal care room.
2.3 Evaluation of neuronal damage in the hippocampal formation (HF)
Brain sections were stained with 0.1% cresyl violet according to Nissl's method (Lee et al., 2004). Briefly, brains tissue from each group were embedded in paraffin and cut at 7 μm. Paraffin sections were deparaffinized and brought to water, then stained with 0.1% cresyl violet for 2 minutes. Slides were washed in running tap water for 2-5 minutes, dehydrated, cleared and then mounted with coverslips. Viable neurons were defined as neurons in which the nucleus appeared normal (a round nucleus showing clear nucleoplasm with a nucleolus). Neurons irreversibly damaged by ischemia exhibit pyknosis and/or nuclear fragments and shrunken cell bodies. Representative sections from the level described in the next paragraph were analyzed for neuronal damage in the hippocampal formation (HF). The HF is formed by two cortical laminae embedded into each other: the cornu ammonis (CA) and the gyrus dentatus (DG). The CA area can be further subdivided into CA1, CA2, CA3, and CA4. The CA1 area is the region most vulnerable to ischemia. A 0-4 neuropathological score (NPS) was used as previously described (Lee et al., 2004): grade 0, no damage to any HF fields; grade 1, scattered ischemic neurons in CA1 area; grade 2, moderate ischemic damage in field CA1 (less than half of pyramidal cells affected); grade 3, severe damage to pyramidal cells in field CA1 (more than half of pyramidal cells affected); grade 4, extensive cell damage in all HF fields.
2.4 Immunohistochemistry (IHC) staining
The expression of TLR4, phosphorylation of NFκB-p65 (p-NFκB-p65) and cleaved caspase-3 activity were examined by IHC as described previously (Hua et al., 2005). The primary antibodies used in the present study were anti-TLR4 (1:1000, gift from R. Medzhitov, Yale University), anti-p-NFκB-p65 (1:50, #3037, Cell Signaling Technology, Inc.) and anti-cleaved caspase-3 (1:50, CP229, Biocare Medical, Concord CA 94520). The biotinylated secondary antibodies and antibody–biotin–avidin–peroxidase complexes (ABC reagent) for TLR4 and p-NFκB-p65 were purchased from Vector Laboratories, Burlingame, CA, USA. The MACH 4 Universal Polymer Detection System for cleaved caspase-3 was purchased from Biocare Medical, Concord CA 94520. Slides were processed without primary antibodies as negative controls. Brain sections were obtained 1.5 mm behind the Bregma in the coronal plane. The slides were imaged in the CA1 and dentate gyrus (DG) fields in the hippocampal formation (HF) at 400 × magnification. The levels of cleaved caspase-3 were determined by an image analyzer in a high power field and calculated as a percentage of the integrated density value (IDV) of positive staining vs. total IDV. The data are expressed as the mean ± SE.
2.5 TUNEL analysis
For the visualization of the DNA fragmentation, TUNEL staining was performed using an In Situ Cell Death Detection Kit (G7130 Promega, Madison, WI 53711) according to the manufacturer's protocol. The TUNEL-positive cells and total cells in the fields of CA1 and DG at 400× magnification were counted. The number of TUNEL-positive cells was expressed as the percentage of total counted cells. The data are expressed as the mean ± SE.
2.6 Electrophoretic mobility shift assay (EMSA)
Nuclear proteins were isolated from hippocampal samples as previously described (Hua et al., 2005; Hua et al., 2007). NFκB binding activity was examined by EMSA in a 15 μl binding reaction mixture containing 15 μg of nuclear proteins and 35 fmol [γ-32P] labeled double-stranded NFκB consensus oligonucleotide.
2.7 Western Blots
Cytoplasmic proteins were prepared from hippocampal tissues and immunoblots were performed as described previously (Hua et al., 2005; Hua et al., 2007). Briefly, the cellular proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred onto Hybond ECL membranes (Amersham Pharmacia, Piscataway, NJ). The ECL membranes were incubated with the appropriate primary antibody followed by incubation with peroxidase-conjugated secondary antibodies (Cell Signaling Technology, Inc.). The signals were detected with the ECL system (Amersham Pharmacia). The same membranes were probed with antibody for glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Biodesign, Saco, Maine) after being washed with stripping buffer. The signals were quantified by scanning densitometry and computer-assisted image analysis. The primary antibodies used in the study were anti-phospho-IκBα, anti-phospo-Akt (p-Akt), anti-phospho-GSK3β (p-GSK3β) (cell signaling technology), anti-interlukin-6 (IL-6), anti tumor necrosis factor α (TNFα), anti-Fas-L (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and anti-high mobility group box 1 (HMGB1, Stressgen).
2.8 Statistical analysis
Continuous scale measurements were summarized by the mean, standard deviation and standard error of the mean (sem) for each group. Groups comparisons for neuronal damage (neuropathological score, NPS) were accomplished with the t-test (applied to score values) and by the chi-square test (applied to frequency counts of the possible NPS categories, Stat-Exact software). ANOVA and the least significant difference procedure were used to assess the effectiveness of intervention (GCI/R vs Sham) and groups (TLR4-/- vs WT) for the expression of cleaved caspase-3, TUNEL positive cells, expression of p-IκB, p-Akt, p-GSK3β, IL-6, TNFα, Fas-L and HMGB1. Probability levels of 0.05 or smaller were used for reporting statistical significance.
3. Results
3.1 Decreased neuronal damage in TLR4-/- mice following GCI/R
Neuronal damage in the HF of WT-I/R and TLR4-/- -I/R mice was evaluated semi-quantitatively as described. Nissl's staining showed neuronal damage in the CA1 and DG fields of the HF in WT-I/R mice characterized by shrunken pyknotic nuclei and shrunken cell bodies (Fig. 1). In the damaged cells of the DG, nuclear fragments characteristic of apoptosis were the usual morphology. The mean NPS of WT-I/R mice was 3.7 ± 0.3 (mean ± SE, all animals but one received a score of 4), which was significantly higher than the 1.7 ± 0.5, observed in TLR4-/- -I/R mice 72 hours after GCI/R (p< 0.05).
Figure 1. TLR4 deficient mice show decreased neuronal damage in the HF after GCI/R.
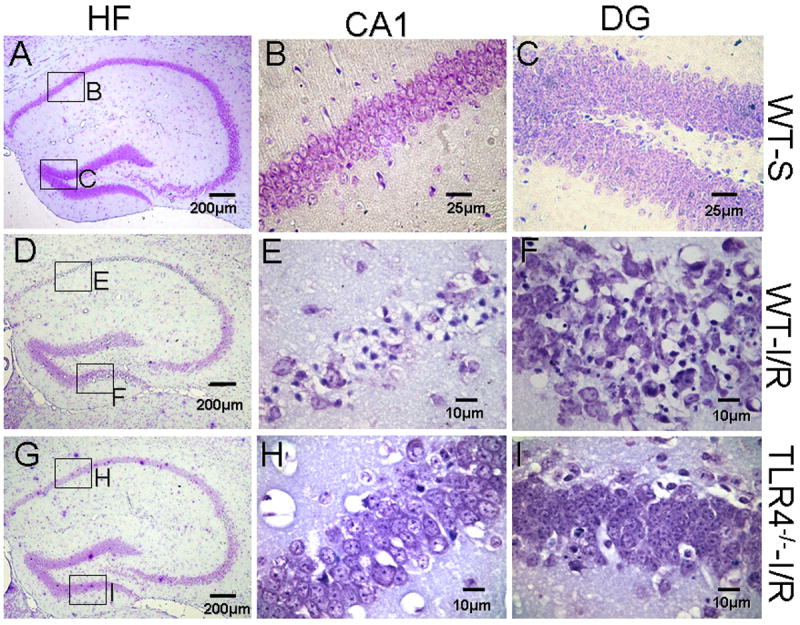
Brain sections were stained with 0.1% cresyl violet. Viable neurons were defined as neurons in which a normal nucleus can be seen. Damaged neurons exhibit features including pyknosis, karyorhexis and shrunken cell bodies. Three days after GCI/R, neuronal damage was prominent in the CA1 and dentate gyrus (DG) of the HF in WT mice. Compared with WT mice, there was less neuronal damage in the HF of TLR4-/- mice. A, D and G: HF; B, E and H: CA1; C, F and I: dentate gyrus (DG). WT-S: wild type sham, WT-I/R: wild type subjected to GCI/R, TLR4-/- -I/R: TLR4-/- mice subjected to GCI/R.
3.2 Reduced neuronal apoptosis in TLR4-/- mice following GCI/R
We evaluated neuronal death using the TUNEL assay in the brains of WT mice and TLR4-/- mice 72 hours after GCI/R. Sham operated mice served as controls. As shown in Figure 2, TUNEL positive cells were detected in the pyramidal cell layer of the CA1 field and in the granular layer of the dentate gyrus (DG) in WT-I/R mice (Fig. 2B and 2E). The numbers of TUNEL positive cells in the CA1 and DG of the HF in TLR4-/--I/R mice (Fig. 2C and 2F) were significantly less compared with WT-I/R mice (p<0.05, Fig. 2M, CA1: 16.8 ± 11.3% vs. 57.8 ± 14.3%, DG: 2.9 ± 1.1% vs. 24.9 ± 8.0%). The presence of cleaved caspase-3, a specific marker for apoptotic cells, was also examined in brain tissue sections with anti-cleaved caspase-3 antibody. As shown in Figure 2, there was no detectable cleaved caspase-3 in WT-S mice (Fig. 2G and 2J). In both WT-I/R and TLR4-/--I/R mice, there was only rare staining for cleaved caspase-3 in CA1 fields (Fig. 2H and 2I), however, the expression of cleaved caspase-3 was significantly greater in the DG fields in WT mice (Fig. 2K and 2L). There was less cleaved caspase-3 expressed in the DG in TLR4-/--I/R mice compared with WT-I/R mice (p < 0.01, Fig. 2N, 1.6 ± 0.4% vs. 10.0 ±1.2%).
Figure 2. TLR4 deficiency results in decreased neuronal apoptosis in the HF in response to GCI/R.
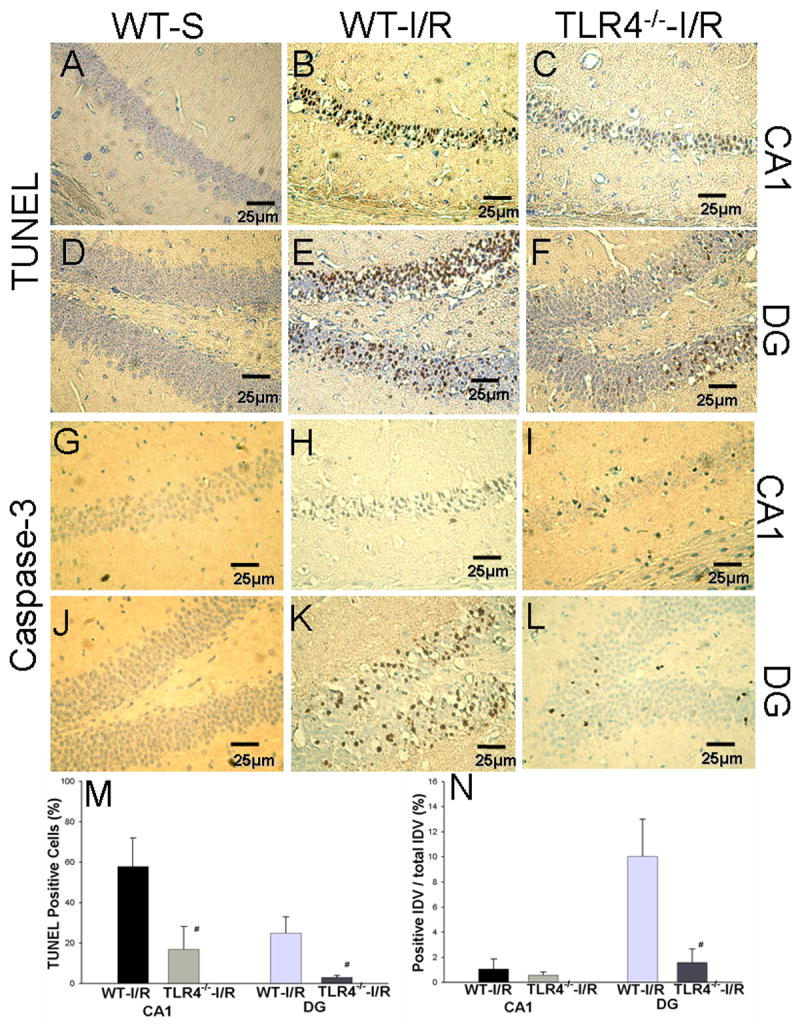
Neuronal apoptosis was confirmed by the methods of TUNEL staining (A to F) and IHC staining for cleaved caspase-3, a specific marker of apoptosis (G to L). The positive staining was shown in brown. Slides were counterstained with hematoxylin. Results show that GCI/R results in increased numbers of TUNEL positive cells in the HF (B and E) in WT mice. In TLR4-/--I/R mice, there were fewer TUNEL positive staining cells in the HF (C and F) compared with WT-I/R mice (M, # p < 0.05). GCI/R results in the expression of cleaved caspase-3 in the DG area of the HF in WT mice three days after GCI/R (K). There was less expression of cleaved caspase-3 in the DG in TLR4-/--I/R mice (L) compared with WT-I/R mice (N, # p < 0.01). In both WT and TLR4-/- mice, there was only light positive staining for cleaved caspase-3 in the CA1 of the HF three days after GCI/R (H and I). A, B, C, G, H and I: CA1; D, E, F, J, K and L: dentate gyrus (DG) of the HF. WT-S: wild type sham, WT-I/R: wild type subjected to GCI/R, TLR4-/- -I/R: TLR4-/- mice subjected to GCI/R.
3.3 Increased expression of TLR4 in WT mice following GCI/R
To investigate the activation of TLR4-mediated signaling, expression of TLR4 protein was detected by IHC. Results showed that TLR4 immunoreactivity was not detectable in cerebral tissue from WT sham (WT-S) (Fig. 3A). In WT-I/R mice, immunoreactivity for TLR4 was consistently demonstrated in the hippocampal formation (HF) and caudate-putamen (CPu) at 72 hours. Less consistent immunoreactivity was present in the retrosplenial granular cortex and other cortical areas, thalamic nuclei, and some white matter tracts such as fimbria-fornix, internal capsule and corpus callosum. Figure 3 illustrates TLR4-positive staining in the CPu (3D), CA1 (3E), dentate gyrus (3F) and cortex (3D) in the WT mice 72 hours after GCI/R.
Figure 3. TLR4 expression in WT mice is increased in brain tissue after GCI/R.
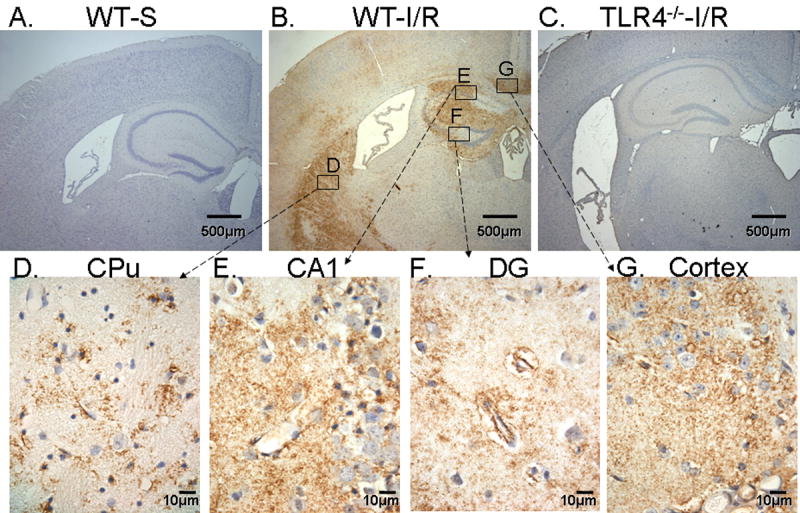
Brain tissue sections were stained by IHC, TLR4 immunoreactivity was indicated by the presence of a brown color. Tissue sections were counterstained with hematoxylin. Results showed that there was no TLR4 expression observed in sham operated WT mice (WT-S) (A). Expression of TLR4 was observed in WT mice brain (B) after GCI/R in the caudate-putamen (CPu, D), CA1 (E), dentate gyrus (DG, F) and cortex (G). WT-S: wild type sham, WT-I/R: wild type subjected to GCI/R. No TLR4 immunoreactivity was observed in TLR4-/- mice subjected to GCI/R (C).
Immunoreactivity for TLR4 generally took the form of small puncta, which were often distributed in neuropil not clearly associated with particular cellular elements. In some instances the immunoreactivity outlined processes around a glial nucleus. Immunoreactivity was also present on or around some but not all endothelial cells. There was some patchy, scattered immunoreactivity present in the pyramidal layer of the CA and the granular layer of the DG; it could not be determined whether this positivity was within neurons or in processes between neurons. In contrast to WT mice subjected to GCI/R, TLR4-/- mice showed no immunoreactivity in brain for TLR4 after GCI/R (Fig. 3C).
3.4 NFκB activation in WT mice following GCI/R
NFκB is an important transcription factor downstream in the TLR4 signaling pathway and in response to a variety of stimuli. We examined the nuclear translocation of NFκB-p65 in the HF of mice following GCI/R. As shown in Figure 4, GCI/R significantly stimulated NFκB p65 nuclear translocation in the HF of WT mice. In contrast, NFκB p65 nuclear translocation in the HF of TLR4-/- mice was significant less compares with WT mice (Fig. 4). EMSA results showed that NFκB DNA binding activity was significantly increased by 94.4% in the HF after GCI/R in WT (Fig. 5A). EMSA also showed GCI/R increased NFκB DNA binding activity in the HF of TLR4-/- mice (Fig. 5A), however, the NFκB DNA binding activity in both sham control and GCI/R in TLR4-/- mice were significantly less compared with WT mice (Fig. 5A). As expected, the levels of p-IκBα were significantly increased by 84.4% in WT mice after GCI/R. In contrast, there was no significant difference of the levels of p-IκBα in the HF between TLR4-/- sham control and TLR4-/- GCI/R mice (Fig. 5B).
Figure 4. Decreased nuclear translocation of phospho-NFκB p65 in the HF of TLR4-/- mice in response to GCI/R.
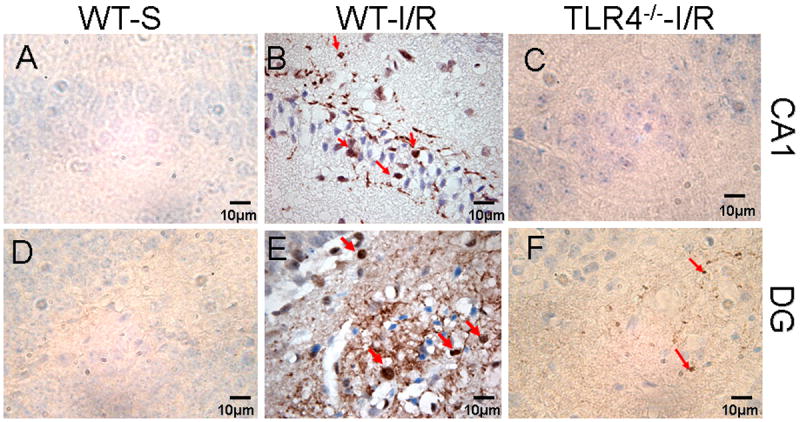
Phospho-NFκB p65 (p-NFκB p65) immunoreactivity was shown as a brown color by IHC staining. Tissue sections were counterstained with hematoxylin. There was no detectable p-NFκB p65 in sham operated mice (A and D). Nuclear translocation of p-NFκB p65 was increased in the HF of WT mice by GCI/R, shown in CA1 (B) and dentate gyrus (DG, E) (arrows). Less p-NFκB p65 was observed in the CA1 and DG of TLR4-/- mice after GCI/R compared with WT mice (B, C, E and F) (arrows). WT-S: wild type sham, WT-I/R: wild type subjected to GCI/R, TLR4-/- -I/R: TLR4-/- mice subjected to GCI/R.
Figure 5. Decreased NFκB DNA binding activity and increased phosphorylation of IκB in the HF of TLR4-/- mice after GCI/R.
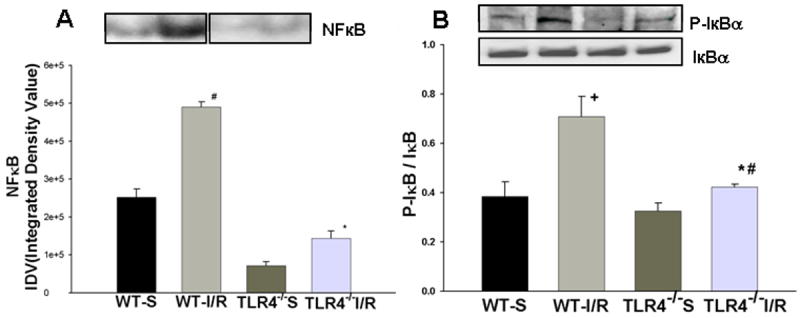
EMSA showed that GCI/R results in increased DNA binding activity of NFκB in the HF of WT mice six hours after GCI/R (A, #: compared with WT-S p< 0.05). The level of NFκB activity was less in TLR4-/- mice compared with WT mice six hours after GCI/R (A, *: compared with WT-I/R, p<0.05). Representative EMSA results are shown at the top. Western Blots showed that the level of phosphorylated IκB increased in the HF of WT mice 6 hours after GCI/R compared with WT control (B, #: compared with WT-S, p < 0.05). There was less p- IκB in the HF of TLR4-/- mice compared with WT mice 6 hours after GCI/R (B, *: compared with WT-I/R, p < 0.05). WT-S: wild type sham, WT-I/R: wild type subjected to GCI/R, TLR4-/--S: TLR4-/- sham, TLR4-/- -I/R: TLR4-/- mice subjected to GCI/R. Results are mean ± SE (n=3 in WT-S and TLR4-/--S groups; n=5 in WT-I/R and TLR4-/-I/R groups). Representative results of Western blots are shown at the top.
3.5 TLR4- deficiency prevents upregulation of IL-6, TNFα, Fas-L and HMGB1 in the HF in response to GCI/R
Activation of NFκB will stimulate the expression of inflammatory cytokines which play a role in the ischemic injury. We examined the expression of inflammatory cytokines in the HF of brains subjected to GCI/R. As shown in Figure 6, the expression of IL-6, TNFα, Fas-L and HMGB1 in the HF of WT mice subjected to 6 hrs of GCI/R were increased by 57.2%, 52.2%, 67.8% and 199.2% (p < 0.05), respectively, compared with sham control. In contrast, GCI/R did not result in significant changes in the expression of IL-6, TNFα, Fas-L and HMGB1 in the HF of TLR4-/- mice. The levels of IL-6, TNFα, Fas-L and HMGB1 in the HF of TLR-/- mice were lower than that in the HF of WT mice following GCI/R (p < 0.05, Fig. 6).
Figure 6. TLR4 dependent increase in IL-6, TNFα, Fas-L and HMGB1 expression in the HF in response to GCI/R.
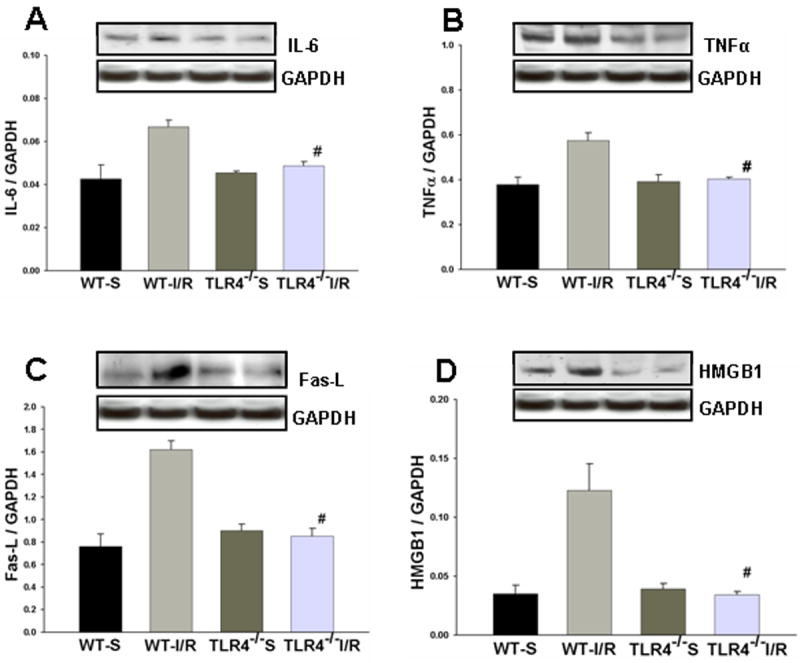
Western Blots showed that IL-6 (A), TNFα (B), Fas-L (C) and HMGB1 (D) levels were increased in the HF of WT mice 6 hours after GCI/R compared with WT sham control (#: compared with WT-S, p < 0.05). IL-6, TNFα, Fas-L and HMGB1 levels in the HF of TLR4-/- mice were not significantly changed after GCI/R and were lower than that in WT mice after GCI/R (*: compared with WT-I/R, p < 0.05). WT-S: wild type sham, WT-I/R: wild type subjected to GCI/R, TLR4-/--S: TLR4-/- sham, TLR4-/- -I/R: TLR4-/- mice subjected to GCI/R. Results are expressed as ratio of integrated density value (IDV) of individual cytokines vs. IDV of GAPDH, mean ± SE (n=3 in WT-S and TLR4-/--S groups; n=5 in WT-I/R and TLR4-/-I/R groups). Representative results of Western blots are shown at the top of each pane.
3.6 Activation of the PI3K/Akt pathway in the HF of TLR4-/- mice after GCI/R
Activation of the PI3K/Akt pathway plays a role in anti-apoptosis and may serve an negative feedback regulation of the TLR4/NFκB activation pathway (Ojaniemi et al., 2003;Romaskova and Makarov, 1999). We examined the levels of phosphorylation of Akt (p-Akt) and GSK3β (p-GSK3β) in HF tissue of TLR4-/- mice following GCI/R. As shown in Figure 7, significantly higher levels of p-Akt and p-GSK3 β were observed in TLR4-/- mice compared with WT mice after GCI/R.
Figure 7. TLR4 deficiency results in increased brain phosphorylation of AKt and GSK3β in response GCI/R.
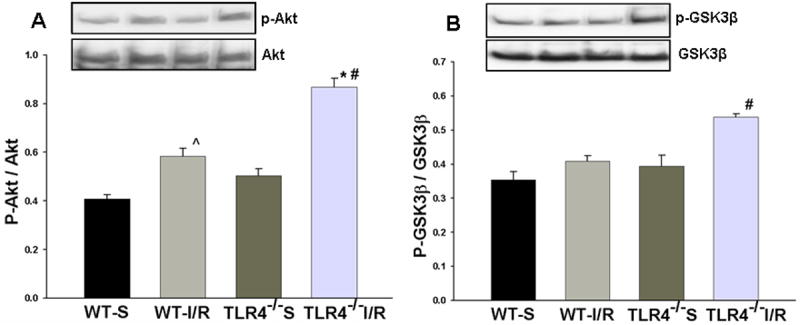
Western Blots showed that the levels of phospho-Akt (p-Akt) (A) and phospho-GSK3β (p- GSK3β, B) in the HF of TLR4-/- mice were significantly higher than those in WT mice at 6 hours after GCI/R (*: compared with WT-I/R, p < 0.05; #: compared with WT-S, p < 0.05). WT-S: wild type sham, WT-I/R: wild type subjected to GCI/R, TLR4-/--S: TLR4-/- sham, TLR4-/- -I/R: TLR4-/- mice subjected to GCI/R. Results are expressed as ratio of integrated density value (IDV) of p-Akt vs. IDV of Akt1, and IDV of p- GSK3β vs. GSK3β, mean ± SE (n=3 in WT-S and TLR4-/--S groups; n=5 in WT-I/R and TLR4-/-I/R groups). Representative results of Western blots are shown at the top of each pane.
4. Discussion
A growing body of evidence suggests a link between innate immune/inflammatory responses and ischemia-induced neuronal damage and TLRs play a role in the induction of innate and adaptive immune responses (Brightbill and Modlin, 2000; Liao et al., 2001; Zhang and Ghosh, 2001). TLR4 has been reported to participate in inflammatory and autoimmune disease in the central nervous system (CNS) (Chakravarty and Herkenham, 2005; Kerfoot et al., 2004). For example, enhanced expression of TLR4 has been observed in inflamed human CNS tissues (Bsibsi et al., 2002), in mouse brain or in organotypic hippocampal culture in response to different cerebral infections, and in neonatal rat brain following hypoxia-ischemia or exposure to alpha-toxin (Maslinska et al., 2004). TLR4 may also contribute the process of neuronal death (Lehnardt et al., 2003). Recent reports indicate that brain infarct size and inflammatory responses are reduced in TLR4 deficient mice after occlusion of the middle cerebral artery (Cao et al., 2006; Caso et al., 2007). However, the mechanisms by which TLR4 signaling in ischemia-induced neuronal death/apoptosis remain unknown.
In the present study, we examined the role and mechanisms of alter TLR4 signaling in ischemia induced hippocampal neuronal death. We used a global cerebral ischemia/reperfusion model which produces a relatively specific insult of neurons that are selectively vulnerable to modest degrees of hypoxia/ischemia. The innate immune and inflammatory responses induced in this model are likely different than those induced by an insult that produces tissue pan-necrosis (i.e., an infarct). Hippocampal neuronal damage was evaluated 72 hours after GCI/R. We observed that GCI/R induced expression of TLR4 in different cerebral areas, including the HF, CPu and cortex (Fig. 3). This is the first report that TLR4 protein increases selectively in different brain regions in adult mice subjected to GCI/R.
We also observed that there was less hippocampal neuronal damage in TLR4-/- mice than in WT mice (Fig. 1 and Fig. 2). Neuronal death in the CA1 region was TUNEL positive with a low level of cleaved caspase-3 and the accompanying morphology, while not specific, is consistent with what has classically been termed necrosis. Death in the DG was TUNEL positive with a higher level of cleaved caspase-3, and the morphology was consistent with the classical description of apoptosis. Interestingly, three days after GCI/R, there were still high levels of cleaved caspase-3 in the dentate gyrus in WT mice. There are two possible explanations for the lack of caspase positivity in CA1. One is that the neurons of CA1 died a caspase-dependent death, but the presence of cleaved caspase-3 in the CA1 pyramidal layer had declined at 3 days after the ischemic insult, since previous studies reported that caspase-3 was activated as early as 30 min and declined at 24 h and afterwards following cerebral ischemia (Cho et al., 2003; Namura et al., 1998). Another explanation is that the majority of the ischemic CA1 neurons follow a caspase-independent necrosis-like death (Muller et al., 2004), while the neurons in DG area follow a caspase-dependent apoptosis pathway. In addition, compared with WT mice, there were fewer TUNEL positive cells and less activation of caspase-3 in HF in TLR4-/- mice subjected to GCI/R (Fig. 2). These data indicated that TLR4-mediated signaling contributes to the hippocampal neuronal death after GCI/R.
NFκB is an important transcription factor downstream in the TLR4-mediated signaling pathway. Activation of TLR4 stimulates IκBα phosphoryation and degradation, resulting in nuclear translocation of NFκB, which initiates transcription of genes associated with innate immune responses and inflammation (Hoshino et al., 2002; Porter and Janicke, 1999; Toshchakov et al., 2002). We observed that that the levels of phosphorylation of IκBα, NFκB nuclear translocation and DNA binding activity were significantly increased in the HF of WT mice after GCI/R, suggesting that GCI/R stimulates activation of the TLR4/NFκB signaling pathway (Fig. 4 and Fig. 5). However, the levels of p-IκBα and NFκB nuclear translocation and DNA binding activity in TLR4-/- mice following GCI/R were lower than in WT mice (Fig. 5). These data are consistent with previous reports that activation of NFκB is regulated through the TLR4-mediated signaling pathway. NFκB is a transcription factor that controls the expression of a variety of target genes including those involved in cell proliferation, apoptosis, and inflammation, which has been shown to be involved in cerebral ischemia (Schneider et al., 1999; Tounai et al., 2007).
TLR4-mediated NFκB signaling regulates the expression of pro-inflammatory cytokines, which possess a wide range of biological actions in brain tissue. Inflammatory processes are known to be associated with the acute cerebral ischemic condition, including the activation of resident brain cells such as microglial cells localized within the ischemic region, and the rapid synthesis of cytokines and chemokines (Barone and Feuerstein, 1999). An increased level of pro-inflammatory cytokines is an early feature of acute brain injury induced by clinical and experimental cerebral ischemia (Berti et al., 2002; Zaremba and Losy, 2004). To investigate whether TLR4 deficiency will prevent the production of inflammatory cytokines following GCI/R, we examined IL-6, TNFα, Fas ligand and high mobility group box 1 (HMGB1) in HF tissue from TLR4-/- and WT mice. TNF-α is one of the key immunomodulatory and pro-inflammatory cytokines upregulated during brain ischemia (Barone et al., 1997). Administration of TNF-α during an ischemic brain insult has been shown to augment the injury, as evidenced by increased tissue damage and neurological deficits (Barone et al., 1997). In addition to inflammation, TNF-α has also been shown to be involved in apoptosis (Wang and Shuaib, 2002). Fas receptor, a surface receptor belonging to the TNF-α receptor family, binds to the Fas ligand (Fas-L). The Fas/Fas-L signaling system activates Fas-associated death domain (FADD) and caspase-8, which in turn activates caspase-3 leading to cell death (Martin-Villalba et al., 1999). Fas-mediated death signaling may play an important role in hippocampal neuronal death after GCI/R (Jin et al., 2001). Recent studies have shown that high mobility group box 1 (HMGB1), originally described as a DNA-binding protein, can also be released extracellularly and functions as a novel proinflammatory cytokine-like factor that connects excitotoxicity-induced acute damage processes and delayed inflammatory processes in the postischemic brain (Kim et al., 2006). Our results demonstrated that HF expression of IL-6, TNFα, Fas-L and HMGB1 was increased at 6 hours after GCI/R in WT mice (Fig. 6) but not in TLR4-deficient mice. We speculate that the lack of inflammatory cytokine up regulation in the HF of TLR4-/- mice after GCI/R may be one mechanism responsible for attenuated neuronal damage. Moreover, it has been suggested that HMGB1 can interact with TLR4 and induce cellular activation and generate inflammatory responses (Park et al., 2006). Indeed, neutralizing antibodies against TLR4 can attenuate HMGB1-induced IL-8 and TNF release (Yu et al., 2006). Whether HMGB1 acts as an endogenous ligand that activates TLR4 signaling in ischemic brain tissue requires further investigation.
Recent evidence suggests that there was cross-talk between TLR signaling and the phosphoinositide 3-kinase (PI3K) /Akt pathway (Ojaniemi et al., 2003; Romaskova and Makarov, 1999). PI3K/Akt has been shown to prevent neuronal apoptosis and protect the hippocampus from ischemic injury (Endo et al., 2006). Akt is an important physiologic mediator of the PI3K pathway (Martin et al., 2005). Phosphorylation of Akt activates the enzyme which modulates cell cycle entry, growth and survival (Cantley, 2002). Activated Akt phosphorylates several downstream targets of the PI3K pathway including glycogen synthase kinase-3β (GSK3β) (Jope and Johnson, 2004; Martin et al., 2005). GSK3β is a crucial regulator of many cellular functions, including cell survival and apoptosis (Jope and Johnson, 2004). GSK3 is a constitutively active enzyme that is inactivated by Akt via phosphorylation (Jope and Johnson, 2004). The PI3K/Akt pathway has been reported to differentially regulate cytokine responses through GSK3β (Martin et al., 2005). Furthermore, inhibition of GSK3β can protect mice from endotoxic shock (Martin et al., 2005). Thus, it is possible that the PI3K/Akt/GSK3β signaling pathway could be responsible, in part, for the neuroprotection observed in TLR4-/- mice following GCI/R. In the present study, we have observed that the levels of phosporylated Akt and GSK3β in HF were higher in TLR4-/- mice compared with WT mice subjected to GCI/R (Fig. 7). Activation of the PI3K/Akt signaling pathway correlates with diminished ischemia-induced hippocampal neuronal damage. Further studies are required to establish a mechanistic link between activation of the PI3K pathway, modulation of TLR4-mediated signaling and neuroprotection in the ischemic brain.
In summary, the present study of GCI/R demonstrates that there was less neuronal damage in the HF of TLR4-/- mice compared to similarly treated WT mice. In WT mice subjected to GCI/R, TLR4-protein expression was increased in areas of the brain selectively vulnerable to transient ischemia, and TLR4-mediated signaling was activated, which correlates and likely contributes to the neuron death in the HF induced by GCI/R. Modulation of TLR4 signaling offers the possibility of attenuating the expression of pro-inflammatory cytokines and enhancing the activation of the PI3K/Akt pathway, thus protecting hippocampal neurons from ischemia injury.
Acknowledgments
This work was supported by AHA postdoctoral fellowship 0625348B to FH; ETSU RDC Grant to RLK; NIH GM53552 to DLW and NIH RO1 HL071837 to CL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- Barone F, Arvin B, White R, Miller A, Webb C, Willette R, Lysko P, Feuerstein G. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- Barone F, Feuerstein G. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Berti R, Williams A, Moffett J, Hale S, Velarde L, Elliott P, Yao C, Dave J, Tortella F. Quantitative real-time RT-PCR analysis of inflammatory gene expression associated with ischemia-reperfusion brain injury. J Cereb Blood Flow Metab. 2002;22:1068–1079. doi: 10.1097/00004647-200209000-00004. [DOI] [PubMed] [Google Scholar]
- Bottcher T, Von Mering M, Ebert S, Meyding-Lamade U, Kuhnt U, Gerber J, Nau R. Differential regulation of Toll-like receptor mRNAs in experimental murine central nervous system infections. Neurosci Lett. 2003;344:17–20. doi: 10.1016/s0304-3940(03)00404-x. [DOI] [PubMed] [Google Scholar]
- Brightbill HD, Modlin RL. Toll-like receptors: molecular mechanisms of the mammalian immune response. Immunology. 2000;101:1–10. doi: 10.1046/j.1365-2567.2000.00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The Phosphoinositide 3-Kinase Pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Cao CX, Yang QW, Ly FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Comm. 2006;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- Caso J, Pradillo J, Hurtado O, Lorenzo P, Moro M, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Liu D, Gonzales C, Zaleska MM, Wood A. Temporal assessment of caspase activation in experimental models of focal and global ischemia. Brain Res. 2003;982:146–155. doi: 10.1016/s0006-8993(03)02846-4. [DOI] [PubMed] [Google Scholar]
- Endo H, Nito C, Kamada H, Nichi T, Chan P. Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1479–1489. doi: 10.1038/sj.jcbfm.9600303. [DOI] [PubMed] [Google Scholar]
- Ha T, Hua F, Li Y, Ma J, Gao X, Kelley J, Zhao A, Haddad GE, Williams DL, Browder IW, Kao RL, Li C. Blockade of MyD88 Attenuates Cardiac Hypertrophy and Decreases Cardiac Myocyte Apoptosis in Pressure Overload Induced Cardiac Hypertrophy in vivo. Am J Physiol Heart Circ Physiol. 2006;290:H985–H994. doi: 10.1152/ajpheart.00720.2005. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-beta in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- Hua F, Ha T, Ma J, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. Blocking the MyD88-dependent pathway protects the myocardium from ischemia/reperfusion injury in rat hearts. Biochem Biophys Res Comm. 2005;338:1118–1125. doi: 10.1016/j.bbrc.2005.10.068. [DOI] [PubMed] [Google Scholar]
- Hua F, Ha T, Ma J, Li Y, Kelley J, Gao X, Browder IW, Kao RL, Williams DL, Li C. Protection against Myocardial Ischemia/Reperfusion Injury in TLR4 Deficient Mice is Mediated through a Phosphoinositide 3-Kinase Dependent Mechanism. J Immunol. 2007;178:7317–7324. doi: 10.4049/jimmunol.178.11.7317. [DOI] [PubMed] [Google Scholar]
- Hua F, Ma J, Li Y, Ha T, Xia Y, Kelley J, Williams DL, Browder IW, Schweitzer JB, Li C. The development of a novel mouse model of transiant global cerebral ischemia. Neuroscience Letters. 2006;400:69–74. doi: 10.1016/j.neulet.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Jin K, Graham S, Mao X, Nagayama T, Simon R, Greenberg D. Fas (CD95) may mediate delayed cell death in hippocampal CA1 sector after global cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1411–1421. doi: 10.1097/00004647-200112000-00005. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:96–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, Bonder C, James WG, Robbins SM, Kubes P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. 2004;173:7070–7077. doi: 10.4049/jimmunol.173.11.7070. [DOI] [PubMed] [Google Scholar]
- Kim BS, Lim SW, Li C, Kim JS, Sun BK, Ahn KO, Han SW, Kim J, Yang CW. Ischemia-reperfusion injury activates innate immunity in rat kidneys. Transplantation. 2005;79:1370–1377. doi: 10.1097/01.tp.0000158355.83327.62. [DOI] [PubMed] [Google Scholar]
- Kim J, Sig Choi J, Yu Y, Nam K, Piao C, Kim S, Lee M, Han P, Park J, Lee J. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26:6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Gregersen R, Meldgaard M, Clausen BH, Heibol EK, Ladeby R, Knudsen J, Frandsen A, Owens T, Finsen B. A role for interferon-gamma in focal cerebral ischemia in mice. J Neuroparthol Exp Neurol. 2004;63:942–955. doi: 10.1093/jnen/63.9.942. [DOI] [PubMed] [Google Scholar]
- Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Ha TZ, Chen Q, Li CF. Role of MyD88-dependent nuclear factor-kappaB signaling pathway in the development of cardiac hypertrophy in vivo. Zonghua Yi Xue Za Zhi. 2005;85:267–272. [PubMed] [Google Scholar]
- Liao SL, Chen WY, Raung SL, Kuo JS, Chen CJ. Association of immune responses and ischemic brain infarction in rat. Neuroreport. 2001;12:1943–1947. doi: 10.1097/00001756-200107030-00034. [DOI] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nature Immunology. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, Schenkel J, Herdegen T, Debatin KM. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslinska D, Laure-Kamionowski M, Maslinski S. Toll-like receptors in rat brains injured by hypoxic-ischaemia or exposed to staphylococcal alpha-toxin. Folia Neuropathol. 2004;42:125–132. [PubMed] [Google Scholar]
- Muller G, Stadelmann C, Bastholm L, Elling F, Lassmann H, Johansen F. Ischemia leads to apoptosis--and necrosis-like neuron death in the ischemic rat hippocampus. Brain Pathol. 2004;14:415–424. doi: 10.1111/j.1750-3639.2004.tb00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskovitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Ychiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- Park J, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim J, Strassheim D, Sohn J, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:917–C924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Romaskova JA, Makarov SS. NF-κB is a target of AKT in anti-apoptotic PDGF signaling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Shimamoto A, Pohlman TH, Shomura S, Tarukawa T, Takao M, Shimpo H. Toll-like receptor 4 mediates lung ischemia-reperfusion injury. Ann Thorac Surg. 2006;82:2017–2023. doi: 10.1016/j.athoracsur.2006.06.079. [DOI] [PubMed] [Google Scholar]
- Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- Tounai H, Hayakawa N, Kato H, Araki T. Immunohistochemical Study on Distribution of NF-kappaB and p53 in Gerbil Hippocampus after Transient Cerebral Ischemia: Effect of Pitavastatin. Metab Brain Dis. 2007;22:89–104. doi: 10.1007/s11011-006-9040-3. [DOI] [PubMed] [Google Scholar]
- Wang C, Shuaib A. Involvement of Inflammatory cytokines in central nervous system injury. Prog Neurobiol. 2002;67:161–172. doi: 10.1016/s0301-0082(02)00010-2. [DOI] [PubMed] [Google Scholar]
- Yu M, Wang H, Ding A, Golenbock D, Latz E, Czura C, Fenton M, Tracey K, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- Zaremba J, Losy J. Cytokines in clinical and experimental ischemic stroke. Neurol Neurochir Pol. 2004;38:S57–S62. [PubMed] [Google Scholar]
- Zhai Y, Shen X, O'Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Cutting Edge: TLR4 Activation Mediates Liver Ischemia/Reperfusion Inflammatory Response via IFN Regulatory Factor 3-Dependent MyD88-Independent Pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- Zhang G, Ghosh S. Toll-like receptor-mediated NF-κB activation: a phylogenetically conserved paradigm in innate immunity. The Journal of Clinical Investigation. 2001;107:13–19. doi: 10.1172/JCI11837. [DOI] [PMC free article] [PubMed] [Google Scholar]