

Abstract
For over 15 years, the peripheral benzodiazepine receptor (PBR), recently named translocator protein 18kDa (TSPO) has been studied as a biomarker of reactive gliosis and inflammation associated with a variety of neuropathological conditions. Early studies documented that in the brain parenchyma, TSPO is exclusively localized in glial cells. Under normal physiological conditions, TSPO levels are low in the brain neuropil but they markedly increase at sites of brain injury and inflammation making it uniquely suited for assessing active gliosis. This research has generated significant efforts from multiple research groups throughout the world to apply TSPO as a marker of “active” brain pathology using in vivo imaging modalities such as Positron Emission Tomography (PET) in experimental animals and humans. Further, in the last few years, there has been an increased interest in understanding the molecular and cellular function(s) of TSPO in glial cells. The latest evidence suggests that TSPO may not only serve as a biomarker of active brain disease but also the use of TSPO-specific ligands may have therapeutic implications in brain injury and repair. This review presents an overview of the history and function of TSPO focusing on studies related to its use as a sensor of active brain disease in experimental animals and in human studies.
Keywords: peripheral benzodiazepine receptor, translocator protein 18kDa, TSPO-brain injury, neurodegeneration, biomarker, PET
1. Reactive Gliosis as a Biomarker of Brain Injury
Reactive gliosis comprises the activation of microglia and astrocytes and is a hallmark response of the CNS to injury (Ladeby et al., 2005; McGraw et al., 2001; Norenberg 2004; Norton et al., 1992; O’Callaghan, 1991; O’Callaghan, 1993; O’Callaghan 2005; Raivich et al., 1999; Sriram and O’Callaghan 2004; Streit et al., 1999; Streit 2000; Streit 2004; Streit et al., 2005). Reactive gliosis has a graded morphological response that is directly associated with the degree of damage in all forms of brain pathology (Raivich et al., 1999).
Reactive gliosis based on morphological examination is a microscopic finding in brain tissue sections and can only be obtained either from invasive biopsy or postmortem autopsy. Therefore, the development and validation of an in vivo biomarker of reactive gliosis is a major advance in the detection of active CNS disease, to monitor disease progression and to assess the effectiveness of therapeutic interventions. To this end, the peripheral benzodiazepine receptor (PBR) or translocator protein (18kDa)(TSPO), a new nomenclature for PBR (Papadopoulos et al., 2006b), is located exclusively in glial cells in the brain parenchyma and has been used as a sensitive biomarker of reactive gliosis and inflammation associated with a variety of brain insults including chemical-induced neurotoxicity (Guilarte et al., 1995; Guilarte et al., 2003; Kuhlmann and Guilarte, 1997; Kuhlmann and Guilarte, 1999; Kuhlmann and Guilarte, 2000; Chen et al., 2004; Chen & Guilarte, 2006), ischemic stroke (Gerhard et al., 2000; Gerhard et al., 2005; Pappata et al., 2000; Stephenson et al., 1995), physical trauma (Miyazawa et al., 1995; Raghavendra Rao et al., 2000), CNS degenerative diseases (Cagnin et al., 2001a; Cagnin et al., 2004; Gerhard et al., 2003; Gerhard et al., 2006; Chen et al., 2007; Henkel et al., 2004; Ouchi et al., 2005; Versijpt et al., 2003) and CNS inflammatory disease (Banati et al., 2000; Debruyne et al., 2003; Hammoud et al., 2005; Mankowski et al., 2003; Vowinckel et al., 1997) to name a few (see Tables 1 and 2). Importantly, increased TSPO levels following brain injury are specific to primary or secondary areas of injury expressing activated glial cells, and TSPO can be visualized and quantified using in vitro and in vivo imaging techniques (Banati et al., 2000; Cagnin et al., 2001a; Cagnin et al., 2001b; Chen et al., 2004; Gerhard et al., 2005; Kuhlmann and Guilarte, 1997; Kuhlmann and Guilarte, 1999; Kuhlmann and Guilarte, 2000; Mankowski et al., 2003; Pappata et al., 2000; Versijpt et al., 2003). Therefore, this approach offers great potential for in vivo imaging of a wide variety of neuropathological conditions.
Table 1.
Reactive gliosis and TSPO expression in experimental animal models.
| Animal models | Reactive gliosis and TSPO expression | Reference |
|---|---|---|
| 6-OHDA induced Parkinsonism | Striatum | Cicchetti et al., 2006 |
| cuprizone-induced demyelination | corpus callosum, fiber bundles in striatum, and cerebellar deep nuclei | Chen et al., 2004 |
| cuprizone-induced demyelination and remyelination | corpus callosum | Chen and Guilarte, 2006 |
| demoic acid neurotoxicity | hippocampus, subiculum, dentate gyrus, amygdala, striatum | Kuhlmann and Guilarte, 1997 |
| experimental autoimmune encephalomyelitis | multiple sclerosis plaques | Vowinckel et al., 1997 |
| experimental autoimmune encephalomyelitis | multiple sclerosis plaques | Banati et al., 2000 |
| facial nerve axotomy | facial nucleus | Banati et al., 1997 |
| facial nerve axotomy | facial nucleus | Gehlert et al., 1997 |
| ischemia | peripheral of infarct zone | Benavides et al., 1990 |
| ischemia | Forebrain | Demerle-Pallardy et al., 1991 |
| ischemia | cerebral cortex | Myers et al., 1991 |
| kainic acid injection | olfactory/limbic | Altar and Baudry, 1990 |
| methamphetamine | striatum, thalamus, hippocampus, dorsal raphe/central gray | Guilarte et al., 2003 |
| MPTP induced Parkinsonism | striatum and substantia nigra | Kuhlmann and Guilarte, 1999 |
| MPTP induced Parkinsonism | cerebral cortex and subcortical regions including caudate/putamen | Chen et al., 2007 (in press) |
| sciatic nerve degeneration and regeneration | sciatic nerve | Lacor et al., 1999 |
| simian immunodeficiency virus encephalitis | frontal cortical white matter | Mankowski et al., 2003 |
| simian immunodeficiency virus encephalitis | frontal cortical white/gray matter, basal ganglion, hippocampus | Venneti et al., 2004 |
| stab wounds | injury site | Miyazawa et al., 1995 |
| transient ischemia | Forebrain | Stephenson et al., 1995 |
| transient ischemia | forebrain | Rao et al., 2001 |
| transient ischemia | infarct core and peripheral zone | Rojas et al., 2007 |
| traumatic brain injury | thalamus | Raghavendra Rao et al., 2000 |
| trimethyltin | hippocampus, olfactory cortex, amygdaloid nucleus, subiculum, and entorhinal cortex | Guilarte et al., 1995 |
| trimethyltin | hippocampus | Kuhlmann and Guilarte, 2000 |
Table 2.
In vivo TSPO expression in human neurological disorder.
| Human neurological disorder | TSPO expression | Reference |
|---|---|---|
| AIDS dementia | thalamus, putamen, frontal, temporal, and occipital cortex | Hammoud et al., 2005 |
| Alzheimer’s dementia | entorhinal, temporoparietal, and cingulate cortex | Cagnin et al., 2001a |
| Alzheimer’s dementia | frontal and mesotemporal cortex | Versijpt et al., 2003 |
| amyotrophic lateral sclerosis | motor cortex, prefrontal cortex, pons, thalamus | Turner et al., 2004 |
| cerebral vasculitis | occipital, temporoparietal cortex | Goerres et al., 2001 |
| corticobasal degeneration | caudate/putamen, substantia nigra, pons, pre- postcentral gyrus, and frontal cortex | Gerhard et al., 2004 |
| corticobasal degeneration | basal ganglia, temporal and parietal cortex | Henkel et al., 2004 |
| frontal temporal dementia | frontal temporal cortex | Cagnin et al., 2004 |
| herpes encephalitis | primary and secondary projected neuron | Cagnin et al., 2001b |
| Huntington’s disease | putamen, frontal cortex | Messmer et al., 1998 |
| Huntington’s disease | caudate/putamen, cortical regions including prefrontal cortex and anterior cingulate | Pavese et al., 2006 |
| Huntington’s disease carrier(presymptomatic) | caudate/putamen, cortical regions | Tai et al., 2007 |
| idiopathic Parkinson’s disease | midbrain | Ouchi et al., 2005 |
| idiopathic Parkinson’s disease | pons, basal ganglia, frontal and temporal cortex | Gerhard et al., 2006 |
| ischemic stroke | cerebral cortex | Ramsay et al., 1992 |
| ischemic stroke | cerebral cortex | Gerhard et al., 2000 |
| ischemic stroke | primary lesion and remote pathological changes following Wallerian degeneration | Gerhard et al., 2005 |
| ischemic stroke | thalamus | Pappata et al., 2000 |
| ischemic stroke | peri-infact zone | Price et al., 2006 |
| multiple sclerosis | multiple sclerosis plaques, cerebral central gray | Banati et al., 2000 |
| multiple sclerosis | normal-appearing white matter | Debruyne et al., 2003 |
| multiple sclerosis | normal-appearing white matter | Versijpt et al., 2005 |
| multiple system atrophy | prefrontal cortex, putamen, pallidum, pons, and substantia nigra | Gerhard et al., 2003 |
| progressive supranuclear palsy | basal ganglia, midbrain, frontal cortex, and cerebellum | Gerhard et al., 2006 |
| Rasmussen’s encephalitis | affected hemisphere | Banati et al., 1999 |
2. The Peripheral Benzodiazepine Receptor/Translocator protein 18kDA-what is it?
Benzodiazepines are one of the most commonly prescribed drugs that have anxiolytic, anticonvulsant, muscle-relaxant, and hypnotic properties. Some of these therapeutic effects are mediated via specific benzodiazepine receptors located in the CNS. The central benzodiazepine receptor (CBR) is coupled to the γ-aminobutyric acid (GABA)A receptor and modulates GABA-regulated opening of Cl− channels and inhibition of neuronal activity (Tallman et al., 1978; Tallman et al., 1980). In addition to CBR, another type of benzodiazepine receptor was identified using rat kidneys as control tissue during CBR binding studies using [3H]-diazepam (Braestrup and Squires, 1977; Schoemaker et al., 1981). This “peripheral” [3H]-diazepam binding site was shown to be abundantly distributed in peripheral tissues and was defined as peripheral-type benzodiazepine binding site (PBBS) or peripheral benzodiazepine receptor (PBR). Subsequently, studies demonstrated the presence of the PBR in glial and in ependymal cells of the brain (Richards and Mohler, 1984; Gavish et al., 1999). It was also determined that this “peripheral” site that binds diazepam was pharmacologically, anatomically, structurally, and physiologically distinct from the CBR (Gavish et al., 1999; Woods and Williams 1996). Despite the fact that for many years, this nomemcleature was used, it was misleading and a group of scientists studying the PBR decided to rename the protein to best represent its function and reached a consensus on a new nomenclature: translocator protein (18 kDa) (TSPO) (Papadopoulos et al., 2006b).
2.1. Molecular Properties of TSPO
TSPO is an 18 kDa protein consisting of 169 amino-acids (Casellas et al., 2002). It is highly hydrophobic and rich in tryptophan (Casellas et al., 2002). The cDNA encoding TSPO has been cloned from various species such as rodents, bovines and humans (Chang et al., 1992; Garnier et al., 1994; Parola et al., 1991; Riond et al., 1991; Sprengel et al., 1989) and there is an 80% sequence homology amongst these species (Casellas et al., 2002). The location of the TSPO gene is in the q13.3 region of the long arm of human chromosome 22 (Riond et al., 1991).
TSPO can form a multimeric complex with the 32 kDa voltage-dependent anion channel (VDAC) also called mitochondrial porin and the 30 kDa adenine nucleotide carrier (ANC) (McEnery et al., 1992) in the outer mitochondrial membrane (Anholt et al., 1986; Bribes et al., 2004). Topographic studies of TSPO reveal that the 18kDa TSPO subunit is organized in clusters of 4–6 molecules associated with one VDAC subunit (Papadopoulos et al., 1990; Papadopoulos et al., 1994; Papadopoulos et al., 1997). Studies using three-dimensional modeling reveal TSPO as a structure with five α-helices spanning one phospholipid layer of the mitochondrial membrane (Bernassau et al., 1993).
There are two other TSPO associated proteins, PBR and protein kinase A associated protein 7 (PAP7) and steroidogenic acute regulatory protein (StAR) localized in steroidogenic tissue and participating in steroid synthesis (Lacapere and Papadopoulos, 2003). StAR can regulate the transport of cholesterol by binding cholesterol in the cytoplasm and transferring it to TSPO on the outer membrane of mitochondria (Lacapere and Papadopoulos, 2003). Phosphorylation of StAR by PKA is facilitated by interaction with PAP7. Furthermore, PAP7 contains an acyl-CoA motif and has sequence identity to diazepine binding inhibitor (DBI), suggesting a common site of interaction between PAP7, DBI and TSPO (Lacapere and Papadopoulos, 2003). A schematic of TSPO and associated proteins is presented in Figure 1.
Figure 1.
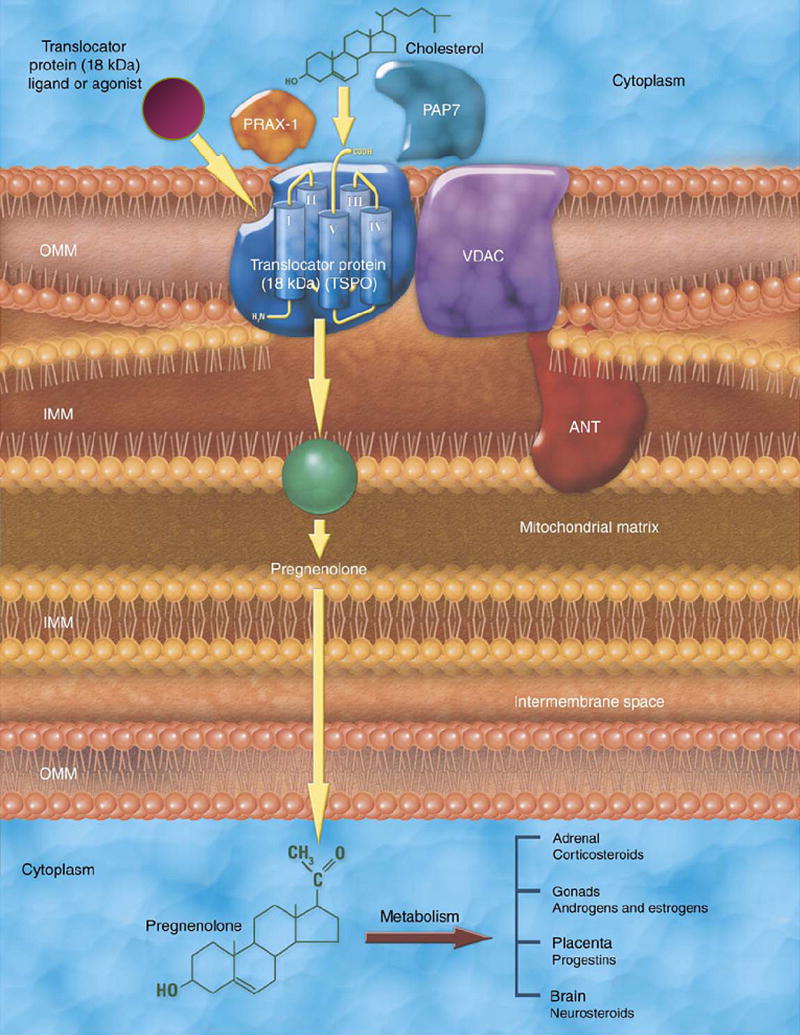
Schematic of TSPO and associated proteins. Published with permission from Elsevier Limited. Taken from Papadopoulos et al., TIPS 27: 402–409, 2006.
2.2. TSPO pharmacology
The TSPO binding site has distinct pharmacological properties from the CBR, although diazepam binds to both classes of receptors with high affinity. Certain benzodiazepines, such as 7-chloro-5-(4-chlorophenyl)-1-methyl-1,3-dihydrobenzo[e][1,4] diazepin-2-one (Ro5-4864), interact weakly with the CBR but have nanomolar affinity for TSPO (Benavides et al., 1983a; Benavides et al., 1983b; Schoemaker et al., 1981). In contrast, clonazepam binds with high affinity to CBR but binds with extremely low affinity to TSPO. On the other hand, 1-(2-chlorophenyl)-N-methyl-(1-methypropyl)-3-isoquinoline carboxamide (PK11195), an isoquinoline carboxamide derivative, is the first non-benzodiazepine ligand found to bind TSPO with nanomolar affinity (Le Fur et al., 1983a; 1983b; 1983c). While the binding affinity of Ro5-4864 for TSPO varies across species (Bolger et al., 1985), PK11195 displays high affinity in all species (Casellas et al., 2002). This suggests that their binding domains are overlapping but not identical (Farges et al., 1993). Thermodynamic analysis indicates that the [3H]-PK11195 binding is entropy driven, whereas the [3H]-RO5-4864 binding is enthalpy driven (Le Fur et al., 1983c). Therefore PK-11195 might be an antagonist of TSPO, and RO5-4864 might be an agonist or a partial agonist (Le Fur et al., 1983c).
Further studies demonstrated that TSPO binds with high affinity to other classes of organic compounds, such as phenoxyphenyl-acetamides, pyrazolopyrimidine, indolacetamide, and imidiazopyridines, which could prospectively act as potential selective TSPO ligands (James et al., 2006 and Section 4.7).
2.3. Endogenous TSPO ligands
A wide variety of endogenous molecules that bind to TSPO have been identified. One putative endogenous ligand is the diazepam binding inhibitor (DBI) or endozepine (Guidotti et al., 1983). As its name suggests, DBI was originally described based on its ability to inhibit the binding of [3H]-diazepam to CBR (Guidotti et al., 1983). DBIs are widely distributed in the CNS (predominantly in glial cells) and in peripheral organs, especially steroidogenic cells (Alho et al., 1991; Alho et al., 1994; Bovolin et al., 1990; Lihrmann et al., 1994; Malagon et al., 1993). DBIs have similar micromolar (μM) affinities for both TSPO and CBR. DBI is a 10-kDa peptide of 86 amino-acids which can be further cleaved into several biologically active fragments including octadecaneuropeptide (ODN or DBI33–50), eicosaneuropeptide (ENP or DBI26–50,) and triakontatetraneuropeptide (TTN or DBI17–50,) (Ferrero et al., 1986; Slobodyansky et al., 1989). TTN has similar affinity for TSPO as DBI but is more selective, whereas the ODN is less potent (Ferrero et al., 1986; Slobodyansky et al., 1989).
DBIs can stimulate steroidogenesis by interacting with TSPO (Besman et al., 1989). It has been found that cultured rat astrocytes contain and release DBI-related peptides especially TTN (Lamacz et al., 1996; Patte et al., 1999). TTN can stimulate neurosteroid synthesis in C6 glioma cells by acting on TSPO (Papadopoulos et al., 1991). Later studies confirm the identical sequences between DBI and the acyl coenzyme-A binding protein (ACBP), suggesting the potential role of DBIs in fatty acid metabolism (Knudsen et al., 1993; Knudsen, 1991).
Cholesterol is also considered an endogenous ligand for TSPO with nanomolar affinity (Lacapere et al., 2001). Cholesterol can bind to the cholesterol recognition/interaction amino acid consensus (CRAC) sequence in the carboxyl terminus of TSPO for transport to the mitochondrial inner-membrane for subsequent steroidogenesis (Li et al., 2001).
There are other potential endogenous ligands such as the porphyrins (protoporphyrin IX, mesoporphyrin IX, deuteroporphyrin IX, hemin), which exhibit a very high (nM) affinity for TSPO but not for CBR (Snyder et al., 1987; Verma et al., 1987). Porphyrins are tetrapyrrolic pigments formed in the biosynthesis pathway of heme, mitochondrial cytochrome, hemoglobin, and other heme proteins (Verma and Snyder, 1989). The concept that porphyrins are endogenous ligands fits with the mitochondrial location of TSPO since the initial and final steps in porphyrin biosynthesis occur within the mitochondria (Verma and Snyder, 1989). This also implicates one of the potential physiological functions of TSPO related to heme biosynthesis (Verma and Snyder, 1989).
2.4. Subcellular location and tissue distribution of TSPO
The subcellular localization of TSPO has been demonstrated primarily in the outer mitochondrial membrane by use of the selective ligand [3H]-PK11195 binding studies in rat adrenal glands (Anholt et al., 1986), as well as in rat testis, lung, kidney, heart, liver, and skeletal muscle (Antkiewicz-Michaluk et al., 1988). Later studies using TSPO immunohistochemistry with confocal microscopy (Garnier et al., 1994) and electron microscopy (Bribes et al., 2004) confirmed the outer mitochondrial membrane localization of TSPO. Although TSPOs are primarily located in the mitochondria, one study demonstrates TSPOs in red blood cells, which are devoid of mitochondria (Olson et al., 1988). This result indicates that TSPOs might also be located in a non-mitochondrial fraction (Olson et al., 1988). Other studies also suggest that small numbers of TSPO may be localized to the plasma membrane of certain peripheral organs, such as liver (O’Beirne et al., 1990; Woods et al., 1996). And there are studies demonstrating that TSPOs are located in the nucleus and perinuclear area in malignant human breast cancer cell lines (Hardwick et al., 1999), glial cells in the CNS (Kuhlmann and Guilarte, 2000), human glioma cell lines (Brown et al., 2000b) and hepatic tumor cells (Corsi et al., 2005).
Radioligand binding assays and autoradiography using either of the selective TSPO tritium labeled ligands Ro5-4864 or PK-11195 have been used to detect the anatomical distribution of TSPO in the body. Glandular and secretory tissues such as the pineal gland, adrenal glands, salivary glands, olfactory epithelium and gonads are particularly abundant in TSPO with renal and myocardial tissue showing intermediate levels (Gavish et al., 1999). In contrast, the liver and brain express relatively low levels of TSPO (Gavish et al., 1999). The tissue distribution of TSPO is not homogeneous within a given organ. In the adrenal glands, the medulla is devoid of TSPO, whereas density in the adrenal cortex is very high (Anholt et al., 1986). In rat liver, the mitochondrial-form of TSPO is located in hepatocytes and the non-mitochondrial-form (plasma) of TSPO is located in biliary epithelial cells (Woods et al., 1996). TSPO is also expressed in circulating blood cells with the highest concentrations in monocytes and polymorphonuclear neutrophils (PMN) (Canat et al., 1993).
2.5. Physiological functions of TSPO
Many physiological functions have been attributed to TSPO, including cell growth and proliferation (Carmel et al., 1999; Lee et al., 2004; Wang et al., 1984), steroidogenesis (Kelly-Hershkovitz et al., 1998; Lacapere and Papadopoulos, 2003; Papadopoulos et al., 1990; Papadopoulos et al., 1991; Papadopoulos, 1998), bile acid synthesis (Lacapere and Papadopoulos, 2003; Woods and Williams, 1996; Woods et al., 1996), calcium flow (Azarashvili et al., 2005; Hong et al., 2006), chemotaxis and cellular immunity (Lenfant et al., 1986; Ruff et al., 1985), heme biosynthesis (Verma and Snyder, 1989), and mitochondrial respiration and apoptosis (Casellas et al., 2002; Hirsch et al., 1989; Hirsch et al., 1998). The fact that TSPO knockout (KO) mice die at an early embryonic stage (Papadopoulos et al., 1997) strongly suggests that TSPO is involved in basic house-keeping functions and is essential for embryonic development.
Amongst the potential physiological functions of TSPO, steroidogenesis is the best characterized. TSPO is abundantly expressed in steroidogenetic tissues where TSPO mediates cholesterol transport into mitochondria (Papadopoulos et al., 1997). TSPO ligands have been shown to stimulate steroidogenesis in adrenal, placental, testicular, ovarian and glial systems (Papadopoulos et al., 1997). TSPO can bind cholesterol and facilitate the transport of cholesterol from the outer to the inner mitochondrial membrane, the rate-determining step of steroidogenesis (Papadopoulos et al., 1997). The side chain cleavage cytochrome P-450 enzyme (P-450scc or CYP11A1), located in the inner mitochondrial membrane, can convert cholesterol to pregnenolone (PREG), a steroid precursor, and initiate steroidogenesis such as neurosteroid production in glial cells (Brown and Papadopoulos, 2001; Brown et al., 2000a; Papadopoulos and Brown, 1995; Papadopoulos, 1998). More recently, studies have also shown that in astrocytes high concentrations of TSPO specific ligands are able to induce changes in intracellular cholesterol trafficking that are not necessarily related to steroid biosynthesis (Falchi et al., 2007). Therefore, it is highly likely that other cholesterol-related functions for TSPO and/or its ligands are yet to be discovered.
Because of its location on the outer membrane of mitochondria and its association with VDAC, TSPO is suggested to be involved in a multimeric protein complex known as the mitochondrial permeability transition (MPT) pore (Casellas et al., 2002; Kinnally et al., 1993). The MPT pore performs like Ca2+, H+ and redox-gated channels with multiple levels of conductance and low selectivity (Kroemer and Reed, 2000). The MPT pore is maintained by the mitochondrial inner membrane potential and the matrix pH, which are regulated by several mitochondrial proteins such as the apoptosis-inhibitory oncoprotein Bcl-2 (Casellas et al., 2002; Hirsch et al., 1998; Kroemer and Reed, 2000). Opening of the MPT pore leads to colloid osmotic swelling of the mitochondrial matrix, defective oxidative phosphorylation, cessation of ATP synthesis and the generation of free radicals (Kroemer and Reed, 2000). The collapse of the mitochondrial inner membrane potential is a critical initiating event for the apoptotic cascades, which include the increase or swelling of the mitochondrial matrix, disruption of the mitochondrial membrane, and the release of cytochrome c and apoptosis inducing factor (AIF) (Casellas et al., 2002; Kroemer and Reed, 2000). AIF can induce nuclear chromatin condensation and DNA fragmentation (Kroemer and Reed, 2000). Cytochrome c can interact with apoptosis activating factor 1(Apaf-1) and pro-caspase 9, cause the activation of caspase 9, which subsequently activates caspase 3 and a series of enzymes causing programmed cell death (Kroemer and Reed, 2000). Myxoma poxvirus M11L is an anti-apoptotic protein localized in the mitochondria and can regulate the MPT pore complex by direct modulation of TSPO, therefore preventing the loss of mitochondrial inner membrane potential in response to induction of apoptosis (Everett et al., 2002). This provides direct evidence that TSPO is involved in the modulation of apoptosis (Everett et al., 2002; Veenman et al., 2007).
3. TSPO: A molecular sensor of active brain disease
Under normal physiological conditions TSPO levels in the CNS are very low and limited to glial cells (astrocytes and microglia). A dramatic increase in TSPO levels occurs in glial cells in response to brain injury or inflammation (Chen et al., 2004; Chen and Guilarte, 2006; Guilarte et al., 1995; Guilarte et al., 2003; Kuhlmann and Guilarte, 1997; Kuhlmann and Guilarte, 1999; Kuhlmann and Guilarte, 2000). Because of the availability of high affinity and selective ligands such as PK-11195, which can be labeled with various radioisotopes (3H, 11C, 123I and 125I), the distribution of TSPO can be visualized and measured using in vitro receptor autoradiography and binding assays as well as in vivo imaging techniques, such as PET or SPECT. The fact that TSPO levels are low in the brain parenchyma and regionally increase in the injured brain makes TSPO an ideal and sensitive marker to detect small changes in the region of injury in comparison to the low levels in normal regions. Time-dependent and region-specific increases of TSPO levels have been measured in areas with neuronal loss or axonal injury following exposure to neurotoxicants (Altar and Baudry, 1990; Benavides et al., 1987; Guilarte et al., 1995; Guilarte et al., 2003; Kuhlmann and Guilarte, 1997; Kuhlmann and Guilarte, 1999; Kuhlmann and Guilarte, 2000) (see Table 1). For example, Figure 2 demonstrates pseudocolor images of [3H]-PK11195 binding to TSPO in rodent brain. Panels A-C represents the temporal TSPO response to the degenerative effects of a single injection of the neurotoxicant TMT, a toxicant whose primary target is the limbic system. In these images, the blue color areas in panel A (control) represent low levels of binding with the notable exception being the lining of the ventricles and choroid plexus that express high levels of binding or red color. In panel B, at 14 days after TMT, there is extensive upregulation of TSPO (red and yellow areas) in the piriform cortex and in the hippocampus, in particular the CA4 region of the dentate gyrus. However, at 6 weeks (panel C), the pattern of TSPO is different with the CA4 and CA1 regions of the hippocampus being more prominent. Also see the progressive increase in TSPO binding in the thalamus (A-C). These changes in TSPO levels following TMT are consistent and correlate with the known neuropathology of this agent in rodent brain (Guilarte et al., 1995). Panels D and E are representative of a control and an animal following seizure elicited by domoic acid, respectively. Note the prominent increase of TSPO in the hippocampus of the domoic acid treated animal. Finally, panels F and G are representative of a control (F) and an animal in which cuprizone, a demyelinating agent was included in the diet for 4 weeks (G). There is increased TSPO in the dorsal hippocampal commisure and in the corpus callosum, brain regions known to exhibit demyelination in this model. These images are just a few examples of the power of using quantitative autoradiography to assess brain injury in rodent models.
Figure 2.

Pseudocolor images of [3H]-R-PK11195 binding to TSPO in rodent brain. Color in images represents levels of [3H]-R-PK11195 binding with blue representing low levels, green-yellow representative of intermediate levels, and red high levels of binding. Panels A, D, F are pseudocolor images from control animals and B, C, E, G are from animals treated with different neurotoxicants. Panels B and C represent levels of TSPO following a single injection (8 mg/kg) of the neurotoxicant trimethyltin. Panel B represents levels of TSPO at 14 days after TMT administration and panel C after 6 weeks following TMT administration. There is a dramatic increase in hippocampal regions and piriform cortex at 14 days with a marked increase in different hippocampal regions at 6 weeks. See progressive increase in the thalamus. Panel E is representative of an animal that had a seizure after domoic acid administration (3 mg/kg). Compare image in E to control in D. Panel G represents a mouse that had been administered cuprizone in the diet for 4 weeks. Increased levels of TSPO are noted in the dorsal hippocampal commisure and in the hippocampus proper, two brain regions that are known to develop dmyelination in this model. D3V = dorsal third ventricle; 3V = third ventricle; LV = lateral ventricle; Th = thalamus; PC = piriform cortex; CA4 = CA4 region of the hippocampus; CA3 = CA3 region of the hippocampus; CA1 = CA1 region of the hippocampus; Ctx = cerebral cortex; Cb = cerebellum; hipp = hippocampus; cc = corpus callosum; dhc = dorsal hippocampal commisure.
Besides its use to assess the temporal and spatial pattern of glial cell activation in neurotoxicant-induced injury, TSPO has been used as a biomarker of brain injury and inflammation in neurodegenerative diseases (Cagnin et al., 2001a; Cagnin et al., 2004; Gerhard et al., 2003; Gerhard et al., 2004; Gerhard et al., 2006; Messmer and Reynolds, 1998; Ouchi et al., 2005; Pavese et al., 2006), in ischemia or stroke (Demerle-Pallardy et al., 1991; Gerhard et al., 2005; Myers et al., 1991; Pappata et al., 2000; Price et al., 2006; Ramsay et al., 1992; Stephenson et al., 1995), in inflammatory diseases such as multiple sclerosis and experimental autoimmune encephalitis (Banati et al., 2000; Debruyne et al., 2002; Debruyne et al., 2003; Mattner et al., 2005; Versijpt et al., 2005; Vowinckel et al., 1997), virus induced encephalitis (Cagnin et al., 2001b; Hammoud et al., 2005; Mankowski et al., 2003; Venneti et al., 2004) and in physical trauma (Miyazawa et al., 1995; Raghavendra Rao et al., 2000; Rao et al., 2001). Therefore, the use of TSPO as a biomarker of brain injury using ex vivo brain tissue is well validated (see Table 1).
The nature of the increase in TSPO measured in these models of brain injury has been studied using radioligand binding studies. Saturation isotherms and Scatchard analysis of [3H]-PK11195 binding to TSPO demonstrate that this service and the increase in binding is due to an increase in the maximal number of [3H]-PK11195 binding sites (Bmax) with no change in the affinity of TSPO to this radioligand (Kd) (See Figure 3)(Banati et al., 2000; Chen et al., 2004; Guilarte et al., 1995; Kuhlmann and Guilarte, 1997 & 1999).
Figure 3.
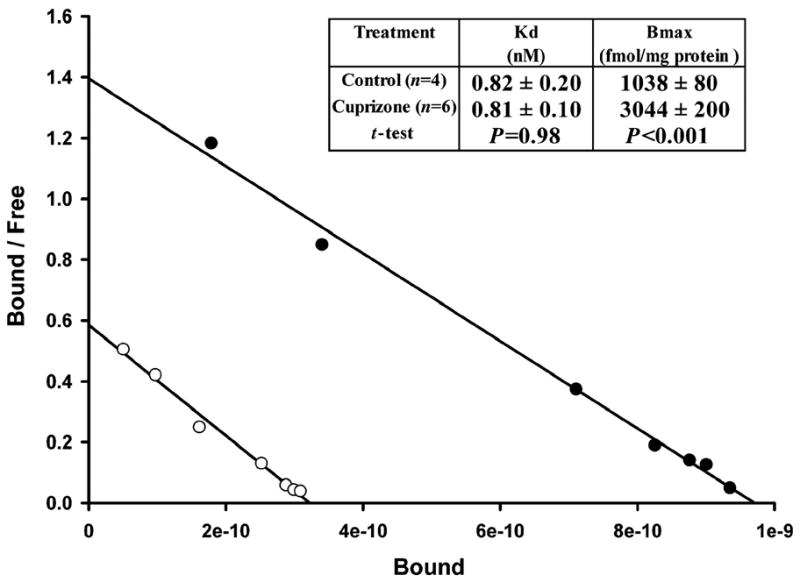
Scatchard analysis of [3H]-R-PK11195 binding to TSPO in the cerebral cortex of animals exposed to cuprizone for 4 weeks. The data clearly show that the effect of cuprizone treatment on [3H]-R-PK11195 specific binding is in the maximal number of binding sies (Bmax) and not in the affinity (Kd). Published with permission from Oxford University Press. Taken from Chen et al., Brain 127: 1379–1392, 2004.
Despite many studies showing an increase of TSPO as a result of brain injury the mechanisms and physiological implications of how and why TSPO responds to brain injury are still not known. This is an area of research that requires a greater amount of attention (See section below).
4. TSPO expression in glial cell types following brain injury
4.1. Temporal pattern of glial cells responses during brain injury and repair
The pattern of glial activation to neuropathological events appears to be a programmed response of the CNS to injury. Microglia respond and become activated within a short amount of time following perturbation of their environment by injury or inflammation (Davalos et al., 2005; Nimmerjahn et al., 2005). The microglia response peaks sometimes after the injury, depending upon the nature of the injury, and it decays with a temporal expression dependent upon the degree and chronicity of the injury. On the other hand, astrocytes become activated sometime after microglia but they appear to have a more protracted period of activation than microglia (Chen et al., 2004; Guilarte 2004; Kuhlmann and Guilarte, 2000; Liberatore et al., 1999; McCann et al., 1996). This pattern of microglia and astrocyte activation appears to be true not only following brain injury but also following recovery from injury (Chen and Guilarte 2006). For example, our laboratory has studied the temporal response of microglia and astrocyte in brain injury models of neurotoxicant exposure (Chen et al., 2004; Kuhlmann and Guilarte, 2000). We observed an early and transient microglial response to injury that eventually decayed to normal levels. Importantly, despite significant reductions in microglia activation there was a late and protracted activation of astrocytes. A similar effect was observed following recovery from a treatment that induces demyelination (Chen and Guilarte, 2006). That is, mice that were administered cuprizone in the diet, a chemical that induces demyelination, can recover if the cuprizone is removed from the diet (Matsushima and Morell, 2001). In this model of demyelination/remyelination we also observed a very rapid decrease in the number of activated microglia with a more protracted decay of the astrocytic response in the corpus callosum during the remyelination or recovery phase (Chen and Guilarte, 2006). In these models of neurotoxicant-induced injury and recovery, TSPO tracks activation of both microglia and astrocytes. Further, these findings were the first to show that TSPO tracks glial cell activation not only as a result of injury but also during recovery from injury.
At present, there is a lack of knowledge on the functional significance of increased TSPO levels in glial cells following neuronal injury or in recovery and whether the enhanced TSPO expression in microglia serves similar functions as in astrocytes. For example, microglia constantly survey their environment and have the ability to respond to brain injury within minutes by directing their ramifications to the site(s) of damage (Nimmerjahn et al., 2005). They proliferate and migrate to the sites of brain injury (Kreutzberg, 1996; Streit et al., 1988, 1999 and 2000), characteristics that are not possessed by astrocytes (Norton et al., 1992; Norenberg 2004). Previous studies indicate that TSPO ligands can influence both the rate of DNA synthesis and the chemotactic potential of breast cancer cell lines (Hardwick et al., 1999), gliomas (Miccoli et al., 1999) and hepatic tumor cell lines (Corsi et al., 2005). Further, TSPO ligands have been shown to modulate chemotaxis and phagocytosis in peripheral monocytes and neutrophils (Ruff et al., 1985; Cosentino et al., 2000; Marino et al., 2001). Since microglia are the monocytes/macrophages of the brain (Kreutzberg, 1996; Streit et al., 1988, 1999, 2000), it is possible that injury–induced upregulation of TSPO in microglia may be associated with their proliferative, migratory and phagocytic capacity, characteristics that are essential for the microglial response to injury. Another potential role of increased TSPO expression in microglia may be related to the secretion of inflammatory cytokines. Choi et al., (2002) have shown that the TSPO antagonist PK11195 can inhibit lipopolysaccharide-induced increases in cyclooxygenase-2 and tumor necrosis factor-α levels in cultured human microglia (Choi et al., 2002). Further, PK11195 can reduce the expression of pro-inflammatory cytokines and neuronal death in the quinolinic acid–injected rat striatum (Ryu et al., 2005).
Astrocytes also increase TSPO level following injury possibly to increase neurosteroid synthesis at the sites of damage. Studies have shown that TSPO activation in astrocytes promotes the synthesis of pregnenolone and progesterone (Le Goascogne et al., 2000), two neurosteroids that possess neurotrophic and neuroprotective activity (Le Goascogne et al., 2000; Schumacher et al., 2000; Veiga et al., 2005). Relevant to a potential difference in the function of the TSPO in microglia and astrocytes, it has been noted that astrocytes but not microglia are capable of synthesizing neurosteroids in culture (Cascio et al., 2000).
4.2. Examination of the TSPO response in microglia and astrocytes in rodent models of brain injury
Since TSPO is present in both microglia and astrocytes, it is important to understand the proportion of the overall TSPO increase after brain injury that is contributed by each of these glial cell types. The cellular localization of TSPO in the injured brain was first studied in rodent models of ischemia. Benavides et al. (1990) reported that TSPO expression in brain cells appeared to have both astrocytic and macrophage-like morphology. However, other studies did not confirm the astrocytic component of the TSPO response in ischemia (Myers et al., 1991; Stephenson et al., 1995). Elevated TSPO levels were consistently correlated with macrophages and microglia rather than astrocytes in the regions of maximal neuronal damage (Myers et al., 1991; Stephenson et al., 1995). Because blood brain barrier (BBB) disruption is a consequence of the ischemia in an, it is likely that the entry of peripheral macrophages and other inflammatory elements contribute to the high TSPO levels (Canat et al., 1993).
Later studies using axotomy models that did not result in disturbance of BBB integrity provided evidence of an association between elevated TSPO levels and microglial markers, with no consistent association with astrocytic markers (Banati et al., 1997; Banati et al., 2000; Gehlert et al., 1997). [3H]-PK11195 emulsion microautoradiography combined with immunohistochemistry provided evidence of colocalization of TSPO and activated microglia but not astrocytes (Banati et al., 1997; Banati et al., 2000). However, these studies did not examine the temporal patterns of expression such as the late time course after the injury. This is an important consideration since it has been shown that the temporal response is very distinct for both microglia and astrocytes. Therefore, if studies only examine the cellular sources of the TSPO response shortly after injury, then a microglia localization and hence a greater association of TSPO with microglial markers is to be expected.
The ability to examine the temporal TSPO response and the glial cell types responsible for the increase in TSPO levels following brain injury was the result of developing methods to assess radioligand binding to TSPO using microautoradiography techniques and immunohistochemical methods in the same cell types (Kuhlmann and Guilarte, 2000; Chen et al., 2004, Chen and Guilarte, 2006). Using this approach, we assessed the TSPO response in rat brains after a single injection of the neurotoxicant trimethyltin, a chemical that targets limbic structures of the brain. This study provided unequivocal evidence of cellular TSPO localization in both activated microglia and astrocytes and it was confirmed by double-labeling fluorescence (Kuhlmann and Guilarte, 2000). It is also noted that in the same study, TSPO expression is abundant not only in the cytoplasm but also in nuclear and perinuclear region in both microglia and astrocytes (Kuhlmann and Guilarte, 2000). The nuclear and perinuclear localization may be associated with the capacity of microglia to differentiate, proliferate, and migrate following a brain insult (Kreutzberg, 1996; Streit et al., 1988). More recent studies from our laboratory using a cuprizone model of demyelination and remyelination also showed that both microglia and astrocytes contributed to the elevation of TSPO levels in areas of demyelination (Chen et al., 2004) and during recovery from demyelinaton (Chen and Guilarte, 2006).
The examination of the glial cell types responsible for the increased density of TSPO following brain injury is now being performed more frequently by several laboratories. For example, recent studies using a transient focal cerebral ischemia rodent model show that the TSPO signal arising from the site of injury derived from TSPO in the infarcted core is primarily associated with microglia. Further, the TSPO signal arising from the rim surrounding the core is primarily from astrocytes (Rojas et al., 2007). Maeda and colleagues (2007) have examined this same question in a model of intrastriatal injection of ethanol. They showed that the astrocytic and microglial expression of TSPO changed longitudinally following injury and revealed increased TSPO levels in both glial cell types. In this particular model, the microglia activation was more persistent than astrocytes, a response that is different from other models of brain injury (Maeda et al., 2007). Nevertheless, these studies indicate that when careful examination of glial cell types is performed in a longitudinal design, brain injury produces increased TSPO levels in both microglia and astrocytes. In many of the studies in which the TSPO response to brain injury is solely attributed to microglia, either only one time point following injury was examined, the contribution from astrocytes was not examined, or double labeling of glial specific immunohistochemical markers with radioligand microautoradiography was not performed. For example, in a recent study examining the TSPO temporal response in an axotomy model of the perforant path to produce a hippocampal lesion, it was concluded that increased TSPO levels in the hippocampus were associated with microglia activation as defined by CD11 immunohistochemistry (Pedersen et al., 2006). However, they did not perform immunohistochemistry with an astrocytic marker such as glial fibrillary acidic protein (GFAP). Therefore, the contribution of astrocytes to the TSPO signal was not studied. Nevertheless, close examination of their data indicates that while at 2 and 5 days following hippocampal lesion there was increased TSPO binding and significant microglia activation, at the latest time point examined (10 days) CD11 immunohistochemistry was normal but [3H]-PK11195 binding to TSPO was still significantly elevated from controls. This suggests that it is likely that increased TSPO levels at the latest time point (10 days) are not contributed by microglia since microglia were of a normal phenotype. Rather, it is likely that at this time point a significant portion of the TSPO signal was associated with astrocytes. However, this was not investigated.
Another study using rat models of intrastriatal injection of lipopolysaccharide (LPS) or 6-hydroxydopamine described a close association of the TSPO response to microglia (Venneti et al., 2007). However, if one examines the correlation of GFAP (an astrocyte marker) and CD68 (a microglia marker) with the binding of the TSPO ligand [3H]-DAA1106 it is clear that the association of [3H]-DAA1106 Bmax and CD68 is mainly driven by two out of the seven data points (see Figure 2-J in Venneti et al., 2007). Further, an increase in [3H]-DAA1106 Bmax from approximately 250–500 fmol/mg was present only when GFAP levels (but not CD68 levels) were elevated based on immunohistochemistry indicating a clear astrocytic response in the absence of microglia. Therefore, the contribution of microglia and astrocytes to the TSPO signal in active brain disease remains a point of contention in the literature.
4.3. Sensitivity of the TSPO response to brain injury
Our laboratory has been interested in determining the degree of brain injury necessary in order to elicit an increase in TSPO levels following exposure to a variety of neurotoxicants. The result indicate that the TSPO response to injury is directly associated with the degree of damage. That is, there is a robust increase in TSPO levels as results of frank neuronal cell loss (Guilarte et al., 1995; Kuhlmann and Guilarte, 1997; Kuhlmann and Guilarte, 2000) and demyelination (Chen et al., 2004; Chen and Guilarte, 2006) with smaller increases measured due to loss of neuronal terminals (Chen et al., 2007; Guilarte et al., 2003; Kuhlmann and Guilarte, 1999). These studies also showed that TSPO is a much more sensitive indicator in detecting brain damage than histological techniques since significant elevations in TSPO levels are measured prior to pathological changes (Chen et al., 2004; Kuhlmann and Guilarte, 1997). Further, increased levels of TSPO are also measured in secondary areas of brain injury resulting from the primary lesion providing a more extensive assessment of damage associated with neural networks (Banati et al., 2000; Cagnin et al., 2001b; Kuhlmann and Guilarte, 1999; Turner et al., 2004). As it will be described below, an important advantage of the TSPO as a biomarker of brain injury is that it can be visualized and quantified using not only in vitro methods such as quantitative autoradiography but also using in vivo imaging techniques such as PET. Therefore, this approach offers great potential for the in vivo imaging of a wide variety of human neurological diseases.
4.4. In vivo assessment of TSPO in animal models of brain injury
While TSPO-PET has been used in several human neurodegenerative disorders (see section 4.5 below), there is a lack of studies showing its utility in small animals such as rodents. Cicchetti and colleagues (2002) investigated the microglial response to degeneration of dopaminergic neurons in vivo using an in-house built PET scanner (PCR-I with resolution 4.5 mm) in a rat model of Parkinson’s disease (PD) by unilateral intrastriatal administration of 6-hydroxydopamine (6-OHDA). Increased [11C]-PK11195 binding ratio in the striatum (67%) and substantia nigra (45%) was demonstrated after 3 weeks of 6-OHDA treatment using the cerebellum as reference region (Cicchetti et al., 2002). Our laboratory began to use small animal PET imaging to demonstrate in vivo changes in TSPO levels in the same animals longitudinally during demyelination and remyelination in a cuprizone-induced demyelination murine model (Chen and Guilarte 2006). The use of TSPO PET imaging in the living mouse brain provides a significant advance in monitoring brain injury during demyelination and remyelination in the same animal. This approach may be useful in studying the function of the TSPO in animal models of chemical-induced neurotoxicity or in models of human neurodegenerative disorders. The use of small-animal imaging provides a novel approach to examine the effects of neurotoxicants on the brain in a longitudinal fashion since the same animal can be imaged repeatedly over time. Further, the use of small-animal imaging significantly reduces the number of animals needed since the same animal can be used at multiple time points. As the improvement of dedicated small animal PET with better resolution (less than 1mm), investigators can confidently evaluate the biochemical changes in the rodent models in vivo and further utilize these models for the development of new pharmaceuticals for diagnosis and therapy.
4.5. In vivo assessment of TSPO in human neurological diseases
TSPO has been widely investigated as an in vivo marker of human neurological diseases using [11C]-(R)-PK11195 PET (see Table 2). These include patients with ischemic stroke (Gerhard et al., 2005; Pappata et al., 2000), multiple sclerosis (Banati et al., 2000; Versijpt et al., 2005; Vowinckel et al., 1997), cerebral vasculitis (Goerres et al., 2001), Rasmussen’s and herpes encephalitis (Banati et al., 1999; Cagnin et al., 2001), HIV encephalitis (Hammoud et al., 2005), Alzheimer’s dementia (Cagnin et al., 2001a; Versijpt et al., 2003), frontotemporal lobe dementia (Cagnin et al., 2004), amyotrophic lateral sclerosis (Turner et al., 2004), corticobasal degeneration (Gerhard et al., 2004; Henkel et al., 2004), multiple system atrophy (Gerhard et al., 2003), Parkinson’s disease (PD) (Gerhard et al., 2006; Ouchi et al., 2005), and Huntington’s disease (HD)(Pavese et al., 2006; Tai et al., 2007). Although most of the human studies demonstrated statistically significant increase of [11C]-(R)-PK11195 binding in either the primary or secondary areas of injury expressing activated glial cells, the degree of increased [11C]-(R)-PK11195 binding varied amongst the different neurological diseases. The degree of the TSPO response measured in PET is closely related to the nature of the insult and the subsequent reactive gliosis. For example, Ouchi and colleagues (2005) have found no significant changes in [11C]-(R)-PK11195 binding to TSPO in the striatum of patients in the early stages of PD, a time in which significant degeneration has already occurred (Ouchi et al., 2005). These authors did find a small but significant increase in TSPO levels in the substantia nigra (SN). On the other hand, Gerhard and colleagues (2006) demonstrated a small but significant increase in [11C]-(R)-PK11195 binding to TSPO in striatum and extrastriatal brain regions including the pallidum, thalamus, pons and cerebral cortex but not in the SN of a group of early and advanced PD patients (Gerhard et al., 2006). It is possible that the glial response elicited by dopaminergic terminal degeneration in the striatum in PD patients may not be sufficiently large to be detected with the sensitivity and resolution of current PET scanners. This notion is consistent with the finding that higher levels of TSPO are measured in the striatum of HD patients (Pavese et al., 2006), a neurodegenerative condition with frank neuronal loss intrinsic to the striatum. The glial TSPO response to the loss of neuronal cell bodies is likely to be more robust and more readily detectible than the response generated from the loss of neuronal terminals as occurs in PD.
4.6. Limitation of in vivo TSPO detection and the development of new classes of radioligands
Most of the work on the validation of TSPO as marker of reactive gliosis has been based on the selective ligand (PK11195) binding to TSPO using either in vitro receptor autoradiography or receptor binding assays. For these types of studies, the experimental conditions are controlled and are maximized to obtain the best signal to noise ratio (i.e., total vs non-specific binding). Thus, increases in radioligand binding to TSPO in the injured brain could be easily identified and visualized compared to the very low levels of TSPO in non-disease areas of the brain. The results from in vitro receptor binding reflects the available receptors/or binding sites in tissues. However, when one applies the TSPO ligands in living experimental animals or humans using PET imaging, one is dealing with a complex environment composed of different kinetic compartments including distribution (blood circulation, permeability through blood brain barrier or plasma membrane of glial cells), metabolism, and excretion. In addition, most of the injected TSPO ligand will bind to TSPO that is abundantly expressed in peripheral steroidogenic organs and only a small portion of the tracer is able to reach the brain. Further, the TSPO-PET imaging is a combination of specific binding, non-specific binding, free [11C]-(R)-PK11195 and its metabolites in brain regions, and [11C]-(R)-PK11195 and its metabolites in the blood pool of the brain. Because of the extremely lipophilic nature of PK11195, there is a significant amount of non-specific binding that contributes to the background levels in TSPO-PET imaging. In addition, the non-specific binding of TSPO ligand in the brain can not be completely determined by pharmacological blocking in vivo (Petit-Taboue et al., 1991, Venneti et al, 2006).
Previous studies have shown an increase of [11C]-PK11195 uptake in the brain after pre-administration of PK11195 or administration of unlabeled PK11195 afterward (Petit-Taboue et al., 1991). The explanation for this phenomenon is that PK11195 blocked TSPO in the peripheral tissues and caused increased [11C]-PK11195 concentration in the blood and more [11C]-PK11195 entered the brain (Petit-Taboue et al., 1991; Venneti et al, 2006). Compartmental or other non-compartmental mathematical modeling with arterial blood input function can help to extrapolate the meaningful biological binding parameters or representative parametric images from dynamic PET images. However, the [11C]-(R)-PK11195 binding is subject to change depending on the model chosen and there is currently no ideal model available to fit the [11C]-(R)-PK11195 PET time activity curves and to provide sensitive detection to the limited increase of TSPO levels from reactive gliosis in neurodegenerative diseases. Therefore, further investigation for the improvement of the TSPO-PET is necessary. The development of new ralioligands for TSPO with lower lipophilicity and higher affinity may solve the problem of high non-specific binding and provide better TSPO-PET imaging for quantification (see section 4.8. below).
4.7. Quantification methodology and new ligands for in vivo TSPO PET imaging
Imaging the TSPO levels with [11C]-(R)-PK11195 PET is currently the best characterized and most widely used radioligand for studies in humans. The most important feature of TSPO as a useful marker of brain injury is the low level of TSPO expression in normal brain. This advantage on the other hand provides some challenges for the modeling of this tracer (Turkheimer et al., 2007). Because of the low TSPO level in the normal brain, the signal from [11C]-(R)-PK11195 in blood vessels and its non-specific binding in tissue becomes predominant (Turkheimer et al., 2007). In addition, as mentioned previously, the abundance of TSPO in peripheral organs also affects the availability of [11C]-(R)-PK11195 for binding in the brain (Turkheimer et al., 2007). Because of the ubiquity of glial cells in the CNS, an ideal reference tissue for mathematical modeling of [11C]-(R)-PK11195 PET is lacking (Turkheimer et al., 2007).
Quantification of [11C]-(R)-PK11195 PET studies has so far been approached either by normalization of the uptake to a reference region or by application of the simplified reference tissue model (SRTM) (Lammertsma and Hume, 1996) with a “reference” region devoid of TSPO derived from cluster analysis (Banati et al., 2000; Pappata et al., 2000; Turkheimer et al., 2000). In brief, Banati and colleagues transformed dynamic TSPO-PET imaging into several clusters of voxels with indistinguishable kinetic behavior and selected one as the “reference” region, which has the fastest clearance and most similarity to the cluster derived from the average of normal controls (Banati et al., 2000; Turkheimer et al., 2000). This approach has some important limitations (Venneti et al., 2006). Recently, full kinetic characterization of [11C]-(R)-PK11195 with measurement of arterial input function has been reported with the application of appropriate 2-tissue compartments, 4-rate-constants model (Kropholler et al., 2005). Further work has shown that blood input modeling provides binding potentials (BP = estimated with the simplified reference region k3/k4) that correlate significantly with those model (Kropholler et al., 2005). Recent work from Turkheimer et al. (2007) provides an alternative approach for the selection of a reference region devoid of TSPO under supervision to prevent the selection of a reference region outside the brain (Turkheimer et al., 2007). The use of a tissue input function may provide practical advantages with no need for extensive arterial blood sampling in patients with brain injury. However, most of the TSPO-PET studies conducted in humans have not been validated by postmortem receptor autoradiography (Venneti et al., 2006).
Another methodological issue in regards to the use of the [11C]-(R)-PK11195 in previous studies is the highly variable kinetic behavior of [11C]-(R)-PK11195 in plasma (Lockhart et al., 2003). Lockhard et al.(2003) measured the binding of racemic [3H]PK11195 to whole human blood and found a low level of binding to blood cells but extensive binding to plasma protein, with strong binding to α1-acid glycoprotein (AGP) and much weaker interaction with albumin. AGP is an acute phase protein, and its levels vary during infection and pathological inflammatory diseases such as multiple sclerosis (Lockhart et al., 2003). This could significantly alter the free plasma concentrations of the ligand and contribute to its variable kinetic behavior. Furthermore, local synthesis of AGP at the site of brain injury may potentially contribute to the [11C]-(R)-PK11195 binding observed in neuroinflammatory diseases (Lockhart et al., 2003). Therefore, the measurement of AGP levels in blood samples or brain tissues is important to future [11C]-(R)-PK11195 PET studies.
In the development of new TSPO ligands, several studies have demonstrated that TSPO binds with high affinity to other classes of organic compounds, such as phenoxyphenyl-acetamides, pyrazolopyrimidine, indolacetamide, and imidiazopyridines, which could act as TSPO ligands in vivo (James et al., 2006). The phenoxyphenyl-acetamide derivative, N-(2,5-dimethoxybenzyl)-N-(5-fluoro-2-phenoxyphenyl) acetamide (DAA1106), has subnanomolar affinity (Ki: 0.043 nM) for TSPO compared to PK11195 (Ki: 0.77nM) when competing with [3H]-DAA1106 (Chaki et al., 1999). [11C]-DAA1106 binding (% injected dose) is four times higher compared to the binding of [11C]-PK11195 in the monkey occipital cortex. Specific binding is estimated as 80% of total binding. Thus, [11C]-DAA1106 might be a good ligand for in vivo imaging of TSPO (Maeda et al., 2004). The most recent work with [11C]-DAA1106 in an animal model of inflammation indicates that this radioligand has higher affinity and greater retention in the brain than [11C]-PK11195 (Venneti et al., 2007).
Another acetamide derivative [11C]-CLINME (2-[6-chloro-2-(4-iodophenyl)-imidazo[1,2-a] pyridin-3-yl]-N-ethyl-N-methyl-acetamide) has recently been shown to perform better than [11C]-PK11195 in terms of specific to nonspecific ratio in vivo using small animal PET in a rat model with intra-striatal injection of AMPA (α-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid)(Boutin et al., 2007b).
The pyrazolopyrimidine, N, N-diethyl-2-[2-(4-methoxyphenyl)-5,7-dimethyl-pyrazolo[1,5-a]pyrimidin-3-yl]-acetamide (DPA-713), has also been reported to be a potent ligand for TSPO, displaying higher affinity than PK11195. [11C]-DPA173 was shown to bind TSPO selectively in the baboon brain with higher brain uptake and relatively slow wash out compared to [11C]-(R)-PK11195 (James et al., 2005). The most recent work with [11C]-DPA173 also demonstrated that this ligand has better signal-to noise ratio than [11C]-PK11195 in vivo (Boutin et al., 2007a). With molecular imaging being increasingly used in medical diagnostics, potential ligands for TSPO will continue to be developed to enhance studies of disease processes. Further improvements in radioligands, methodology for imaging analysis, and instrumentation for imaging acquisition are required in order to provide greater sensitivity and accuracy.
4.8. TSPO ligands as potential therapeutic agents for brain injury and inflammation
Emerging evidence suggests that the administration of TSPO selective ligands may be useful in the treatment of inflammatory conditions (Torres et al., 2000) as well as attenuation of seizures and brain injury (Ferzaz et al., 2002; Ryu et al., 2005; Veenman et al., 2002; Veiga et al., 2005). The exact mechanisms by which TSPO specific ligands confer protection are not precisely known. However, associations between TSPO activation and stimulation of neurosteroid synthesis have been noted (Lacapere and Papadopoulos, 2003). Cascio et al., (2000) have shown a correlation between TSPO expression, steroid synthesis, myelination and oligodendrocyte differentiation. Thus, TSPO activation may assist in the recovery from injury that produces demyelination. Consistent with this hypothesis, Lacor et al., (1999) have shown that TSPO levels and DBI (the putative endogenous ligand of TSPO) are increased after peripheral nerve injury. Following regeneration, TSPO and DBI levels decreased to normal levels and in the absence of regeneration, TSPO and DBI remained elevated. In the same studies, they also showed that TSPO activation by an exogenous ligand Ro5-4864 increased pregnenolone levels in the injured tissue. These findings strongly suggest that TSPO plays an important role in regeneration and neurosteroid synthesis and may be a trophic factor in recovery from brain injury.
4.9. New probes for studying the cellular and sub-cellular localization of TSPO in injured brain tissue
The ability to selectively detect and measure the cellular and sub-cellular distribution of TSPO in cultured cells and in vivo prompted the development of TSPO ligands labeled with fluorescence and MR probes. TSPO ligands with fluorescent probes will become popular for the in vitro detection of TSPO in living cells. To this aim, Kozikowski et al. (1997) reported the first fluorescent high-affinity TSPO ligand 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)-labeled FGIN-1–27 (N,N-dihexyl-2-(4-fluorophenly)-indole-3-acetamide) and provided a tool allowing the direct imaging by fluorescence microscopy of the 18 kDa TSPO proteins in living cells (Kozikowski et al., 1997). However, NBD is not the ideal fluorescent dye due to its low sensitivity and limited signal to noise ratio related to its potential autofluorescence and scatter in tissues and cells (Manning et al., 2006).
Another TSPO ligand, a lanthanide chelated PK-11195 (Ln-PK-11195) with the potential to provide both an optical and MR signature depending on the metal ion that is chelated (i.e. Eu3+ for optical and Gd3+ for MR), was developed by Manning et al. (2004). A fluorescence dye lissamine conjugated form of PK11195 (Liss-Con-PK-11195), which has a relatively high molar extinction coefficient, offers improved sensitivity for microscopy imaging and high-throughput screening detection in multiwell plates (Manning et al., 2006). Liss-Con-PK11195 specifically binds to TSPO and displays attractive optical properties for live cell fluorescence microscopic imaging and high-throughput plate reader assays (Manning et al., 2006). However, the permeability of Gd-PK11195 through the BBB is questionable and this may limit the use of MRI with Gd-PK11195 in the CNS.
5. Concluding Remarks
In vivo imaging of TSPO as a biomarker of reactive gliosis has gained a great deal of attention in the last decade. This is based on the fact that imaging and quantitation of TSPO levels provides an excellent approach for the detection of active brain disease, to study the progression of neurodegeneration, and to monitor the effects of therapeutic strategies. Current efforts to improve the in vivo characteristics of TSPO specific radioligands, the development of improved mathematical models for image analysis, and the development of PET instrumentation with greater resolution and sampling acquisition are required in order to make TSPO imaging useful in the clinical and research settings. Lastly, it is critically important to undestand the function and how TSPO expression is regulated in the different glial cell types in order to gain a better understanding of this rather unique protein and its expression in health and disease.
Acknowledgments
This work was supported by NIEHS grant number ES07062 to T.R.G. The work with the cuprizone model of demyelination was performed in partial fulfillment of doctoral degree requirements for M-K.C.
List of Abbreviations
- ACBP
acyl coenzyme-A binding protein
- AD
Alzheimer’s disease
- AGP
α1-acid glycoprotein
- AIF
apoptosis inducing factor
- ANC
adenine nucleotide carrier
- Apaf-1
apoptosis activating factor 1
- BBB
blood brain barrier
- Bmax
maximal number of binding sites
- CBR
central benzodiazepine receptors
- CNS
central nervous system
- CRAC
cholesterol recognition/interaction amino acid consensus
- DA
dopamine
- DAA1106
N-(2,5-dimethoxybenzyl)-N-(5-fluoro-2-phenoxyphenyl) acetamide
- DHEA
dehydroepiandrosterone
- DPA173
N,N-diethyl-2-[2-(4-methoxyphenyl)-5,7-dimethyl-pyrazolo[1,5-a]pyrimidin-3-yl]-acetamide
- ENP
eicosaneuropeptide
- FGIN-1-27
N,N-dihexyl-2-(4-fluorophenly) indole-3-acetamide
- GABA
gamma-aminobutyric acid
- GFAP
glial fibrillary acidic protein
- IL
interleukin
- Kd
dissociation constant
- kDa
kilodaltons
- KO
knockout
- Liss-Con-PK-11195
lissamine conjugated form of PK11195
- Ln-PK-11195
lanthanide chelated PK-11195
- MPT
mitochondrial permeability transition
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MS
multiple sclerosis
- NBD
7-nitrobenz-2-oxa-1,3-diazol-4-yl
- NO
nitric oxide
- ODN
octadecaneuropeptide
- P-450scc
side chain cleavage cytochrome P-450 enzyme
- PAF
platelet activating factor
- PAP7
peripheral benzodiazepine receptor and protein kinase A associated protein 7
- PBR
peripheral benzodiazepine receptor
- PD
Parkinson’s disease
- PET
positron emission tomography
- PK 11195
1-(2-chlorophenyl)-N-methyl-(1-methypropyl)-3-isoquinoline Carboxamide
- PKA
protein kinase A
- PMN
polymorphonuclear neutrophil
- PRAX-1
peripheral benzodiazepine receptor associated protein-1
- PREG
pregnenolone
- PROG
progesterone
- RO5-4864
7-chloro-5-(4-chlorophenyl)-1-methyl-1,3-dihydrobenzo[e][1,4] diazepin-2-one
- ROS
reactive oxygen species
- SPECT
single photon emission computer tomography
- SRTM
simplified reference tissue model
- StAR
steroidogenic acute regulatory protein
- TMT
trimethyltin
- TNF
tumor necrosis factor
- TSPO
translocator protein (18kDa)
- TTN
triakontatetraneuropeptide
- VDAC
voltage-dependent anion channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alho H, Harjuntausta T, Schultz R, Pelto-Huikko M, Bovolin P. Immunohistochemistry of diazepam binding inhibitor (DBI) in the central nervous system and peripheral organs: its possible role as an endogenous regulator of different types of benzodiazepine receptors. Neuropharmacology. 1991;30:1381–1386. doi: 10.1016/s0028-3908(11)80005-5. [DOI] [PubMed] [Google Scholar]
- Alho H, Varga V, Krueger KE. Expression of mitochondrial benzodiazepine receptor and its putative endogenous ligand diazepam binding inhibitor in cultured primary astrocytes and C-6 cells: relation to cell growth. Cell Growth Differ. 1994;5:1005–1014. [PubMed] [Google Scholar]
- Altar CA, Baudry M. Systemic injection of kainic acid: gliosis in olfactory and limbic brain regions quantified with [3H]-PK 11195 binding autoradiography. Exp Neurol. 1990;109:333–341. doi: 10.1016/s0014-4886(05)80024-x. [DOI] [PubMed] [Google Scholar]
- Anholt RR, Pedersen PL, De Souza EB, Snyder SH. The peripheral-type benzodiazepine receptor. Localization to the mitochondrial outer membrane. J Biol Chem. 1986;261:576–583. [PubMed] [Google Scholar]
- Antkiewicz-Michaluk L, Guidotti A, Krueger KE. Molecular characterization and mitochondrial density of a recognition site for peripheral-type benzodiazepine ligands. Mol Pharmacol. 1988;34:272–278. [PubMed] [Google Scholar]
- Azarashvili T, Krestinina O, Yurkov I, Evtodienko Y, Reiser G. High-affinity peripheral benzodiazepine receptor ligand, PK11195, regulates protein phosphorylation in rat brain mitochondria under control of Ca(2+) J Neurochem. 2005;94:1054–1062. doi: 10.1111/j.1471-4159.2005.03260.x. [DOI] [PubMed] [Google Scholar]
- Banati RB, Myers R, Kreutzberg GW. PK (‘peripheral benzodiazepine’)--binding sites in the CNS indicate early and discrete brain lesions: microautoradiographic detection of [3H]PK11195 binding to activated microglia. J Neurocytol. 1997;26:77–82. doi: 10.1023/a:1018567510105. [DOI] [PubMed] [Google Scholar]
- Banati RB, Goerres GW, Myers R, Gunn RN, Turkheimer FE, Kreutzberg GW, et al. [11C](R)-PK11195 positron emission tomography imaging of activated microglia in vivo in Rasmussen’s encephalitis. Neurology. 1999;53:2199–2203. doi: 10.1212/wnl.53.9.2199. [DOI] [PubMed] [Google Scholar]
- Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123:2321–2337. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- Braestrup C, Squires RF. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc Natl Acad Sci U S A. 1977;74:3805–3809. doi: 10.1073/pnas.74.9.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides J, Capdeville C, Dauphin F, Dubois A, Duverger D, Fage D, et al. The quantification of brain lesions with an omega 3 site ligand: a critical analysis of animal models of cerebral ischaemia and neurodegeneration. Brain Res. 1990;522:275–289. doi: 10.1016/0006-8993(90)91472-s. [DOI] [PubMed] [Google Scholar]
- Benavides J, Fage D, Carter C, Scatton B. Peripheral type benzodiazepine binding sites are a sensitive indirect index of neuronal damage. Brain Res. 1987;421:167–172. doi: 10.1016/0006-8993(87)91287-x. [DOI] [PubMed] [Google Scholar]
- Benavides J, Malgouris C, Imbault F, Begassat F, Uzan A, Renault C, et al. Peripheral type” benzodiazepine binding sites in rat adrenals: binding studies with [3H]-PK11195 and autoradiographic localization. Arch Int Pharmacodyn Ther. 1983a;266:38–49. [PubMed] [Google Scholar]
- Benavides J, Quarteronet D, Imbault F, Malgouris C, Uzan A, Renault C, et al. Labelling of “peripheral-type” benzodiazepine binding sites in the rat brain by using [3H]-PK11195, an isoquinoline carboxamide derivative: kinetic studies and autoradiographic localization. J Neurochem. 1983b;41:1744–1750. doi: 10.1111/j.1471-4159.1983.tb00888.x. [DOI] [PubMed] [Google Scholar]
- Bernassau JM, Reversat JL, Ferrara P, Caput D, Lefur G. A 3D model of the peripheral benzodiazepine receptor and its implication in intra mitochondrial cholesterol transport. J Mol Graph. 1993;11:236–44. 235. doi: 10.1016/0263-7855(93)80003-a. [DOI] [PubMed] [Google Scholar]
- Besman MJ, Yanagibashi K, Lee TD, Kawamura M, Hall PF, Shively JE. Identification of des-(Gly-Ile)-endozepine as an effector of corticotropin-dependent adrenal steroidogenesis: stimulation of cholesterol delivery is mediated by the peripheral benzodiazepine receptor. Proc Natl Acad Sci U S A. 1989;86:4897–4901. doi: 10.1073/pnas.86.13.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger GT, Weissman BA, Lueddens H, Basile AS, Mantione CR, Barrett JE, et al. Late evolutionary appearance of ‘peripheral-type’ binding sites for benzodiazepines. Brain Res. 1985;338:366–370. doi: 10.1016/0006-8993(85)90170-2. [DOI] [PubMed] [Google Scholar]
- Boutin H, Chauveau F, Thominiaux C, Gregoire MC, James ML, Trebossen R, et al. 11C-DPA-713: a novel peripheral benzodiazepine receptor PET ligand for in vivo imaging of neuroinflammation. J Nucl Med. 2007a;48:573–581. doi: 10.2967/jnumed.106.036764. [DOI] [PubMed] [Google Scholar]
- Boutin H, Chauveau F, Thominiaux C, Kuhnast B, Gregoire MC, Jan S, et al. A. In vivo imaging of brain lesions with [(11)C]CLINME, a new PET radioligand of peripheral benzodiazepine receptors. Glia. 2007b;55:1459–1468. doi: 10.1002/glia.20562. [DOI] [PubMed] [Google Scholar]
- Bovolin P, Schlichting J, Miyata M, Ferrarese C, Guidotti A, Alho H. Distribution and characterization of diazepam binding inhibitor (DBI) in peripheral tissues of rat. Regul Pept. 1990;29:267–281. doi: 10.1016/0167-0115(90)90089-f. [DOI] [PubMed] [Google Scholar]
- Braestrup C, Squires RF. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)-diazepam binding. Proc Natl Acad Sci U S A. 1977;74:3805–9. doi: 10.1073/pnas.74.9.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bribes E, Carriere D, Goubet C, Galiegue S, Casellas P, Simony-Lafontaine J. Immunohistochemical assessment of the peripheral benzodiazepine receptor in human tissues. J Histochem Cytochem. 2004;52:19–28. doi: 10.1177/002215540405200103. [DOI] [PubMed] [Google Scholar]
- Brown RC, Cascio C, Papadopoulos V. Pathways of neurosteroid biosynthesis in cell lines from human brain: regulation of dehydroepiandrosterone formation by oxidative stress and beta-amyloid peptide. J Neurochem. 2000a;74:847–859. doi: 10.1046/j.1471-4159.2000.740847.x. [DOI] [PubMed] [Google Scholar]
- Brown RC, Degenhardt B, Kotoula M, Papadopoulous V. Location-dependent role of the human glioma cell peripheral-type benzodiazepine receptor in proliferation and steroid biosynthesis. Cancer Lett. 2000b;156:125–132. doi: 10.1016/s0304-3835(00)00451-1. [DOI] [PubMed] [Google Scholar]
- Brown RC, Papadopoulos V. Role of the peripheral-type benzodiazepine receptor in adrenal and brain steroidogenesis. Int Rev Neurobiol. 2001;46:117–143. doi: 10.1016/s0074-7742(01)46061-2. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001a;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Myers R, Gunn RN, Lawrence AD, Stevens T, Kreutzberg GW, et al. In vivo visualization of activated glia by [11C]-(R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain. 2001b;124:2014–2027. doi: 10.1093/brain/124.10.2014. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Rossor M, Sampson EL, Mackinnon T, Banati RB. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol. 2004;56:894–897. doi: 10.1002/ana.20332. [DOI] [PubMed] [Google Scholar]
- Canat X, Carayon P, Bouaboula M, Cahard D, Shire D, Roque C, et al. Distribution profile and properties of peripheral-type benzodiazepine receptors on human hemopoietic cells. Life Sci. 1993;52:107–118. doi: 10.1016/0024-3205(93)90293-c. [DOI] [PubMed] [Google Scholar]
- Carmel I, Fares FA, Leschiner S, Scherubl H, Weisinger G, Gavish M. Peripheral-type benzodiazepine receptors in the regulation of proliferation of MCF-7 human breast carcinoma cell line. Biochem Pharmacol. 1999;58:273–278. doi: 10.1016/s0006-2952(99)00093-3. [DOI] [PubMed] [Google Scholar]
- Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40:475–486. doi: 10.1016/s0197-0186(01)00118-8. [DOI] [PubMed] [Google Scholar]
- Cascio C, Brown RC, Liu Y, Han Z, Hales DB, Papadopoulos V. Pathways of dehydroepiandrosterone formation in rat brain glia. J Steroid Biochem Mol Biol. 2000;75:177–186. doi: 10.1016/s0960-0760(00)00163-1. [DOI] [PubMed] [Google Scholar]
- Chaki S, Funakoshi T, Yoshikawa R, Okuyama S, Okubo T, Nakazato A, et al. Binding characteristics of [3H]-DAA1106, a novel and selective ligand for peripheral benzodiazepine receptors. Eur J Pharmacol. 1999;371:197–204. doi: 10.1016/s0014-2999(99)00118-1. [DOI] [PubMed] [Google Scholar]
- Chang YJ, McCabe RT, Rennert H, Budarf ML, Sayegh R, Emanuel BS, et al. The human “peripheral-type” benzodiazepine receptor: regional mapping of the gene and characterization of the receptor expressed from cDNA. DNA Cell Biol. 1992;11:471–480. doi: 10.1089/dna.1992.11.471. [DOI] [PubMed] [Google Scholar]
- Chen MK, Baidoo K, Verina T, Guilarte TR. Peripheral benzodiazepine receptor imaging in CNS demyelination: functional implications of anatomical and cellular localization. Brain. 2004;127:1379–1392. doi: 10.1093/brain/awh161. [DOI] [PubMed] [Google Scholar]
- Chen MK, Guilarte TR. Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol Sci. 2006;91:532–539. doi: 10.1093/toxsci/kfj172. [DOI] [PubMed] [Google Scholar]
- Chen M-K, Kuwabara H, Zhou Y, Adams RJ, Braši JR, McGlothan JL, et al. VMAT2 and dopamine neuron loss in a primate model of Parkinson’s disease. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.05108.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HB, Khoo C, Ryu JK, van Breemen E, Kim SU, McLarnon JG. Inhibition of lipopolysaccharide-induced cyclooxygenase-2, tumor necrosis factor-alpha and [Ca2+]i responses in human microglia by the peripheral benzodiazepine receptor ligand PK11195. J Neurochem. 2002;83:546–55. doi: 10.1046/j.1471-4159.2002.01122.x. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- Corsi L, Geminiani E, Avallone R, Baraldi M. Nuclear location-dependent role of peripheral benzodiazepine receptor (PBR) in hepatic tumoral cell lines proliferation. Life Sci. 2005;76:2523–2533. doi: 10.1016/j.lfs.2004.08.040. [DOI] [PubMed] [Google Scholar]
- Cosentino M, Marino F, Cattaneo S, Di Grazia L, Francioli C, Fietta AM, et al. Diazepam-binding inhibitor-derived peptides induce intracellular calcium changes and modulate human neutrophil function. J Leukoc Biol. 2000;67:637–643. doi: 10.1002/jlb.67.5.637. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Debruyne JC, Van Laere KJ, Versijpt J, De Vos F, Eng JK, Strijckmans K, et al. Semiquantification of the peripheral-type benzodiazepine ligand [11C]-PK11195 in normal human brain and application in multiple sclerosis patients. Acta Neurol Belg. 2002;102:127–135. [PubMed] [Google Scholar]
- Debruyne JC, Versijpt J, Van Laere KJ, De Vos F, Keppens J, Strijckmans K, et al. PET visualization of microglia in multiple sclerosis patients using [11C]-PK11195. Eur J Neurol. 2003;10:257–264. doi: 10.1046/j.1468-1331.2003.00571.x. [DOI] [PubMed] [Google Scholar]
- Demerle-Pallardy C, Duverger D, Spinnewyn B, Pirotzky E, Braquet P. Peripheral type benzodiazepine binding sites following transient forebrain ischemia in the rat: effect of neuroprotective drugs. Brain Res. 1991;565:312–320. doi: 10.1016/0006-8993(91)91663-l. [DOI] [PubMed] [Google Scholar]
- Everett H, Barry M, Sun X, Lee SF, Frantz C, Berthiaume LG, et al. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J Exp Med. 2002;196:1127–1139. doi: 10.1084/jem.20011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farges R, Joseph-Liauzun E, Shire D, Caput D, Le Fur G, Loison G, et al. Molecular basis for the different binding properties of benzodiazepines to human and bovine peripheral-type benzodiazepine receptors. FEBS Lett. 1993;335:305–308. doi: 10.1016/0014-5793(93)80407-l. [DOI] [PubMed] [Google Scholar]
- Falchi AM, Battetta B, Sanna F, Piludu M, Sogos V, Serra M, et al. Intracellular cholesterol changes induced by translocator protein (18 kDa)TSPO/PBR ligands. Neuropharmacology. 2007;53:318–329. doi: 10.1016/j.neuropharm.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Ferrero P, Santi MR, Conti-Tronconi B, Costa E, Guidotti A. Study of an octadecaneuropeptide derived from diazepam binding inhibitor (DBI): biological activity and presence in rat brain. Proc Natl Acad Sci U S A. 1986;83:827–831. doi: 10.1073/pnas.83.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferzaz B, Brault E, Bourliaud G, Robert JP, Poughon G, Claustre Y, et al. SSR180575 (7-chloro-N,N,5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5-b]indole-1-acetamide), a peripheral benzodiazepine receptor ligand, promotes neuronal survival and repair. J Pharmacol Exp Ther. 2002;301:1067–1078. doi: 10.1124/jpet.301.3.1067. [DOI] [PubMed] [Google Scholar]
- Garnier M, Dimchev AB, Boujrad N, Price JM, Musto NA, Papadopoulos V. In vitro reconstitution of a functional peripheral-type benzodiazepine receptor from mouse Leydig tumor cells. Mol Pharmacol. 1994;45:201–211. [PubMed] [Google Scholar]
- Gavish M, Bachman I, Shoukrun R, Katz Y, Veenman L, Weisinger G, et al. Enigma of the peripheral benzodiazepine receptor. Pharmacol Rev. 1999;51:629–650. [PubMed] [Google Scholar]
- Gehlert DR, Stephenson DT, Schober DA, Rash K, Clemens JA. Increased expression of peripheral benzodiazepine receptors in the facial nucleus following motor neuron axotomy. Neurochem Int. 1997;31:705–713. doi: 10.1016/s0197-0186(97)00007-7. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Banati RB, Goerres GB, Cagnin A, Myers R, Gunn RN, et al. [11C]-(R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology. 2003;61:686–9. doi: 10.1212/01.wnl.0000078192.95645.e6. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Neumaier B, Elitok E, Glatting G, Ries V, Tomczak R, et al. In vivo imaging of activated microglia using [11C]-PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport. 2000;11:2957–2960. doi: 10.1097/00001756-200009110-00025. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C]-(R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Schwarz J, Myers R, Wise R, Banati RB. Evolution of microglial activation in patients after ischemic stroke: a [11C]-(R)-PK11195 PET study. Neuroimage. 2005;24:591–595. doi: 10.1016/j.neuroimage.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21:89–93. doi: 10.1002/mds.20668. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Watts J, Trender-Gerhard I, Turkheimer F, Banati RB, Bhatia K, et al. In vivo imaging of microglial activation with [11C]-(R)-PK11195 PET in corticobasal degeneration. Mov Disord. 2004;19:1221–1226. doi: 10.1002/mds.20162. [DOI] [PubMed] [Google Scholar]
- Goerres GW, Revesz T, Duncan J, Banati RB. Imaging cerebral vasculitis in refractory epilepsy using [11C]-(R)-PK11195 positron emission tomography. AJR Am J Roentgenol. 2001;176:1016–1018. doi: 10.2214/ajr.176.4.1761016. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Forchetti CM, Corda MG, Konkel D, Bennett CD, Costa E. Isolation, characterization, and purification to homogeneity of an endogenous polypeptide with agonistic action on benzodiazepine receptors. Proc Natl Acad Sci U S A. 1983;80:3531–3535. doi: 10.1073/pnas.80.11.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilarte TR. Peripheral Benzodiazepine Receptor Imaging in Glial Cells: Molecular Sensors of Brain Pathology. In: Aschner M, Costa L, editors. The Role of Glia in Neurotoxicity. 2. chapter 12. CRC press; 2004. pp. 207–219. [Google Scholar]
- Guilarte TR, Kuhlmann AC, O’Callaghan JP, Miceli RC. Enhanced expression of peripheral benzodiazepine receptors in trimethyltin-exposed rat brain: a biomarker of neurotoxicity. Neurotoxicology. 1995;16:441–450. [PubMed] [Google Scholar]
- Guilarte TR, Nihei MK, McGlothan JL, Howard AS. Methamphetamine-induced deficits of brain monoaminergic neuronal markers: distal axotomy or neuronal plasticity. Neuroscience. 2003;122:499–513. doi: 10.1016/s0306-4522(03)00476-7. [DOI] [PubMed] [Google Scholar]
- Hammoud DA, Endres CJ, Chander AR, Guilarte TR, Wong DF, Sacktor NC, et al. Imaging glial cell activation with [11C]-(R)-PK11195 in patients with AIDS. J Neurovirol. 2005;11:346–355. doi: 10.1080/13550280500187351. [DOI] [PubMed] [Google Scholar]
- Hardwick M, Fertikh D, Culty M, Li H, Vidic B, Papadopoulos V. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999;59:831–842. [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Henkel K, Karitzky J, Schmid M, Mader I, Glatting G, Unger JW, et al. Imaging of activated microglia with PET and [11C]-PK11195 in corticobasal degeneration. Mov Disord. 2004;19:817–821. doi: 10.1002/mds.20040. [DOI] [PubMed] [Google Scholar]
- Hirsch JD, Beyer CF, Malkowitz L, Beer B, Blume AJ. Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Mol Pharmacol. 1989;35:157–163. [PubMed] [Google Scholar]
- Hirsch T, Decaudin D, Susin SA, Marchetti P, Larochette N, Resche-Rigon M, et al. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp Cell Res. 1998;241:426–434. doi: 10.1006/excr.1998.4084. [DOI] [PubMed] [Google Scholar]
- Hong SH, Choi HB, Kim SU, McLarnon JG. Mitochondrial ligand inhibits store-operated calcium influx and COX-2 production in human microglia. J Neurosci Res. 2006;83:1293–1298. doi: 10.1002/jnr.20829. [DOI] [PubMed] [Google Scholar]
- Hurley SD, O’Banion MK, Song DD, Arana FS, Olschowka JA, Haber SN. Microglial response is poorly correlated with neurodegeneration following chronic, low-dose MPTP administration in monkeys. Exp Neurol. 2003;184:659–668. doi: 10.1016/S0014-4886(03)00273-5. [DOI] [PubMed] [Google Scholar]
- James ML, Fulton RR, Henderson DJ, Eberl S, Meikle SR, Thomson S, et al. Synthesis and in vivo evaluation of a novel peripheral benzodiazepine receptor PET radioligand. Bioorg Med Chem. 2005;13:6188–6194. doi: 10.1016/j.bmc.2005.06.030. [DOI] [PubMed] [Google Scholar]
- James ML, Selleri S, Kassiou M. Development of ligands for the peripheral benzodiazepine receptor. Curr Med Chem. 2006;13:1991–2001. doi: 10.2174/092986706777584979. [DOI] [PubMed] [Google Scholar]
- Joseph-Liauzun E, Delmas P, Shire D, Ferrara P. Topological analysis of the peripheral benzodiazepine receptor in yeast mitochondrial membranes supports a five-transmembrane structure. J Biol Chem. 1998;273:2146–2152. doi: 10.1074/jbc.273.4.2146. [DOI] [PubMed] [Google Scholar]
- Kelly-Hershkovitz E, Weizman R, Spanier I, Leschiner S, Lahav M, Weisinger G, et al. Effects of peripheral-type benzodiazepine receptor antisense knockout on MA-10 Leydig cell proliferation and steroidogenesis. J Biol Chem. 1998;273:5478–5483. doi: 10.1074/jbc.273.10.5478. [DOI] [PubMed] [Google Scholar]
- Kinnally KW, Zorov DB, Antonenko YN, Snyder SH, McEnery MW, Tedeschi H. Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc Natl Acad Sci U S A. 1993;90:1374–1378. doi: 10.1073/pnas.90.4.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen J. Acyl-CoA-binding and transport, an alternative function for diazepam binding inhibitor (DBI), which is identical with acyl-CoA-binding protein. Neuropharmacology. 1991;30:1405–1410. doi: 10.1016/s0028-3908(11)80009-2. [DOI] [PubMed] [Google Scholar]
- Knudsen J, Mandrup S, Rasmussen JT, Andreasen PH, Poulsen F, Kristiansen K. The function of acyl-CoA-binding protein (ACBP)/diazepam binding inhibitor (DBI) Mol Cell Biochem. 1993;123:129–138. doi: 10.1007/BF01076484. [DOI] [PubMed] [Google Scholar]
- Kozikowski AP, Kotoula M, Ma D, Boujrad N, Tuckmantel W, Papadopoulos V. Synthesis and biology of a 7-nitro-2,1,3-benzoxadiazol-4-yl derivative of 2-phenylindole-3-acetamide: a fluorescent probe for the peripheral-type benzodiazepine receptor. J Med Chem. 1997;40:2435–2439. doi: 10.1021/jm970220w. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Kropholler MA, Boellaard R, Schuitemaker A, van Berckel BN, Luurtsema G, Windhorst AD, et al. Development of a tracer kinetic plasma input model for (R)-[11C]PK11195 brain studies. J Cereb Blood Flow Metab. 2005;25:842–851. doi: 10.1038/sj.jcbfm.9600092. [DOI] [PubMed] [Google Scholar]
- Kuhlmann AC, Guilarte TR. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J Neurochem. 2000;74:1694–1704. doi: 10.1046/j.1471-4159.2000.0741694.x. [DOI] [PubMed] [Google Scholar]
- Kuhlmann AC, Guilarte TR. Regional and temporal expression of the peripheral benzodiazepine receptor in MPTP neurotoxicity. Toxicol Sci. 1999;48:107–116. doi: 10.1093/toxsci/48.1.107. [DOI] [PubMed] [Google Scholar]
- Kuhlmann AC, Guilarte TR. The peripheral benzodiazepine receptor is a sensitive indicator of domoic acid neurotoxicity. Brain Res. 1997;751:281–288. doi: 10.1016/s0006-8993(96)01409-6. [DOI] [PubMed] [Google Scholar]
- Lacapere JJ, Delavoie F, Li H, Peranzi G, Maccario J, Papadopoulos V, et al. Structural and functional study of reconstituted peripheral benzodiazepine receptor. Biochem Biophys Res Commun. 2001;284:536–541. doi: 10.1006/bbrc.2001.4975. [DOI] [PubMed] [Google Scholar]
- Lacapere JJ, Papadopoulos V. Peripheral-type benzodiazepine receptor: structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids. 2003;68:569–585. doi: 10.1016/s0039-128x(03)00101-6. [DOI] [PubMed] [Google Scholar]
- Lacor P, Gandolfo P, Tonon MC, Brault E, Dalibert I, Schumacher M, et al. Regulation of the expression of peripheral benzodiazepine receptors and their endogenous ligands during rat sciatic nerve degeneration and regeneration: a role for PBR in neurosteroidogenesis. Brain Res. 1999;815:70–80. doi: 10.1016/s0006-8993(98)01105-6. [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, et al. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Lamacz M, Tonon MC, Smih-Rouet F, Patte C, Gasque P, Fontaine M, et al. The endogenous benzodiazepine receptor ligand ODN increases cytosolic calcium in cultured rat astrocytes. Brain Res Mol Brain Res. 1996;37:290–296. doi: 10.1016/0169-328x(95)00330-u. [DOI] [PubMed] [Google Scholar]
- Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- Le Fur G, Guilloux F, Rufat P, Benavides J, Uzan A, Renault C, et al. Peripheral benzodiazepine binding sites: effect of PK 11195, 1-(2-chlorophenyl)-N-methyl-(1-methylpropyl)-3 isoquinolinecarboxamide. II. In vivo studies. Life Sci. 1983a;32:1849–1856. doi: 10.1016/0024-3205(83)90063-2. [DOI] [PubMed] [Google Scholar]
- Le Fur G, Perrier ML, Vaucher N, Imbault F, Flamier A, Benavides J, et al. Peripheral benzodiazepine binding sites: effect of PK 11195, 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide. I. In vitro studies. Life Sci. 1983b;32:1839–1847. doi: 10.1016/0024-3205(83)90062-0. [DOI] [PubMed] [Google Scholar]
- Le Fur G, Vaucher N, Perrier ML, Flamier A, Benavides J, Renault C, et al. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]-RO5-4864 and [3H]-PK 11195, by thermodynamic studies. Life Sci. 1983c;33:449–457. doi: 10.1016/0024-3205(83)90794-4. [DOI] [PubMed] [Google Scholar]
- Le Goascogne C, Eychenne B, Tonon MC, Lachapelle F, Baumann N, Robel P. Neurosteroid progesterone is up-regulated in the brain of jimpy and shiverer mice. Glia. 2000;29:14–24. doi: 10.1002/(sici)1098-1136(20000101)29:1<14::aid-glia2>3.3.co;2-e. [DOI] [PubMed] [Google Scholar]
- Lee DH, Kang SK, Lee RH, Ryu JM, Park HY, Choi HS, et al. Effects of peripheral benzodiazepine receptor ligands on proliferation and differentiation of human mesenchymal stem cells. J Cell Physiol. 2004;198:91–99. doi: 10.1002/jcp.10391. [DOI] [PubMed] [Google Scholar]
- Lenfant M, Haumont J, Zavala F. In vivo immunomodulating activity of PK-11195, a structurally unrelated ligand for “peripheral” benzodiazepine binding sites--I. Potentiation in mice of the humoral response to sheep red blood cells. Int J Immunopharmacol. 1986;8:825–828. doi: 10.1016/0192-0561(86)90021-4. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Davis B, Matthews JC, Rahmoune H, Hong G, Gee A, et al. The peripheral benzodiazepine receptor ligand PK11195 binds with high affinity to the acute phase reactant alpha1-acid glycoprotein: implications for the use of the ligand as a CNS inflammatory marker. Nucl Med Biol. 2003;30:199–206. doi: 10.1016/s0969-8051(02)00410-9. [DOI] [PubMed] [Google Scholar]
- Li H, Degenhardt B, Tobin D, Yao ZX, Tasken K, Papadopoulos V. Identification, localization, and function in steroidogenesis of PAP7: a peripheral-type benzodiazepine receptor- and PKA (RIalpha)-associated protein. Mol Endocrinol. 2001;15:2211–2228. doi: 10.1210/mend.15.12.0736. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Lihrmann I, Plaquevent JC, Tostivint H, Raijmakers R, Tonon MC, Conlon JM, et al. Frog diazepam-binding inhibitor: peptide sequence, cDNA cloning, and expression in the brain. Proc Natl Acad Sci U S A. 1994;91:6899–6903. doi: 10.1073/pnas.91.15.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann MJ, O’Callaghan JP, Martin PM, Bertram T, Streit WJ. Differential activation of microglia and astrocytes following trimethyltin-induced neurodegeneration. Neuroscience. 1996;72:273–281. doi: 10.1016/0306-4522(95)00526-9. [DOI] [PubMed] [Google Scholar]
- Maeda J, Suhara T, Zhang MR, Okauchi T, Yasuno F, Ikoma Y, et al. Novel peripheral benzodiazepine receptor ligand [11C]-DAA1106 for PET: an imaging tool for glial cells in the brain. Synapse. 2004;52:283–291. doi: 10.1002/syn.20027. [DOI] [PubMed] [Google Scholar]
- Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, et al. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007;1157:100–111. doi: 10.1016/j.brainres.2007.04.054. [DOI] [PubMed] [Google Scholar]
- Malagon M, Vaudry H, Van Strien F, Pelletier G, Gracia-Navarro F, Tonon MC. Ontogeny of diazepam-binding inhibitor-related peptides (endozepines) in the rat brain. Neuroscience. 1993;57:777–786. doi: 10.1016/0306-4522(93)90023-9. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Queen SE, Tarwater PJ, Adams RJ, Guilarte TR. Elevated peripheral benzodiazepine receptor expression in simian immunodeficiency virus encephalitis. J Neurovirol. 2003;9:94–100. doi: 10.1080/13550280390173283. [DOI] [PubMed] [Google Scholar]
- Manning HC, Goebel T, Thompson RC, Price RR, Lee H, Bornhop DJ. Targeted molecular imaging agents for cellular-scale bimodal imaging. Bioconjug Chem. 2004;15:1488–1495. doi: 10.1021/bc049904q. [DOI] [PubMed] [Google Scholar]
- Manning HC, Smith SM, Sexton M, Haviland S, Bai M, Cederquist K, et al. A peripheral benzodiazepine receptor targeted agent for in vitro imaging and screening. Bioconjug Chem. 2006;17:735–740. doi: 10.1021/bc060020b. [DOI] [PubMed] [Google Scholar]
- Marino F, Cattaneo S, Cosentino M, Rasini E, Di Grazia L, Fietta AM, et al. Diazepam stimulates migration and phagocytosis of human neutrophils: possible contribution of peripheral-type benzodiazepine receptors and intracellular calcium. Pharmacol. 2001;63:42–49. doi: 10.1159/000056111. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattner F, Katsifis A, Staykova M, Ballantyne P, Willenborg DO. Evaluation of a radiolabelled peripheral benzodiazepine receptor ligand in the central nervous system inflammation of experimental autoimmune encephalomyelitis: a possible probe for imaging multiple sclerosis. Eur J Nucl Med Mol Imaging. 2005;32:557–563. doi: 10.1007/s00259-004-1690-y. [DOI] [PubMed] [Google Scholar]
- McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci U S A. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63:109–115. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Messmer K, Reynolds GP. Increased peripheral benzodiazepine binding sites in the brain of patients with Huntington’s disease. Neurosci Lett. 1998;241:53–56. doi: 10.1016/s0304-3940(97)00967-1. [DOI] [PubMed] [Google Scholar]
- Miccoli L, Oudard S, Beurdeley-Thomas A, Dutrillaux B, Poupon MF. Effect of 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide (PK11195), a specific ligand of the peripheral benzodiazepine receptor, on the lipid fluidity of mitochondria in human glioma cells. Biochem Pharmacol. 1999;58:715–721. doi: 10.1016/s0006-2952(99)00151-3. [DOI] [PubMed] [Google Scholar]
- Miyazawa N, Diksic M, Yamamoto Y. Chronological study of peripheral benzodiazepine binding sites in the rat brain stab wounds using [3H] PK-11195 as a marker for gliosis. Acta Neurochir (Wien) 1995;137:207–216. doi: 10.1007/BF02187195. [DOI] [PubMed] [Google Scholar]
- Myers R, Manjil LG, Cullen BM, Price GW, Frackowiak RS, Cremer JE. Macrophage and astrocyte populations in relation to [3H]-PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J Cereb Blood Flow Metab. 1991;11:314–322. doi: 10.1038/jcbfm.1991.64. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Norenberg MD. The reactive astrocyte. In: Aschner M, Costa L, editors. The Role of Glia in Neurotoxicity. 2. chapter 5. CRC press; 2004. pp. 73–93. [Google Scholar]
- Norton WT, Aquino DA, Hozumi I, Chiu FC, Brosnan CF. Quantitative aspects of reactive gliosis: a review. Neurochem Res. 1992;17:877–885. doi: 10.1007/BF00993263. [DOI] [PubMed] [Google Scholar]
- O’Beirne GB, Woods MJ, Williams DC. Two subcellular locations for peripheral-type benzodiazepine acceptors in rat liver. Eur J Biochem. 1990;188:131–138. doi: 10.1111/j.1432-1033.1990.tb15380.x. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Sriram K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin Drug Saf. 2005;4:433–442. doi: 10.1517/14740338.4.3.433. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP. Quantitative features of reactive gliosis following toxicant-induced damage of the CNS. Ann N Y Acad Sci. 1993;679:195–210. doi: 10.1111/j.1749-6632.1993.tb18299.x. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP. Assessment of neurotoxicity: use of glial fibrillary acidic protein as a biomarker. Biomed Environ Sci. 1991;4:197–206. [PubMed] [Google Scholar]
- Olson JM, Ciliax BJ, Mancini WR, Young AB. Presence of peripheral-type benzodiazepine binding sites on human erythrocyte membranes. Eur J Pharmacol. 1988;152:47–53. doi: 10.1016/0014-2999(88)90834-5. [DOI] [PubMed] [Google Scholar]
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V. Structure and function of the peripheral-type benzodiazepine receptor in steroidogenic cells. Proc Soc Exp Biol Med. 1998;217:130–142. doi: 10.3181/00379727-217-44215. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Amri H, Boujrad N, Cascio C, Culty M, Garnier M, et al. Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids. 1997;62:21–28. doi: 10.1016/s0039-128x(96)00154-7. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006b;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Berkovich A, Krueger KE, Costa E, Guidotti A. Diazepam binding inhibitor and its processing products stimulate mitochondrial steroid biosynthesis via an interaction with mitochondrial benzodiazepine receptors. Endocrinology. 1991;129:1481–1488. doi: 10.1210/endo-129-3-1481. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Boujrad N, Ikonomovic MD, Ferrara P, Vidic B. Topography of the Leydig cell mitochondrial peripheral-type benzodiazepine receptor. Mol Cell Endocrinol. 1994;104:R5–R9. doi: 10.1016/0303-7207(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Brown AS. Role of the peripheral-type benzodiazepine receptor and the polypeptide diazepam binding inhibitor in steroidogenesis. J Steroid Biochem Mol Biol. 1995;53:103–110. doi: 10.1016/0960-0760(95)00027-w. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Mukhin AG, Costa E, Krueger KE. The peripheral-type benzodiazepine receptor is functionally linked to Leydig cell steroidogenesis. J Biol Chem. 1990;265:3772–3779. [PubMed] [Google Scholar]
- Pappata S, Levasseur M, Gunn RN, Myers R, Crouzel C, Syrota A, et al. Thalamic microglial activation in ischemic stroke detected in vivo by PET and [11C]-PK1195. Neurology. 2000;55:1052–1054. doi: 10.1212/wnl.55.7.1052. [DOI] [PubMed] [Google Scholar]
- Parola AL, Stump DG, Pepperl DJ, Krueger KE, Regan JW, Laird HE., 2nd Cloning and expression of a pharmacologically unique bovine peripheral-type benzodiazepine receptor isoquinoline binding protein. J Biol Chem. 1991;266:14082–14087. [PubMed] [Google Scholar]
- Patte C, Gandolfo P, Leprince J, Thoumas JL, Fontaine M, Vaudry H, et al. GABA inhibits endozepine release from cultured rat astrocytes. Glia. 1999;25:404–411. [PubMed] [Google Scholar]
- Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- Pedersen MD, Minuzzi L, Wirenfeldt M, Meldgaard M, Slidsborg C, Cumming P, et al. Up-regulation of PK11195 binding in areas of axonal degeneration coincides with early microglial activation in mouse brain. Eur J Neurosci. 2006;24:991–1000. doi: 10.1111/j.1460-9568.2006.04975.x. [DOI] [PubMed] [Google Scholar]
- Petit-Taboue MC, Baron JC, Barre L, Travere JM, Speckel D, Camsonne R, et al. Brain kinetics and specific binding of [11C]-PK 11195 to omega 3 sites in baboons: positron emission tomography study. Eur J Pharmacol. 1991;200:347–351. doi: 10.1016/0014-2999(91)90594-g. [DOI] [PubMed] [Google Scholar]
- Price CJ, Wang D, Menon DK, Guadagno JV, Cleij M, Fryer T, et al. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke. 2006;37:1749–1753. doi: 10.1161/01.STR.0000226980.95389.0b. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp Neurol. 2000;161:102–114. doi: 10.1006/exnr.1999.7269. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Ramsay SC, Weiller C, Myers R, Cremer JE, Luthra SK, Lammertsma AA, et al. Monitoring by PET of macrophage accumulation in brain after ischaemic stroke. Lancet. 1992;339:1054–1055. doi: 10.1016/0140-6736(92)90576-o. [DOI] [PubMed] [Google Scholar]
- Rao VL, Bowen KK, Rao AM, Dempsey RJ. Up-regulation of the peripheral-type benzodiazepine receptor expression and [3H]-PK11195 binding in gerbil hippocampus after transient forebrain ischemia. J Neurosci Res. 2001;64:493–500. doi: 10.1002/jnr.1101. [DOI] [PubMed] [Google Scholar]
- Richards JG, Mohler H. Benzodiazepine receptors. Neuropharmacology. 1984;23:233–242. doi: 10.1016/0028-3908(84)90064-9. [DOI] [PubMed] [Google Scholar]
- Riond J, Mattei MG, Kaghad M, Dumont X, Guillemot JC, Le Fur G, et al. Molecular cloning and chromosomal localization of a human peripheral-type benzodiazepine receptor. Eur J Biochem. 1991;195:305–311. doi: 10.1111/j.1432-1033.1991.tb15707.x. [DOI] [PubMed] [Google Scholar]
- Rojas S, Martin A, Arranz MJ, Pareto D, Purroy J, Verdaguer E, et al. Imaging brain inflammation with [11C]-PK11195 by PET and induction of the peripheral-type benzodiazepine receptor after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600500. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Ruff MR, Pert CB, Weber RJ, Wahl LM, Wahl SM, Paul SM. Benzodiazepine receptor-mediated chemotaxis of human monocytes. Science. 1985;229:1281–1283. doi: 10.1126/science.2994216. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Choi HB, McLarnon JG. Peripheral benzodiazepine receptor ligand PK11195 reduces microglial activation and neuronal death in quinolinic acid-injected rat striatum. Neurobiol Dis. 2005;20:550–561. doi: 10.1016/j.nbd.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Schoemaker H, Bliss M, Yamamura HI. Specific high-affinity saturable binding of [3H]-Ro5-4864 to benzodiazepine binding sites in the rat cerebral cortex. Eur J Pharmacol. 1981;71:173–175. doi: 10.1016/0014-2999(81)90405-2. [DOI] [PubMed] [Google Scholar]
- Schumacher M, Akwa Y, Guennoun R, Robert F, Labombarda F, Desarnaud F, et al. Steroid synthesis and metabolism in the nervous system: trophic and protective effects. J Neurocytol. 2000;29:307–326. doi: 10.1023/a:1007152904926. [DOI] [PubMed] [Google Scholar]
- Slobodyansky E, Guidotti A, Wambebe C, Berkovich A, Costa E. Isolation and characterization of a rat brain triakontatetraneuropeptide, a posttranslational product of diazepam binding inhibitor: specific action at the Ro5-4864 recognition site. J Neurochem. 1989;53:1276–1284. doi: 10.1111/j.1471-4159.1989.tb07425.x. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Verma A, Trifiletti RR. The peripheral-type benzodiazepine receptor: a protein of mitochondrial outer membranes utilizing porphyrins as endogenous ligands. FASEB J. 1987;1:282–288. doi: 10.1096/fasebj.1.4.2820823. [DOI] [PubMed] [Google Scholar]
- Sprengel R, Werner P, Seeburg PH, Mukhin AG, Santi MR, Grayson DR, et al. Molecular cloning and expression of cDNA encoding a peripheral-type benzodiazepine receptor. J Biol Chem. 1989;264:20415–20421. [PubMed] [Google Scholar]
- Sriram K, O’Callaghan JP. Signaling mechanisms underlying toxicant-induced gliosis. In: Aschner M, Costa L, editors. The Role of Glia in Neurotoxicity. 2. chapter 9. CRC press; 2004. pp. 141–171. [Google Scholar]
- Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15:5263–5274. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL. Role of microglia in the central nervous system’s immune response. Neurol Res. 2005;27:685–691. doi: 10.1179/016164105X49463a. [DOI] [PubMed] [Google Scholar]
- Streit WJ. The role of microglia in neurotoxicity. In: Aschner M, Costa L, editors. The Role of Glia in Neurotoxicity. 2. chapter 2. CRC press; 2004. pp. 29–41. [Google Scholar]
- Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol Pathol. 2000;28:28–30. doi: 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1:301–307. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–1766. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- Tallman JF, Paul SM, Skolnick P, Gallager DW. Receptors for the age of anxiety: pharmacology of the benzodiazepines. Science. 1980;207:274–281. doi: 10.1126/science.6101294. [DOI] [PubMed] [Google Scholar]
- Tallman JF, Thomas JW, Gallager DW. GABAergic modulation of benzodiazepine binding site sensitivity. Nature. 1978;274:383–385. doi: 10.1038/274383a0. [DOI] [PubMed] [Google Scholar]
- Torres SR, Frode TS, Nardi GM, Vita N, Reeb R, Ferrara P, et al. Anti-inflammatory effects of peripheral benzodiazepine receptor ligands in two mouse models of inflammation. Eur J Pharmacol. 2000;408:199–211. doi: 10.1016/s0014-2999(00)00760-3. [DOI] [PubMed] [Google Scholar]
- Turkheimer FE, Banati RB, Visvikis D, Aston JA, Gunn RN, Cunningham VJ. Modeling dynamic PET-SPECT studies in the wavelet domain. J Cereb Blood Flow Metab. 2000;20:879–893. doi: 10.1097/00004647-200005000-00015. [DOI] [PubMed] [Google Scholar]
- Turkheimer FE, Edison P, Pavese N, Roncaroli F, Anderson AN, Hammers A, et al. Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med. 2007;48:158–167. [PubMed] [Google Scholar]
- Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C]-(R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–609. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Unger JW. Glial reaction in aging and Alzheimer’s disease. Microsc Res Tech. 1998;43:24–28. doi: 10.1002/(SICI)1097-0029(19981001)43:1<24::AID-JEMT4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Veenman L, Papadopoulos V, Gavish M. Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des. 2007;13:2385–2405. doi: 10.2174/138161207781368710. [DOI] [PubMed] [Google Scholar]
- Veenman L, Leschiner S, Spanier I, Weisinger G, Weizman A, Gavish M. PK 11195 attenuates kainic acid-induced seizures and alterations in peripheral-type benzodiazepine receptor (PBR) protein components in the rat brain. J Neurochem. 2002;80:917–927. doi: 10.1046/j.0022-3042.2002.00769.x. [DOI] [PubMed] [Google Scholar]
- Veiga S, Azcoitia I, Garcia-Segura LM. Ro5-4864, a peripheral benzodiazepine receptor ligand, reduces reactive gliosis and protects hippocampal hilar neurons from kainic acid excitotoxicity. J Neurosci Res. 2005;80:129–137. doi: 10.1002/jnr.20430. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wang G, Bissel SJ, Mathis CA, Meltzer CC, et al. PET imaging of brain macrophages using the peripheral benzodiazepine receptor in a macaque model of neuroAIDS. J Clin Invest. 2004;113:981–989. doi: 10.1172/JCI20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006;80:308–322. doi: 10.1016/j.pneurobio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wang G, Slagel SL, Mason NS, Mathis CA, et al. A comparison of the high-affinity peripheral benzodiazepine receptor ligands DAA1106 and (R)-PK11195 in rat models of neuroinflammation: implications for PET imaging of microglial activation. J Neurochem. 2007;102:2118–2131. doi: 10.1111/j.1471-4159.2007.04690.x. [DOI] [PubMed] [Google Scholar]
- Verma A, Nye JS, Snyder SH. Porphyrins are endogenous ligands for the mitochondrial (peripheral-type) benzodiazepine receptor. Proc Natl Acad Sci U S A. 1987;84:2256–2260. doi: 10.1073/pnas.84.8.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Snyder SH. Peripheral type benzodiazepine receptors. Annu Rev Pharmacol Toxicol. 1989;29:307–322. doi: 10.1146/annurev.pa.29.040189.001515. [DOI] [PubMed] [Google Scholar]
- Versijpt J, Debruyne JC, Van Laere KJ, De Vos F, Keppens J, Strijckmans K, et al. Microglial imaging with positron emission tomography and atrophy measurements with magnetic resonance imaging in multiple sclerosis: a correlative study. Mult Scler. 2005;11:127–134. doi: 10.1191/1352458505ms1140oa. [DOI] [PubMed] [Google Scholar]
- Versijpt JJ, Dumont F, Van Laere KJ, Decoo D, Santens P, Audenaert K, et al. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. A pilot study. Eur Neurol. 2003;50:39–47. doi: 10.1159/000070857. [DOI] [PubMed] [Google Scholar]
- Vowinckel E, Reutens D, Becher B, Verge G, Evans A, Owens T, et al. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res. 1997;50:345–353. doi: 10.1002/(SICI)1097-4547(19971015)50:2<345::AID-JNR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Wang JK, Morgan JI, Spector S. Benzodiazepines that bind at peripheral sites inhibit cell proliferation. Proc Natl Acad Sci U S A. 1984;81:753–756. doi: 10.1073/pnas.81.3.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman BA, Raveh L. Peripheral benzodiazepine receptors: on mice and human brain imaging. J Neurochem. 2003;84:432–437. doi: 10.1046/j.1471-4159.2003.01568.x. [DOI] [PubMed] [Google Scholar]
- Woods MJ, Williams DC. Multiple forms and locations for the peripheral-type benzodiazepine receptor. Biochem Pharmacol. 1996;52:1805–1814. doi: 10.1016/s0006-2952(96)00558-8. [DOI] [PubMed] [Google Scholar]
- Woods MJ, Zisterer DM, Williams DC. Two cellular and subcellular locations for the peripheral-type benzodiazepine receptor in rat liver. Biochem Pharmacol. 1996;51:1283–1292. doi: 10.1016/0006-2952(96)00034-2. [DOI] [PubMed] [Google Scholar]