

Abstract
Single-minded 1 (Sim1) encodes a transcription factor essential for formation of the hypothalamic paraventricular nucleus (PVN). Sim1 haploinsufficiency is associated with hyperphagic obesity and increased linear growth in humans and mice, similar to the phenotype of melanocortin 4 receptor (Mc4r) mutations. PVN neurons in Sim1+/− mice are hyporesponsive to the melanocortin agonist melanotan II. PVN neuropeptides oxytocin (Oxt), TRH and CRH inhibit feeding when administered centrally. Consequently, we hypothesized that altered PVN neuropeptide expression mediates the hyperphagia of Sim1+/− mice. To test this hypothesis, we measured hypothalamic expression of PVN neuropeptides in Sim1+/− and wild-type mice. Oxt mRNA and peptide were decreased by 80% in Sim1+/− mice, whereas TRH, CRH, arginine vasopressin (Avp), and somatostatin mRNAs were decreased by 20–40%. Sim1+/− mice also showed abnormal regulation of Oxt but not CRH mRNA in response to feeding state. A selective Mc4r agonist activated PVN Oxt neurons in wild-type mice, supporting involvement of these neurons in melanocortin feeding circuits. To test whether Oxt itself regulates feeding, we measured the effects of central administration of an Oxt receptor antagonist or repeated doses of Oxt on food intake of Sim1+/− and wild-type mice. Sim1+/− mice were hypersensitive to the orexigenic effect of the Oxt receptor antagonist. Oxt decreased the food intake and weight gain of Sim1+/− mice at a dose that did not affect wild-type mice. Our results support the importance of Oxt neurons in feeding regulation and suggest that reduced Oxt neuropeptide is one mechanism mediating the hyperphagic obesity of Sim1+/− mice.
SINGLE-MINDED 1 (SIM1) encodes a member of the basic helix-loop-helix Per Arnt Sim family of nuclear transcription factors (1). SIM1 is one of six genes implicated in human monogenic obesity (2). A balanced chromosomal translocation that disrupts one allele of SIM1 was identified in a girl with severe early-onset obesity, hyperphagia, and accelerated linear growth (3). Recently, nonsynonymous SIM1 sequence variants were found in 6 of 379 obese adults vs. 0/378 lean controls (4), similar to the frequencies of nonsynonymous MC4R mutations (eight in obese group vs. two in lean group in the same study), the most common monogenic cause of early-onset morbid obesity and also associated with tall stature (5).
Heterozygous inactivation of the murine Sim1 gene (Sim1+/−) leads to an analogous phenotype of hyperphagic obesity and increased linear growth, with enhanced sensitivity to diet-induced obesity (6,7). Homozygous Sim1 knockout mice die shortly after birth and exhibit failure of terminal migration and differentiation of the neurons of the paraventricular (PVN), supraoptic (SON), and anterior periventricular nuclei of the hypothalamus (8,9), which produce the neuropeptides arginine vasopressin (Avp), oxytocin (Oxt), CRH, TRH, and somatostatin (Sst). Michaud et al. (7) reported that Sim1+/− mice have a 24% reduction in PVN cellularity measured by Nissl staining, with no specific neuropeptide subtype affected, leading them to propose that the phenotype of hyperphagic obesity is due to a hypothalamic developmental defect. By contrast, we found no difference in the number of PVN Sim1 neurons in Sim1+/− vs. Sim1+/+ mice, using a Sim1-enhanced green fluorescent protein (GFP) transgenic reporter to mark these neurons (10). In the absence of a gross neuroanatomic defect in Sim1+/− mice, it is unclear which PVN neurons are important for the regulation of feeding behavior by Sim1. Of the neuropeptides produced in the PVN, Oxt (11,12,13,14), TRH (15,16), and CRH (17,18,19,20) have consistently been shown to have an anorectic effect when given centrally to rodents.
Sim1 is expressed postdevelopmentally in the PVN and supraoptic nucleus (SON) as well as the basomedial amygdala and a subset of lateral hypothalamic neurons (6). The Sim1+/− phenotype is suggestive of defective hypothalamic melanocortin signaling (21), and we hypothesized that Sim1 physiologically regulates body weight by modulating PVN Mc4r signaling (6). Consistent with this hypothesis, PVN neurons of Sim1+/− mice showed impaired activation in response to peripheral injection of melanotan II, a melanocortin receptor agonist (10). Furthermore, transgenic overexpression of human SIM1 partially suppressed both diet-induced and Agouti yellow (Ay) obesity by reducing feeding (22). Moreover, viral-mediated overexpression of Sim1 in the PVN of adult mice reduced food intake, whereas small interfering RNA-mediated inhibition of Sim1 expression increased food consumption (23). These findings suggest that Sim1 acts postdevelopmentally to regulate feeding.
Leptin secreted by adipocytes activates receptors in the arcuate nucleus (ARC), increasing proopiomelanocortin expression and stimulating release of α-MSH by ARC neurons that project to the PVN and LH (24). This hypothalamic adiposity signal is integrated with hindbrain satiety signals such as cholecystokinin (CCK), mediated by vagal afferents (25,26,27,28,29). Neuroanatomic evidence suggests that the PVN is central to this interaction (30,31). In rats, the neurons responsible for this interaction may be parvocellular oxytocinergic neurons in the posterior PVN projecting to the hindbrain and spinal cord (32,33,34,35). Fourth ventricular injection of an Oxt receptor antagonist attenuated the effect of leptin on food intake, suggesting that Oxt itself is an important signal in addition to classical neurotransmitters (36). Interestingly, patients with Prader-Willi syndrome, a human genetic disorder characterized by severe obesity, have a 42% reduction of parvocellular PVN Oxt neurons (37). Additionally, the levels of circulating Oxt in these patients are abnormally low for their degree of obesity (38). The gene regulatory network that controls Oxt cell development is conserved in vertebrate species as diverse as zebrafish, chicks, and mice (39,40). Zebrafish Sim1 is required for the development of neurons that produce isotocin, the fish homolog of Oxt (39). It is not known whether the Oxt gene is also a transcriptional target of Sim1.
To further examine the effect of Sim1 haploinsufficiency on PVN neuronal function, we evaluated expression of PVN neuropeptides in Sim1+/− mice. We also compared the regulation of Oxt and CRH in these mice in response to fasting and refeeding. We further investigated the effect of an oxytocin receptor antagonist, OVT (d(CH2)5, Tyr(Me)2,Orn8-Oxytocin) on food intake in Sim1+/− vs. wild-type mice and tested whether PVN Oxt neurons in wild-type mice are activated by an Mc4r-selective agonist. Finally, we measured the effect of chronic intracerebroventricular (icv) Oxt injections on food intake and body weight of Sim1+/− and wild-type mice. Our findings support the role of PVN Oxt neurons and Oxt itself in the regulation of feeding and suggest a neuroendocrine mechanism for the hyperphagia of Sim1+/− mice.
RESULTS
Sim1+/− Mice Exhibit Reduced Expression of PVN Oxt
To determine which PVN neurons are sensitive to reduced Sim1 dosage, we compared the hypothalamic mRNA levels of the neuropeptides CRH, TRH, Oxt, Sst, and Avp in Sim1+/− and wild-type mice. All mRNAs were decreased in Sim1+/− mice, with the greatest reduction (−80%) in Oxt (Fig. 1). We next performed immunostaining for Oxt and CRH, for which suitable antibodies were available, to determine whether the reductions in hypothalamic mRNAs resulted in reduced PVN neuropeptide levels. Oxt but not CRH immunoreactivity was demonstrably reduced (Fig. 2). Although difficult to determine in the absence of another marker of Oxt neurons, the marked reduction in Oxt immunoreactivity may reflect a decreased number of Oxt neurons rather than reduced staining within existing neurons (Fig. 2). The reduction in Oxt immunoreactivity was evident throughout the PVN. There was a similar reduction in Oxt peptide expression in the SON (data not shown).
Figure 1.
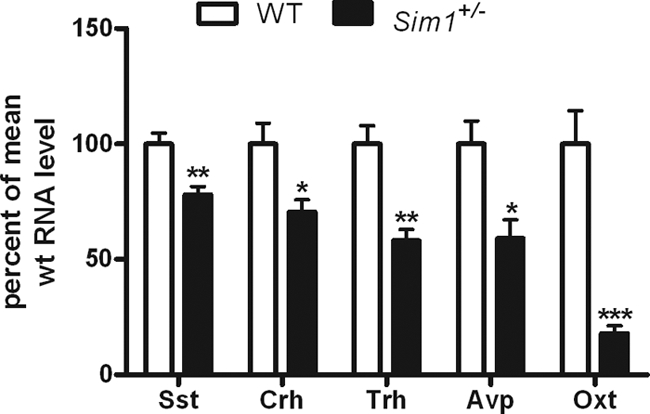
Sim1+/− Mice Exhibit Reduced mRNA Expression of PVN Neuropeptides
Real-time PCR showing hypothalamic expression of Sst, CRH, TRH, Avp, and Oxt mRNA in Sim1+/− mice compared with wild-type mice (n = 5 for each group). Groups were compared using a two-tailed unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars indicate sem. WT, Wild type.
Figure 2.

Sim1+/− Mice Exhibit Reduced Expression of PVN Oxt But Not CRH Peptide
A, Representative images of immunohistochemical analysis of Oxt and CRH peptides in PVN of Sim1+/− mice compared with wild-type mice (captured with ×10 and ×20 objectives). B, Cell counts and densitometry measurements of Oxt and CRH immunohistochemistry. For B, groups were compared using a two-tailed unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars indicate sem. Numbers of animals are shown in the figure. WT, Wild type.
Sim1+/− Mice Fail to Regulate Hypothalamic Oxt mRNA in Response to Feeding State
Wild-type mice showed reduced Oxt mRNA levels in the hypothalamus with fasting that were restored with refeeding, whereas CRH mRNA displayed the opposite pattern (Fig. 3). This was consistent with previous studies (41,42). By contrast, Oxt mRNA in Sim1+/− mice was not changed by fasting or refeeding, although these mice showed normal regulation of CRH expression in response to feeding state (Fig. 3).
Figure 3.
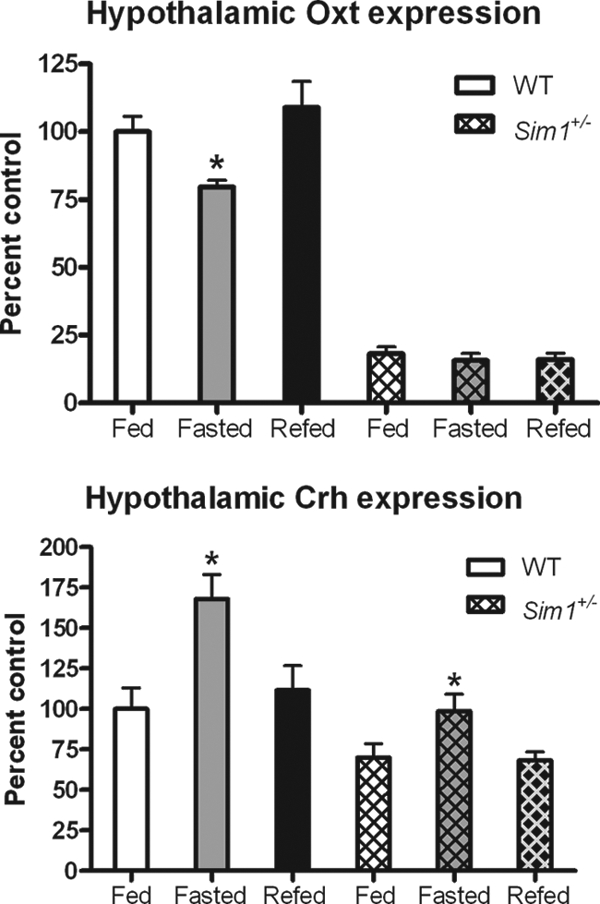
Sim1+/− Mice Fail to Regulate Oxt mRNA Expression in Response to Feeding State
Real-time PCR showing hypothalamic expression of CRH and Oxt mRNAs in fed, fasted, and refed Sim1+/− mice vs. wild-type mice (n = 5 for all groups except wild type fasted and Sim1+/− refed, where n = 4). Each subgroup (e.g. wild-type Oxt) was analyzed using one-way ANOVA with Newman-Keuls multiple comparison post-test. (*, P < 0.05 indicates that the fasted group is significantly different from the fed or refed groups). WT, Wild type.
Oxt Is Colocalized in a Subset of Sim1 Neurons in the PVN
To determine whether PVN Sim1 neurons coexpress Oxt, we used a Sim1-GFP transgenic mouse line as reported previously (10). Figure 4A shows two color immunofluorescent detection of Oxt (red ) and GFP (green) in PVN sections harvested from a wild-type mouse not pretreated with colchicine. The result shows that Oxt is colocalized with GFP in a subset of Sim1 neurons in the PVN.
Figure 4.

Oxt Is Colocalized with Sim1 in PVN Neurons and PVN Oxt Neurons Are Activated by Central Mc4r-Selective Agonist Injection
A, Representative ×40 images showing colocalization of Oxt (red) and Sim1-GFP (green). B, Colocalization of Oxt (green) and c-Fos (red) after aCSF or Mc4r-selective agonist (3 μg) icv (×40). After Mc4r-selective agonist treatment, most c-Fos+ neurons in the region shown are Oxt+, and many Oxt+ neurons are c-Fos+ (arrows).
PVN Oxt Neurons Are Activated by Central Injection of an Mc4r-Selective Agonist
To determine whether PVN Oxt neurons might be germane to the defective melanocortin response of Sim1+/− mice, we treated wild-type mice icv with the Mc4r agonist cyclo(β-Ala-His-D-Phe-Arg-Trp-Glu)-NH2 and tested the response of Oxt neurons using induction of c-Fos immunoreactivity as a marker of neuronal activation. This agonist is highly selective for the Mc4r (>90-fold selectivity over Mc3r and >3400-fold selectivity over Mc5r) (43). In mice it acts centrally to reduce 2-h food intake by 50% at a dose of 1 μg (44). Figure 4B shows two color immunofluorescent detection of c-Fos (red) and Oxt (green) in hypothalamic sections harvested 6 h after icv injection of the Mc4r agonist or artificial cerebrospinal fluid (aCSF) from a wild-type mouse pretreated with colchicine. The result shows that c-Fos was robustly induced in many PVN Oxt neurons at this time point. A control colchicine-pretreated animal that received aCSF vehicle showed negligible c-Fos immunoreactivity. PVN Oxt immunoreactivity decreased after Mc4r agonist treatment in both wild-type and Sim1+/− mice (data not shown). This decrease, coupled with the already low level of Oxt immunoreactivity in Sim1+/− mice, precluded simultaneous detection or quantitation of c-Fos- and Oxt-positive neurons in these animals.
Central Oxt Receptor Antagonist Administration Exacerbates Hyperphagia of Sim1+/− Mice
We hypothesized that reduced Oxt neuropeptide levels in Sim1+/− mice might render these mice hypersensitive to further inhibition of Oxt signaling. To test this hypothesis, we treated Sim1+/− mice and wild-type littermates icv with the Oxt receptor antagonist OVT (d(CH2)5, Tyr(Me)2,Orn8-Oxytocin). An OVT dose of 0.5 μg did not affect feeding of wild-type mice but increased the food intake of the already hyperphagic Sim1+/− mice by approximately 50% (Fig. 5).
Figure 5.

Central Administration of Oxt Receptor Antagonist OVT Exacerbates Hyperphagia of Sim1+/− Mice at a Dose that Does Not Affect Food Intake of Wild-Type Mice
After 1 wk of habituation to daily icv injection, mice were injected at the onset of the dark cycle with aCSF on d 1 and 0.5 μg OVT on d 2. OVT food intake was normalized to aCSF food intake for each mouse. Means were calculated for each treatment and compared using a paired t test (n = 14 for wild-type group; n = 11 for Sim1+/− group; ***, P < 0.001). WT, Wild type.
Central Oxt Injection Rescues Hyperphagic Obesity of Sim1+/− Mice
To further substantiate the role of Oxt deficiency in the phenotype of Sim1+/− mice, we tested whether Oxt replacement could rescue their hyperphagia and obesity. Sim1+/− mice and wild-type littermates were injected twice daily icv with either 10 ng of Oxt or vehicle (aCSF). Daily food intake and body weight were measured over 12 d. Oxt normalized food intake of Sim1+/− mice but had no effect on food intake of wild-type mice (Fig. 6A), arguing against a nonspecific anorectic effect such as nausea. Repeated Oxt injections also led to decreased weight gain compared with vehicle treatment in Sim1+/− mice but not their wild-type littermates (Fig. 6B). Mice were weighed again after a 12-d washout period after completion of icv injections. Sim1+/− mice that had previously received Oxt or aCSF gained a similar amount of weight (Fig. 6C).
Figure 6.
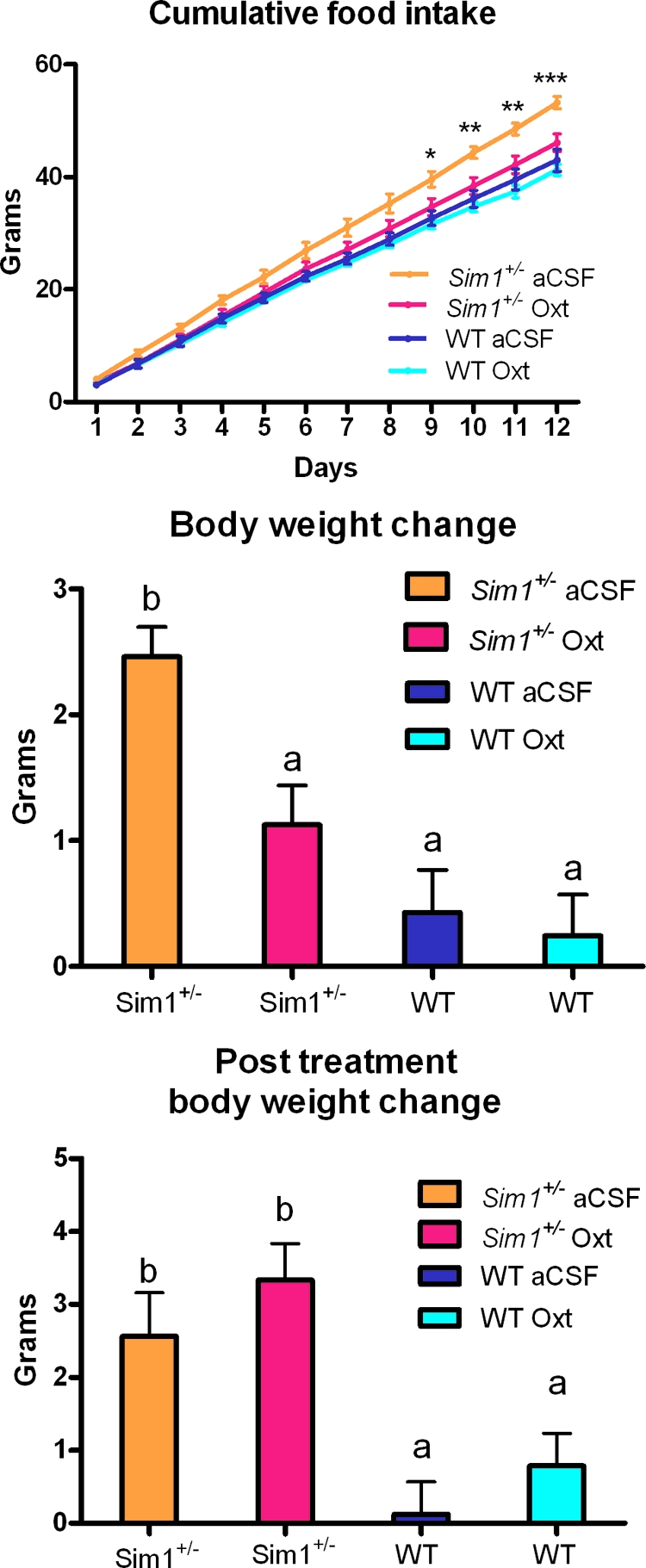
Central Oxt Injection Rescues Hyperphagia and Reduces Weight Gain of Sim1+/− Mice but Does Not Affect Food Intake or Weight Gain of Wild-Type Mice
A and B, After 1 wk of habituation to twice daily icv injection, mice were injected twice daily with either aCSF or 10 ng Oxt. Food intake and body weight were measured daily for 12 d. Food intake was analyzed by two-way ANOVA (group vs. time) and body weight change was analyzed with one-way ANOVA (group). Both analyses were done with a Bonferroni posttest to determine intergroup significance. C, Body weight was measured again and compared with body weight 12 d after the last injection (d 24) on d 12. Groups were compared using one-way ANOVA with a Bonferroni posttest. For A: *, P < 0.05; **, P < 0.01; ***, P < 0.001. For B, and C, groups with different letters are statistically different (P < 0.05). n = 7 for wild-type aCSF; n = 5 for Sim1+/− aCSF; n = 7 for wild-type Oxt; n = 8 for Het Oxt. WT, Wild type.
Wild-Type Mice Are Insensitive to High Doses of icv Oxt but Respond to the Oxt Receptor Antagonist OVT
Having shown that food intake of Sim1+/− mice was sensitive to both OVT and Oxt at doses that did not change food intake of wild-type mice, we sought to investigate the dose-response of wild-type mice to both agents. ICV Oxt has been consistently shown to reduce food intake of rats at doses of 1–4 nmol (∼1–4 μg) (11,12,13) but not mice (45). ICV OVT blocks anorexia in rats at doses of 9 nmol (∼9 μg). Doses of Oxt as high as 1 μg failed to inhibit food intake in wild-type mice (Fig. 7A). On the other hand, OVT was capable of increasing food intake in wild-type mice at a dose of 1.5 μg (Fig. 7B).
Figure 7.
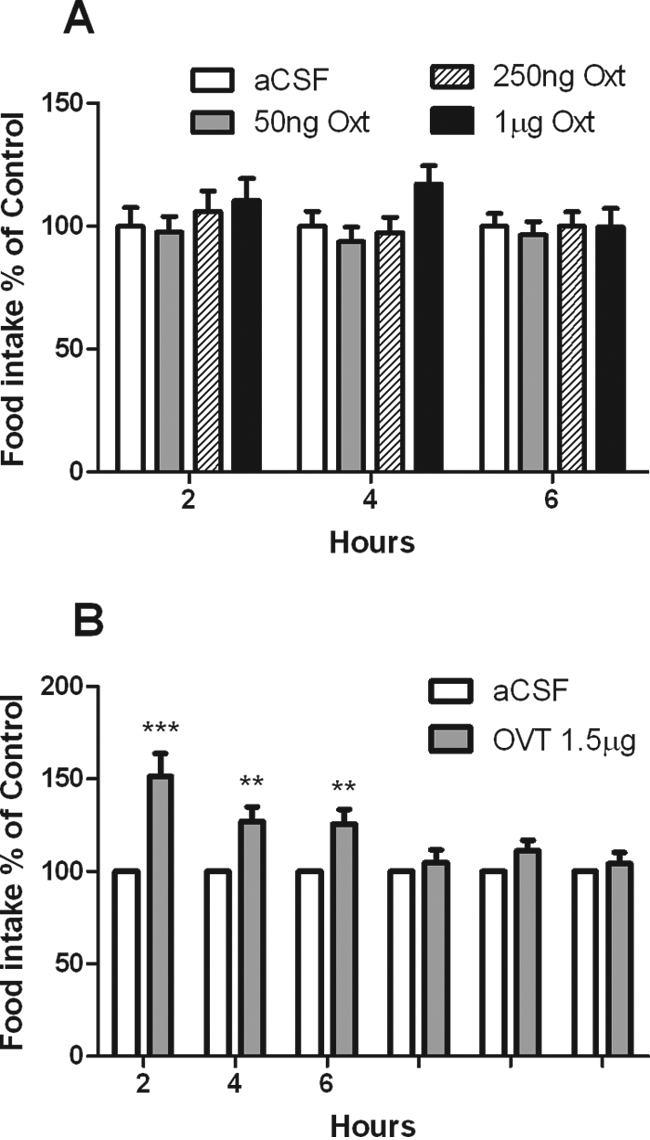
Wild-Type Mice Are Insensitive to High Doses of Intracerebroventricular Oxt but Are Sensitive to the Oxt Receptor Antagonist OVT
A, After 1 wk of habituation to daily icv injection, mice were injected at the onset of the dark cycle with either aCSF or 50 ng, 250 ng, or 1 μg of Oxt. Food intake was normalized to aCSF food intake for each mouse. Means were calculated for each treatment, and the groups at each time point were compared using one-way ANOVA (n = 9, P = NS). B, After 1 wk of habituation to daily icv injection, mice were injected at the onset of the dark cycle with aCSF on d 1 and 1.5 μg OVT on d 2. Food intake was normalized to aCSF food intake for each mouse. Means were calculated for each treatment, and the groups at each time point were compared using a paired t test. (n = 11; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
DISCUSSION
We previously showed that Sim1+/− mice have normal hypothalamic Mc4r mRNA levels but blunted anorexia and activation of PVN neurons in response to the Mc3r/Mc4r agonist melanotan II (10). The total number of PVN neurons was unchanged in Sim1+/− mice, and these neurons appeared to induce c-Fos normally in response to another stimulus, hypertonic saline. We also showed that overexpression of SIM1 rescued hyperphagia of Ay mice, with no effect on energy expenditure (22). These findings led us to propose that Sim1 functions downstream of Mc4r in the PVN.
Sim1 is necessary for the development of all PVN neurons. Given that mouse Sim1 is required for the development of neurons of the PVN, SON, and anterior periventricular nucleus expressing AVP, Oxt, CRH, TRH, and Sst, we hypothesized that one or more of these neuronal populations may be affected by Sim1 haploinsufficiency and that this may mediate the hyperphagic obesity of Sim1+/− mice. Microarray expression profiling revealed that Oxt mRNA was reduced in the hypothalamus of Sim1+/− mice compared with controls (data not shown). These results were confirmed in the present study using quantitative RT-PCR, revealing a marked (∼80%) reduction in Oxt expression in Sim1+/− mice, with a similar decrease in Oxt peptide as measured by immunohistochemistry. The reduction in Oxt peptide in the PVN appeared to be global and likely affected both magnocellular and parvocellular neurons, although these populations cannot be distinguished morphologically or anatomically in mice (Ref. 46 and J. Elmquist, personal communication). Other PVN neuropeptide mRNAs were reduced in Sim1+/− mice, but to a much lesser degree than Oxt.
It is unclear whether reduced Oxt expression in Sim1+/− mice was due to a decreased number of Oxt neurons or to decreased Oxt expression in most of these neurons below the threshold for immunodetection. Presently we have no way other than Oxt expression to mark these neurons. By contrast, CRH neurons did not show measurable reduction in either number or peptide expression, suggesting that Sim1 haploinsufficiency preferentially affects Oxt neurons. These findings are consistent with a conserved role for Sim1 in development of Oxt (or isotocin in zebrafish) neuronal lineages (39).
The Oxt promoter has not been well characterized, even in the well-known context of regulation by estrogen (47,48). Inspection of the upstream or downstream 5 kb of genomic sequence with Regulatory VISTA (rVISTA) (49) and the UCSC Genome Browser (50) did not reveal any conserved binding sites for Sim1 or its heterodimer partner Arnt2, suggesting that if Sim1 physiologically regulates Oxt expression, the mechanism is indirect.
There is a large body of literature supporting the role of Oxt and Oxt neurons in regulation of feeding in both humans and rodents. Patients with Prader-Willi syndrome have a 42% reduction of parvocellular PVN Oxt neurons (37). Plasma Oxt levels are elevated in subjects with common obesity and decrease after gastric banding (51), suggesting that there may be Oxt resistance analogous to leptin resistance. Centrally administered Oxt has been shown to reduce food intake in rats, and this effect is blocked by Oxt receptor antagonists (11,12,13). Oxt is secreted into the bloodstream by magnocellular neurons in response to exogenous CCK, and serum Oxt levels are directly proportional to the degree of inhibition of food intake by this treatment (42). Despite these observations, peripheral administration of Oxt at physiological levels does not modulate food intake in rats, suggesting that peripheral release takes place in parallel with central effects on feeding. Peripheral administration of Oxt in rats at high doses has been shown to reduce food intake by some investigators (12,13) but not others (52).
Neuroanatomic and pharmacological studies in rats point to parvocellular PVN Oxt neurons that project monosynaptically to the hindbrain as being important in satiety. Oxt receptor expression in the mouse hindbrain is greatest in the nucleus of the solitary tract (NTS). A known satiety center (53), and there is evidence that Oxt preferentially regulates food intake in the hindbrain (36). Furthermore, oxytocinergic projections to the nucleus of the solitary tract in newborn rats come solely from the PVN (33). Disruption of PVN oxytocinergic fibers projecting to the hindbrain leads to hyperphagic obesity (34). Furthermore, PVN Oxt neuron axonal projections interact with NTS neurons that are activated by CCK (35). Finally, Baskin and colleagues (35,36) characterized a subset of parvocellular PVN Oxt neurons that both respond to leptin and project to the NTS and showed that injection of OVT into the fourth ventricle attenuated the effect of leptin on food intake. These results support the notion that PVN parvocellular Oxt neurons transmit hypothalamic adiposity signals to the NTS, where they are integrated with gut satiety signals. Furthermore, there is evidence that the parvocellular PVN itself also integrates adiposity signals such as leptin with satiety signals from the NTS. This integration is then followed by modulation of NTS neurons that reduce meal size. This evidence comes from the work of Moran and colleagues (54), who showed that leptin modulates CCK-induced c-Fos in both the PVN and NTS, as well as the work of Verbalis and colleagues (55), who showed that CCK activates oxytocinergic parvocellular PVN neurons that then project to the dorsal vagal complex (dorsal motor nucleus of the vagus and NTS).
Further evidence for the role of PVN Oxt neurons in feeding regulation comes from a report showing an effect of ghrelin on these neurons (56). In this study, Levine and colleagues showed that the Oxt receptor antagonist OVT exacerbated ghrelin-induced hyperphagia. Others have shown that parvocellular and magnocellular PVN Oxt neurons are activated by insulin (57). Hypothalamic Oxt has also been implicated in the regulation of food intake during pregnancy (58).
Studies of a Sim1-GFP transgenic mouse showed that essentially all cells in the PVN expressing NeuN, a marker of neurons but not glial cells, were also GFP positive (our unpublished results). Based on these and other data (59), we proposed that all adult mouse PVN neurons express Sim1 (10). Here we showed that Oxt colocalized in a subset of Sim1 neurons (Fig. 4A). Colocalization of Oxt and Mc4r has been previously demonstrated in mouse PVN neurons (32). It is also clear from the work of Balthasar et al. (59) that Mc4r is expressed in PVN Sim1 neurons. Together, the data indicate that a subset of PVN neurons coexpress Sim1, Mc4r, and Oxt. Our previous results suggest that Sim1 functions downstream of Mc4r (10), and we show here that proper Oxt expression is dependent upon Sim1. Thus, there may exist a molecular pathway from Mc4r to Sim1 to Oxt within PVN neurons, although the notion of a linear relationship is likely an oversimplification of the relevant circuits.
Our results further bolster the relevance of PVN Oxt neurons in the melanocortin feeding circuitry by showing that these neurons are activated by a centrally administered Mc4r-selective agonist (Fig. 4B). Regulation of Oxt neurons by melanocortin agonists is not limited to the PVN but has also been shown in the SON, where α-MSH induces the release of Oxt from the dendrites of magnocellular neurons while inhibiting its secretion from nerve terminals in the posterior pituitary (60,61).
A key question is whether reduced expression of Oxt is causally related to the hyperphagia of Sim1+/− mice. To answer this question, we examined the effect of both an Oxt receptor antagonist and Oxt on food intake in Sim1+/− vs. wild-type. We reasoned that if Oxt simply marks absent or defective PVN neurons in Sim1+/− mice but is not itself involved in feeding regulation, then treatment with Oxt or an Oxt receptor antagonist should not differentially affect food intake of Sim1+/− vs. wild-type mice. On the other hand, if Oxt deficiency is mechanistically related to the hyperphagia of Sim1+/− mice, then administration of an Oxt receptor antagonist might exacerbate and administration of Oxt might ameliorate their hyperphagia.
The results of our pharmacological experiments clearly support the conclusion that Oxt neuropeptide deficiency per se contributes to the hyperphagic obesity in Sim1+/− mice. Further experiments are required to address the neuronal mechanism of Oxt action. For instance, Oxt may act as a neuromodulator of synaptic signaling by classical neurotransmitters released in the NTS by parvocellular PVN neuronal projections.
Despite the large body of evidence implicating Oxt in food intake regulation, wild-type and Oxt−/− mice ingest similar amounts of standard chow ad libitum, after overnight food deprivation when drinking water is available, and after systemic administration of either CCK or d-fenfluramine (62,63). On the other hand, Oxt−/− mice display an increased intake of both sweet and nonsweet carbohydrate solutions (64). Oxt receptor-deficient mice have been generated, but no characterization of their food intake or body weight has been published (65). There are several possible explanations for the apparent discrepancy between genetic models and anatomic and pharmacological data. Oxt may mark the identity of neurons projecting from the PVN to the NTS but not be critical for their action in meal termination, which could be mediated by classical neurotransmitters such as GABA or glutamate. Our results argue against this possibility. Alternatively, Oxt may be an important physiological regulator of feeding in normal mice, but there could be developmental mechanisms that compensate for its absence in Oxt−/− mice. Functional and developmental compensation by hypothalamic neurons has been demonstrated, most notably in Npy/Agrp neurons. Despite compelling pharmacological evidence for a prominent role for these two peptides in energy homeostasis, mice deficient in Npy, Agrp, or both have no demonstrable feeding phenotype (66). On the other hand, partial ablation of Npy/Agrp neurons postnatally leads to the expected lean phenotype, and complete ablation in adulthood leads to starvation (66,67,68,69,70). Developmental compensation by these neurons appears to take place postnatally, because neonatal ablation had minimal effects on body weight or feeding regulation (67). We hypothesize that similar developmental compensation explains the absence of a feeding phenotype in Oxt−/− mice. These compensatory mechanisms may involve compensation by other PVN neuropeptides implicated in feeding regulation, i.e. CRH or TRH. These compensatory mechanisms may be intact in Oxt−/−mice and impaired in Sim1+/− mice, which show moderately decreased mRNA levels of CRH and TRH.
Another explanation for the lack of a feeding phenotype in Oxt−/−mice may be species differences. Our results do not support this possibility. Whereas icv Oxt consistently reduces food intake of rats (11,12,13), no such hypophagic effect has been demonstrated in mice (45). We too could not find such an effect of icv Oxt on wild-type mice. On the other hand, we were able to demonstrate that OVT increased food intake in wild-type mice. These results in wild-type mice, coupled with our results in Sim1+/− mice, support the hypothesis that Oxt exerts a tonic inhibition of feeding in mice. This is consistent with the work of Blevins et al. (35) in rats, who also concluded that Oxt exerts a tonic stimulatory effect on NTS neurons that reduce meal size by showing that fourth ventricular administration of OVT blunted CCK-induced satiety.
Further experiments are needed to determine the site of action of Oxt in rescuing the hyperphagia of Sim1+/− mice and whether Sim1 haploinsufficiency leads to a developmental reduction in Oxt neurons or a postdevelopmental reduction of Oxt expression. Any reduction in the number of PVN neurons must be subtype specific. Because there is no difference in the total PVN Sim1 neuron count of Sim1+/− mice vs. wild-type mice (10), a reduction in the number of Oxt neurons may be due to fate switching. Additional experimental approaches, such as conditional postnatal Sim1 inactivation, are needed to determine whether the decrease in Oxt expression in Sim1+/− mice is developmental or regulatory. Regardless, our results support the importance of the oxytocinergic pathway from the PVN to the NTS in feeding regulation and suggest that reduced Oxt expression is responsible for much of the hyperphagia of Sim1+/− mice (Fig. 8). Further study of the PVN to NTS oxytocinergic pathway may be relevant to our understanding of other genetic causes of human obesity such as Prader-Willi syndrome or MC4R mutations.
Figure 8.

Model Showing ARC Adiposity Signals, e.g., Leptin Relayed via α-MSH to PVN Parvocellular Oxt/Sim1 Neurons Projecting to the Hindbrain, Where They Are Integrated with Satiety Signals
PVN Sim1 neurons may also integrate satiety signals from reciprocal projections from the NTS (not shown) with adiposity signals from the ARC and elsewhere in the brain. POMC, Proopiomelanocortin.
MATERIALS AND METHODS
Animal Care
C57BL6 mice (6–8 wk of age) from the National Cancer Institute were used unless otherwise stated. Mice were kept on a 12-h light, 12-h dark cycle and fed a low-fat chow diet (Global diet 2016; Teklad, Madison, WI) ad libitum. Generation, breeding, and genotyping of Sim1+/− mice were previously described (6). All experimental protocols were approved by the University of Texas Southwestern Institutional Animal Care and Use Committee.
Real-Time PCR
Hypothalami from fresh brains were dissected with a block (David Kopf Instruments, Tujunga, CA) as described (71), using the following landmarks: posteriorly, posterior aspect of median eminence; anteriorly, 5 mm anterior to the median eminence; dorsally, the thalamus; laterally, medial to the dentate gyrus. Total RNA was extracted using Tripure reagent (Roche Applied Science, Indianapolis, IN). Quantitative real-time PCR was performed using an ABI 3700 instrument (Applied Biosystems, Foster City, CA) and the QuantiTect HotStart SYBR green qPCR kit (QIAGEN, Valencia, CA). Neuropeptide measurements were normalized to β-actin or GAPDH mRNA levels. Primers sequences were 5′-GAGGACCTGCGACTAGACTGAC-3′ and 5′-CAGCAGCTCTGCCAAGAAGTA-3′ (Sst); 5′-CGTTGAGAGACTGAAGAGAAAGG-3′ and 5′-GGACGACAGAGCCACCAG-3′ (CRH); 5′-CTTTGATCTTCGTGCTAACTGGT-3′ and 5′-CTTCAACGTCTTCCTCCTTCTC-3′ (mTRH); 5′-CTCTGACATGGAGCTGAGACAG-3′ and 5′-AGGGCAGGTAGTTCTCCTCCT-3′ (AVP); 5′-TGGCTTACTGGCTCTGACCT-3′ and 5′-AGGCAGGTAGTTCTCCTCCTG-3′ (Oxt).
Taqman primers were used for determining Oxt and CRH mRNA levels for feeding state experiments (4331182 and 4351372; Applied BioSytems, Foster City, CA).
5′-GAC GAT GCT CCC CGG GCT GTA TTC-3′ and 5′-TCT CTT GCT CTG GGC CTC GTC ACC-3′ (β-actin) and GAPDH (4352339E, Applied BioSytems) were used for normalization. All reactions were performed at 53 C annealing temperature.
Cannulation (icv)
Mice were anesthetized with ketamine (117 mg/kg) and xylazine (7.92 mg/kg). After shaving the head and applying a microbicide, each animal was placed in a stereotaxic chamber (Stoelting, Wood Dale, IL). A sagittal section was cut and the skin was clipped back. Bregma coordinates were visually determined, and the location of the lateral ventricle was calculated (0.2 mm posterior; 1.0 mm lateral; 2.1 mm deep to skull surface). The site of cannulation on the skull surface was marked. The skull was punctured using an engraving Dremel bit. The guide cannula (C315GS-4/spc; Plastics One, Roanoke, VA) was inserted and glued to the skull surface with dual cure paste (031458550; Den-mat, Santa Maria, CA). A dummy cannula (or dust cap) was placed inside the guide cannula (C315DCS-4/spc; Plastics One). The scalp was closed with dissolvable sutures. Upon waking, the animal was injected ip with 0.1 mg/kg of Buprenex (buprenorphine hydrochloride) for analgesia. Mice were then allowed to recover for 1 wk. Cannula placement was tested by examining the effect of 0.2 μg of angiotensin II on drinking behavior as described elsewhere (72).
Injections (icv)
Individually housed mice were habituated to handling and injections with aCSF for at least 1 wk before experimentation. Single injections were made manually using an internal cannula (C315IS-4/spc; Plastics One) connected to PE-50 tubing and a Hamilton syringe. Drug or vehicle (2 μl) was injected over a 30-sec period.
OXT Wild-Type vs. Sim1+/−.
Female mice (6–12 wk of age) were equally divided into the following groups matched for age and weight (wild-type aCSF, n = 7; wild-type Oxt, n = 7; Sim1+/− aCSF, n = 5; and Sim1+/− Oxt, n = 8). After 1 wk of habituation to twice daily icv injection, mice were injected with 10 ng of Oxt (H2510; Bachem Bioscience, Inc., King of Prussia, PA) or vehicle, twice a day for 12 d (at the onset of the dark cycle and 6 h later). Food intake and body weight were recorded daily. Food intake was analyzed by two-way ANOVA (group vs. time) and body weight change was analyzed with one-way ANOVA (group). Both analyses were done with a Bonferroni posttest to determine intergroup significance. On d 24, 12 d after the last injection, body weight was measured again and compared with body weight on d 12. Posttreatment body weight change for the groups was compared using one-way ANOVA with a Bonferroni posttest.
Wild-Type OVT vs. Sim1+/−.
Mice were icv injected at the onset of the dark cycle with 2 μl aCSF to determine baseline food intake. Mice were injected 24 h later with 0.5 μg OVT [d(CH2)5, Tyr(Me)2,Orn8-Oxytocin; H4928, Bachem]. Food intake was measured at 1-h intervals for 6 h. The experiment was repeated the following week. Food intake from the two aCSF measurement days was averaged for each mouse, and food intake from the two OVT measurement days was averaged for each mouse. OVT food intake was normalized to aCSF food intake for each mouse. Means were calculated for each treatment and compared using a paired t test.
Wild-Type Oxt.
After 1 wk of habituation to daily icv injection, 6- to 10-wk-old female wild-type mice were injected at the onset of the dark cycle with aCSF, 50 ng, 250 ng or 1 μg of OXT. Food intake was normalized to aCSF food intake. Means were calculated for each treatment, and the groups at each time point were compared using one-way ANOVA.
Wild-Type OVT.
After 1 wk of habituation to daily icv injection, 6- to 10-wk-old female wild-type mice were injected at the onset of the dark cycle with aCSF on d 1 and 1.5 μg OVT on d 2. Food intake was normalized to aCSF food intake for each mouse. Means were calculated for each treatment, and the groups at each time point were compared using a paired t test.
Immunohistochemistry
For colocalization of c-Fos and Oxt, 8-wk-old female C57BL/6 mice received 1 μg colchicine icv 24 h before subsequent experimental procedures. For c-Fos experiments, mice were icv injected with 3 μg of the Mc4r-selective agonist or vehicle (aCSF) 4 h before intracardiac perfusion. The Mc4r-selective agonist cyclo(β-Ala-His-D-Phe-Arg-Trp-Glu)-NH2 was purchased from Phoenix Pharmaceuticals (Burlingame, CA). Sim1 GFP transgenic mice were not colchicine treated, and immunohistochemistry was performed as described previously (10). Mice were deeply anesthetized with pentobarbital (7.5 mg /0.15 ml, ip) and transcardially perfused with 10 ml of heparinized saline (10 U/ml, 2 ml/min) followed by 10 ml of phosphate-buffered 4% paraformaldehyde (2 ml/min). Brains were removed, postfixed for 24 h in 4% paraformaldehyde, and then equilibrated in 30% sucrose in PBS for 72 h. Immunohistochemistry was performed as described elsewhere (10,70). Briefly, brains were coronally sectioned (35 μm) on a freezing microtome (Leica SM 2000R; Wetzlar, Germany) and stored in PBS at 4 C. Sections were incubated for 16 h in mouse anti-Oxt antiserum (MAB5296, 1:5000, Millipore Corp., Billerica, MA) or rabbit anti-CRH antiserum (AB-02, 1:800; Advanced Targeting Systems, San Diego, CA) and then incubated with either Cy-3 affiniPure goat antimouse IgG secondary antiserum or Cy-3 affiniPure goat antirabbit IgG secondary antiserum (115–165-166, 111–165-003, 1:400; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) for 2 h at room temperature. Sections were placed in 4′,6-diamidino-2-phenylindole (DAPI) (0.2 μg /ml, 236276; Roche, Indianapolis, IN) and then mounted and coverslipped using Vectashield (H-1000; Vector Laboratories, Burlingame, CA). Images of sections containing PVN were captured on an Olympus BX61 microscope using Cytovision software (Applied Imaging Corp., San Jose, CA). For fluorescent c-Fos and Oxt double labeling, sections were incubated for 16 h in rabbit anti-Fos antiserum (Ab-5; 1:3000 dilution; Calbiochem, La Jolla, CA) and mouse anti-Oxt antiserum. Sections were then incubated with Cy-3 goat antirabbit IgG secondary antiserum and Cy-2 affiniPure goat antimouse IgG secondary antiserum (115–225-166, 1:400; Jackson ImmunoResearch) for 2 h at room temperature. Sections were treated with DAPI and mounted as described above. For fluorescent GFP and Oxt double labeling, sections were incubated for 48 h in rabbit anti-GFP antiserum (A6455, 1:5000; Molecular Probes, Eugene, OR) and mouse anti-Oxt antiserum. Sections were then incubated with fluorescein isothiocyanate goat antirabbit IgG secondary antiserum and Cy-2 affiniPure goat antimouse IgG secondary antiserum (115-225-166, 1:400; Jackson ImmunoResearch) for 2 h at room temperature. Sections were treated with DAPI and mounted as described above. Cell counts and densitometry were determined using ImageJ software (National Institutes of Health, Bethesda, MD). Each side of a section was counted separately, and counts from six sides were averaged for each animal. Densitometry was performed on images that were captured with identical settings using the Integrated Density function after background subtraction.
Data Analysis
Data were analyzed using Microsoft Excel and plotted using Prism software (GraphPad Software, San Diego, CA). Values are means ± sem. Unless otherwise noted, means were compared using two-tailed t tests, with Welch’s correction if F test indicated unequal sample variances. Differences were considered statistically significant if P < 0.05.
Acknowledgments
We thank Lane Jaeckle Santos, Elizabeth Keohane Bhoj, Clay Williams, John Shelton, James Richardson, Laurent Gautron, Toshiro Kishi, and Joel Elmquist for their assistance and Michael Schwartz for helpful discussions.
Footnotes
This work was supported by grants from the American Diabetes Association (1-05-RA-154) and National Institutes of Health (5K08DK073228 and 1RL1DK081185).
Disclosure Statement: B.K., T.G., K.T., Y.U., and A.Z. have nothing to declare.
First Published Online May 1, 2008
Abbreviations: ARC, Arcuate nucleus; Avp, arginine vasopressin; CCK, cholecystokinin; DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; NTS, nucleus of the solitary tract; OVT, d(CH2)5, Tyr(Me)2,Orn8-oxytocin; Oxt, oxytocin; PVN, paraventricular nucleus; SON, supraoptic nucleus; Sst, somatostatin.
References
- Ward MP, Mosher JT, Crews ST 1998 Regulation of bHLH-PAS protein subcellular localization during Drosophila embryogenesis. Development 125:1599–1608 [DOI] [PubMed] [Google Scholar]
- Hung CC, Luan J, Sims M, Keogh JM, Hall C, Wareham NJ, O'Rahilly S, Farooqi IS 2007 Studies of the SIM1 gene in relation to human obesity and obesity-related traits. Int J Obes 31:429–434 [DOI] [PubMed] [Google Scholar]
- Holder Jr JL, Butte NF, Zinn AR 2000 Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet 9:101–108 [DOI] [PubMed] [Google Scholar]
- Ahituv N, Kavaslar N, Schackwitz W, Ustaszewska A, Martin J, Hebert S, Doelle H, Ersoy B, Kryukov G, Schmidt S, Yosef N, Ruppin E, Sharan R, Vaisse C, Sunyaev S, Dent R, Cohen J, McPherson R, Pennacchio LA 2007 Medical sequencing at the extremes of human body mass. Am J Hum Genet 80:779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S 2003 Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 348:1085–1095 [DOI] [PubMed] [Google Scholar]
- Holder Jr JL, Zhang L, Kublaoui BM, DiLeone RJ, Oz OK, Bair CH, Lee YH, Zinn AR 2004 Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am J Physiol Endocrinol Metab 287:E105–E113 [DOI] [PubMed] [Google Scholar]
- Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Levy E, Mitchell GA, Himms-Hagen J, Fan CM 2001 Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet 10:1465–1473 [DOI] [PubMed] [Google Scholar]
- Michaud JL, Rosenquist T, May NR, Fan CM 1998 Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev 12:3264–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Fan CM 2007 Allocation of paraventricular and supraoptic neurons requires Sim1 function: a role for a Sim1 downstream gene PlexinC1. Mol Endocrinol 21:1234–1245 [DOI] [PubMed] [Google Scholar]
- Kublaoui BM, Holder Jr JL, Gemelli T, Zinn AR 2006 Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Mol Endocrinol 20:2483–2492 [DOI] [PubMed] [Google Scholar]
- Olson BR, Drutarosky MD, Chow MS, Hruby VJ, Stricker EM, Verbalis JG 1991 Oxytocin and an oxytocin agonist administered centrally decrease food intake in rats. Peptides 12:113–118 [DOI] [PubMed] [Google Scholar]
- Arletti R, Benelli A, Bertolini A 1989 Influence of oxytocin on feeding behavior in the rat. Peptides 10:89–93 [DOI] [PubMed] [Google Scholar]
- Arletti R, Benelli A, Bertolini A 1990 Oxytocin inhibits food and fluid intake in rats. Physiol Behav 48:825–830 [DOI] [PubMed] [Google Scholar]
- Olson BR, Drutarosky MD, Stricker EM, Verbalis JG 1991 Brain oxytocin receptors mediate corticotropin-releasing hormone-induced anorexia. Am J Physiol 260:R448–R452 [DOI] [PubMed] [Google Scholar]
- Choi YH, Hartzell D, Azain MJ, Baile CA 2002 TRH decreases food intake and increases water intake and body temperature in rats. Physiol Behav 77:1–4 [DOI] [PubMed] [Google Scholar]
- Steward CA, Horan TL, Schuhler S, Bennett GW, Ebling FJ 2003 Central administration of thyrotropin releasing hormone (TRH) and related peptides inhibits feeding behavior in the Siberian hamster. Neuroreport 14:687–691 [DOI] [PubMed] [Google Scholar]
- Krahn DD, Gosnell BA, Grace M, Levine AS 1986 CRF antagonist partially reverses CRF- and stress-induced effects on feeding. Brain Res Bull 17:285–289 [DOI] [PubMed] [Google Scholar]
- Krahn DD, Gosnell BA, Majchrzak MJ 1990 The anorectic effects of CRH and restraint stress decrease with repeated exposures. Biol Psychiatry 27:1094–1102 [DOI] [PubMed] [Google Scholar]
- Vettor R, Fabris R, Pagano C, Federspil G 2002 Neuroendocrine regulation of eating behavior. J Endocrinol Invest 25:836–854 [DOI] [PubMed] [Google Scholar]
- Valassi E, Scacchi M, Cavagnini F 2008 Neuroendocrine control of food intake. Nutr Metab Cardiovasc Dis 18:158–168 [DOI] [PubMed] [Google Scholar]
- Wisse BE, Schwartz MW 2003 The skinny on neurotrophins. Nat Neurosci 6:655–656 [DOI] [PubMed] [Google Scholar]
- Kublaoui BM, Holder Jr JL, Tolson KP, Gemelli T, Zinn AR 2006 SIM1 overexpression partially rescues agouti yellow and diet-induced obesity by normalizing food intake. Endocrinology 147:4542–4549 [DOI] [PubMed] [Google Scholar]
- Yang C, Gagnon D, Vachon P, Tremblay A, Levy E, Massie B, Michaud JL 2006 Adenoviral-mediated modulation of Sim1 expression in the paraventricular nucleus affects food intake. J Neurosci 26:7116–7120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG 1997 Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes 46:2119–2123 [DOI] [PubMed] [Google Scholar]
- Morton GJ, Blevins JE, Williams DL, Niswender KD, Gelling RW, Rhodes CJ, Baskin DG, Schwartz MW 2005 Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest 115:703–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson CA, Reid DF, Ritter RC 2002 Daily CCK injection enhances reduction of body weight by chronic intracerebroventricular leptin infusion. Am J Physiol 282:R1368–R1373 [DOI] [PubMed] [Google Scholar]
- Matson CA, Reid DF, Cannon TA, Ritter RC 2000 Cholecystokinin and leptin act synergistically to reduce body weight. Am J Physiol 278:R882–R890 [DOI] [PubMed] [Google Scholar]
- Matson CA, Ritter RC 1999 Long-term CCK-leptin synergy suggests a role for CCK in the regulation of body weight. Am J Physiol 276:R1038–R1045 [DOI] [PubMed] [Google Scholar]
- Matson CA, Wiater MF, Kuijper JL, Weigle DS 1997 Synergy between leptin and cholecystokinin (CCK) to control daily caloric intake. Peptides 18:1275–1278 [DOI] [PubMed] [Google Scholar]
- Baskin DG, Blevins JE, Schwartz MW 2001 How the brain regulates food intake and body weight: the role of leptin. J Pediatr Endocrinol Metab 14(Suppl 6): 1417–1429 [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DG 2000 Central nervous system control of food intake. Nature 404:661–671 [DOI] [PubMed] [Google Scholar]
- Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK 2003 Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci 23:7143–7154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L 1998 Oxytocinergic inputs to the nucleus of the solitary tract and dorsal motor nucleus of the vagus in neonatal rats. J Comp Neurol 399:101–109 [DOI] [PubMed] [Google Scholar]
- Kirchgessner AL, Sclafani A, Nilaver G 1988 Histochemical identification of a PVN-hindbrain feeding pathway. Physiol Behav 42:529–543 [DOI] [PubMed] [Google Scholar]
- Blevins JE, Eakin TJ, Murphy JA, Schwartz MW, Baskin DG 2003 Oxytocin innervation of caudal brainstem nuclei activated by cholecystokinin. Brain Res 993:30–41 [DOI] [PubMed] [Google Scholar]
- Blevins JE, Schwartz MW, Baskin DG 2004 Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol 287:R87–R96 [DOI] [PubMed] [Google Scholar]
- Swaab DF, Purba JS, Hofman MA 1995 Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol Metab 80:573–579 [DOI] [PubMed] [Google Scholar]
- Hoybye C 2004 Endocrine and metabolic aspects of adult Prader-Willi syndrome with special emphasis on the effect of growth hormone treatment. Growth Horm IGF Res 14:1–15 [DOI] [PubMed] [Google Scholar]
- Eaton JL, Glasgow E 2006 The zebrafish bHLH PAS transcriptional regulator, single-minded 1 (sim1), is required for isotocin cell development. Dev Dyn 235:2071–2082 [DOI] [PubMed] [Google Scholar]
- Caqueret A, Coumailleau P, Michaud JL 2005 Regionalization of the anterior hypothalamus in the chick embryo. Dev Dyn 233:652–658 [DOI] [PubMed] [Google Scholar]
- Jang M, Romsos DR 1998 Neuropeptide Y and corticotropin-releasing hormone concentrations within specific hypothalamic regions of lean but not ob/ob mice respond to food-deprivation and refeeding. J Nutr 128:2520–2525 [DOI] [PubMed] [Google Scholar]
- Verbalis JG, McCann MJ, McHale CM, Stricker EM 1986 Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science 232:1417–1419 [DOI] [PubMed] [Google Scholar]
- Bednarek MA, MacNeil T, Tang R, Kalyani RN, Van der Ploeg LH, Weinberg DH 2001 Potent and selective peptide agonists of α-melanotropin action at human melanocortin receptor 4: their synthesis and biological evaluation in vitro. Biochem Biophys Res Commun 286:641–645 [DOI] [PubMed] [Google Scholar]
- Navarro M, Cubero I, Chen AS, Chen HY, Knapp DJ, Breese GR, Marsh DJ, Thiele TE 2005 Effects of melanocortin receptor activation and blockade on ethanol intake: a possible role for the melanocortin-4 receptor. Alcohol Clin Exp Res 29:949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpl G, Fahrenholz F 2001 The oxytocin receptor system: structure, function, and regulation. Physiol Rev 81:629–683 [DOI] [PubMed] [Google Scholar]
- Castel M, Morris JF 1988 The neurophysin-containing innervation of the forebrain of the mouse. Neuroscience 24:937–966 [DOI] [PubMed] [Google Scholar]
- Ivell R, Walther N 1999 The role of sex steroids in the oxytocin hormone system. Mol Cell Endocrinol 151:95–101 [DOI] [PubMed] [Google Scholar]
- Koohi MK, Ivell R, Walther N 2005 Transcriptional activation of the oxytocin promoter by oestrogens uses a novel non-classical mechanism of oestrogen receptor action. J Neuroendocrinol 17:197–207 [DOI] [PubMed] [Google Scholar]
- Loots GG, Ovcharenko I, Pachter L, Dubchak I, Rubin EM 2002 rVISTA for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res 12:832–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002 The human genome browser at UCSC. Genome Res 12:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock S, Granstrom L, Backman L, Matthiesen AS, Uvnas-Moberg K 1989 Elevated plasma levels of oxytocin in obese subjects before and after gastric banding. Int J Obes 13:213–222 [PubMed] [Google Scholar]
- Verbalis JG, Blackburn RE, Olson BR, Stricker EM 1993 Central oxytocin inhibition of food and salt ingestion: a mechanism for intake regulation of solute homeostasis. Regul Pept 45:149–154 [DOI] [PubMed] [Google Scholar]
- Gould BR, Zingg HH 2003 Mapping oxytocin receptor gene expression in the mouse brain and mammary gland using an oxytocin receptor-LacZ reporter mouse. Neuroscience 122:155–167 [DOI] [PubMed] [Google Scholar]
- Emond M, Schwartz GJ, Ladenheim EE, Moran TH 1999 Central leptin modulates behavioral and neural responsivity to CCK. Am J Physiol 276:R1545–R1549 [DOI] [PubMed] [Google Scholar]
- Olson BR, Hoffman GE, Sved AF, Stricker EM, Verbalis JG 1992 Cholecystokinin induces c-fos expression in hypothalamic oxytocinergic neurons projecting to the dorsal vagal complex. Brain Res 569:238–248 [DOI] [PubMed] [Google Scholar]
- Olszewski PK, Bomberg EM, Martell A, Grace MK, Levine AS 2007 Intraventricular ghrelin activates oxytocin neurons: implications in feeding behavior. Neuroreport 18:499–503 [DOI] [PubMed] [Google Scholar]
- Griffond B, Deray A, Bahjaoui-Bouhaddi M, Colard C, Bugnon C, Fellmann D 1994 Induction of Fos-like immunoreactivity in rat oxytocin neurons following insulin injections. Neurosci Lett 178:119–123 [DOI] [PubMed] [Google Scholar]
- Douglas AJ, Johnstone LE, Leng G 2007 Neuroendocrine mechanisms of change in food intake during pregnancy: a potential role for brain oxytocin. Physiol Behav 91:352–365 [DOI] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB 2005 Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123:493–505 [DOI] [PubMed] [Google Scholar]
- Sabatier N 2006 α-Melanocyte-stimulating hormone and oxytocin: a peptide signalling cascade in the hypothalamus. J Neuroendocrinol 18:703–710 [DOI] [PubMed] [Google Scholar]
- Sabatier N, Caquineau C, Dayanithi G, Bull P, Douglas AJ, Guan XM, Jiang M, Van der Ploeg L, Leng G 2003 α-Melanocyte-stimulating hormone stimulates oxytocin release from the dendrites of hypothalamic neurons while inhibiting oxytocin release from their terminals in the neurohypophysis. J Neurosci 23: 10351–10358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L, Vollmer RR, Karam J, Phillips D, Li X, Amico JA 2005 Dehydration anorexia is attenuated in oxytocin-deficient mice. Am J Physiol 288:R1791–R1799 [DOI] [PubMed] [Google Scholar]
- Mantella RC, Rinaman L, Vollmer RR, Amico JA 2003 Cholecystokinin and D-fenfluramine inhibit food intake in oxytocin-deficient mice. Am J Physiol 285:R1037–R1045 [DOI] [PubMed] [Google Scholar]
- Sclafani A, Rinaman L, Vollmer RR, Amico JA 2007 Oxytocin knockout mice demonstrate enhanced intake of sweet and nonsweet carbohydrate solutions. Am J Physiol 292:R1828–R1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi Y, Yoshida M, Bielsky IF, Ross HE, Kawamata M, Onaka T, Yanagisawa T, Kimura T, Matzuk MM, Young LJ, Nishimori K 2005 Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc Natl Acad Sci USA 102:16096–16101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, Chen A, Camacho RE, Shearman LP, Gopal-Truter S, MacNeil DJ, Van der Ploeg LH, Marsh DJ 2002 Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol 22:5027–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD 2005 NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 310:683–685 [DOI] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Bruning JC 2005 Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci 8:1289–1291 [DOI] [PubMed] [Google Scholar]
- Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, Ghatei MA, Bloom SR 2005 Post-embryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J 19:1680–1682 [DOI] [PubMed] [Google Scholar]
- Beuckmann CT, Sinton CM, Williams SC, Richardson JA, Hammer RE, Sakurai T, Yanagisawa M 2004 Expression of a poly-glutamine-ataxin-3 transgene in orexin neurons induces narcolepsy-cataplexy in the rat. J Neurosci 24:4469–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinyama H, Masuzaki H, Fang H, Flier JS 2003 Regulation of melanocortin-4 receptor signaling: agonist-mediated desensitization and internalization. Endocrinology 144:1301–1314 [DOI] [PubMed] [Google Scholar]
- Johnson AK, Epstein AN 1975 The cerebral ventricles as the avenue for the dipsogenic action of intracranial angiotensin. Brain Res 86:399–418 [DOI] [PubMed] [Google Scholar]