

Abstract
OBJECTIVE—This study determined the effects of high glucose exposure and cytokine treatment on generation of reactive oxygen species (ROS) and activation of inflammatory and apoptotic pathways in human retinal endothelial cells (HRECs).
RESEARCH DESIGN AND METHODS—Glucose consumption of HRECs, human retinal pigment epithelial cells (HRPEs), and human Müller cells (HMCs) under elevated glucose conditions was measured and compared with cytokine treatment. Production of ROS in HRECs was examined using 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), spin-trap electron paramagnetic resonance, and MitoTracker Red staining after high glucose and cytokine treatment. The activation of different signaling cascades, including the mitogen-activated protein kinase pathways, tyrosine phosphorylation pathways, and apoptosis by high glucose and cytokines in HRECs, was determined.
RESULTS—HRECs, in contrast to HRPEs and HMCs, did not increase glucose consumption in response to increasing glucose concentrations. Exposure of HRECs to 25 mmol/l glucose did not stimulate endogenous ROS production, activation of nuclear factor-κB (NF-κB), extracellular signal–related kinase (ERK), p38 and Jun NH2-terminal kinase (JNK), tyrosine phosphorylation, interleukin (IL)-1β, or tumor necrosis factor-α (TNF-α) production and only slightly affected apoptotic cell death pathways compared with normal glucose (5 mmol/l). In marked contrast, exposure of HRECs to proinflammatory cytokines IL-1β or TNF-α increased glucose consumption, mitochondrial superoxide production, ERK and JNK phosphorylation, tyrosine phosphorylation, NF-κB activation, and caspase activation.
CONCLUSIONS—Our in vitro results indicate that HRECs respond to cytokines rather than high glucose, suggesting that in vivo diabetes–related endothelial injury in the retina may be due to glucose-induced cytokine release by other retinal cells and not a direct effect of high glucose.
Hyperglycemia is a major causative factor in the development of diabetic retinopathy. Several biochemical pathways have been associated with hyperglycemia, including glucose-mediated increases in reactive oxygen species (ROS) (1), diacylglycerol production, and the subsequent activation of the protein kinase C pathway (2), flux through the polyol metabolic pathway (3), accumulation of advanced glycation end products (4), and cytokine secretion (5). Most of these results are based on studies using macrovascular cells (6), and these studies were never confirmed using microvascular endothelial cells from the end organs affected by diabetic microvascular complications. Furthermore, although diabetic animal studies have supported that chronic hyperglycemia generates ROS in the vascular environment, the precise cellular origin of the ROS has not been established (7).
It is well recognized that mitochondria are the main source of endogenous ROS in most mammalian cell types (8). Of the oxygen consumed by mitochondria, ∼1–5% is converted to ROS as byproducts of oxidation-reduction reactions in the respiratory chain. Small proportions of ROS that escape antioxidant defenses lead to chronic low-level oxidative damage to mitochondria, which accumulates throughout life. This accumulation is intensified in chronic disease states, such as diabetes, and during acute stress and/or inflammation (9).
In addition to mitochondria, small quantities of ROS can also be generated from other intracellular sources. For example, neutrophil-type NADPH oxidase (10) and NADPH-like enzymes (NOX) generate small quantities of ROS in the absence of a specific stimulus in endothelial cells (11). Cytokines, including tumor necrosis factor-α (TNF-α), generate ROS that serve as essential second messengers in normal intracellular signaling pathways, but their overproduction can result in pathology (12).
The initiation and progression of diabetic retinopathy has been attributed not only to hyperglycemia and ROS but also to inflammation. The inflammatory response in early diabetes includes cytokine release, which activates key transcriptional regulators, including nuclear factor-κB (NF-κB). NF-κB in turn regulates expression of proinflammatory molecules, including inducible nitric oxide synthase, cyclooxygenase (COX)2, and intracellular adhesion molecule (ICAM). Increased expression of cell adhesion molecules, such as ICAM, promotes leukostasis. These conditions promote vascular permeability and ultimately endothelial cell death (13).
When endothelial cells are exposed to interleukin (IL)-1β, TNF-α, and interferon-γ (IFN-γ), they produce additional cytokines, such as IL-8, monocyte chemotactic protein-1 (MCP-1), and RANTES (regulated upon activation, normal T-cell expressed and secreted), among others, that appear to be endothelial cell-type specific. Human trials and animal studies using aspirin and other anti-inflammatory agents demonstrate both prevention and improvement of diabetic retinopathy, providing additional support for the pathogenic role of cytokine-induced leukocyte activation (14).
Although cytokine production, oxidative stress, and glucose-induced vascular toxicity have all been implicated as key causes of diabetic retinopathy, no detailed studies have been performed to date to determine whether retinal endothelial cell dysfunction is mediated primarily by direct glucose toxicity or by direct cytokine toxicity. This study was designed to address these critical questions and to separate the vascular effects of high glucose from the vascular effects of proinflammatory cytokines on human retinal endothelial cells (HRECs).
RESEARCH DESIGN AND METHODS
Cell culture media, Alexa Fluor 488 acetylated LDL, and MitoTracker Red were obtained from Invitrogen (Carlsbad, CA). Endothelial cell growth supplement (ECGS) was from Upstate Biotechnology (Lake Placid, NY), and insulin transferrin selenium mix (ITS), 7-amino-4-trifluoro-methylcoumarin (AFC), and commonly used chemicals and reagents were from Sigma-Aldrich (St. Louis, MO). TNF-α and IL-1β were from R&D Systems (Minneapolis, MN). The following antibodies were used: rabbit anti-human Von Willebrand factor from Dako (Carpinteria, CA); rabbit anti–phospho-IκBα (Ser32), rabbit anti–inhibitor of κB (IκB) α, and rabbit anti–phosphor-p44/42 mitogen-activated protein kinase (MAPK) (Thr202/Tyr204) from Cell Signaling Technology (Danvers, MA); rabbit anti–phosphor–stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) (T183/Y185), rabbit anti–phosphor-p38(TGpY) from Promega (Madison, WI); mouse anti-phosphotyrosine (4G10) from Upstate Biotechnology; and mouse anti-actin and rabbit anti–c-FLIP antibody from Sigma-Aldrich. Caspase substrates (AFC coupled) were from Calbiochem (San Diego, CA); IL-1β ELISA assays were from Pierce/Endogen (Rockford, IL).
Cell culture
Tissue was provided by the National Disease Research Interchange (Philadelphia, PA), and primary HRECs, human retinal pigment epithelial cell (HRPEs), and human Müller cells (HMCs) were prepared and cultured as previously described (15,16). Primary cultures of HRECs were obtained from at least three separate donors. Passages 3–6 were used in the experiments. Purity of the cells was determined using acetylated LDL uptake and Von Willebrand factor staining. Only 99% and higher purity HREC preparations were used in the study. HRECs were grown in six-well plates coated with 0.1% gelatin in 2 ml growth medium/well consisting of Dulbecco's modified Eagle's medium/F12 (1:1 ratio, 5 mmol/l glucose) supplemented with 10% fetal bovine serum, 5% ECGS, 1% penicillin/streptomycin, and 1× ITS at 37°C in humidified 95% air and 5% CO2 until fully confluent. For experimental conditions, serum content was decreased to 2%. Cells treated with 5 mmol/l glucose and 20 mmol/l mannitol or l-glucose were used as an osmotic control. Medium was changed every 72 h or every 12 h for experiments with glucose fluctuations from 5 to 25 mmol/l concentrations. For cytokine treatment, HRECs were switched to serum-free media for 12 h and treated with 0–10 ng/ml TNF-α or 0–2 ng/ml IL-1β for the times indicated in results.
For co-culture experiments, HRECs (1 × 106) were plated in six-well plates containing growth medium. HMCs were plated in a HMC:HREC 1:50 ratio. The co-culture was treated in medium containing 5 or 25 mmol/l glucose. After 48 h of treatment, the cells were trypsinized with 0.05 mg/ml trypsin:EDTA for 30 s to selectively collect HRECs. HRECs were lyzed in 100 mmol/l HEPES buffer, pH 7.5, containing 10% sucrose, 0.1% CHAPS, and the general protease inhibitors 1 mmol/l EDTA, 1 mmol/l phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin; sonicated for 10 s; and centrifuged at 9,000g for 5 min at 4°C, followed by protein measurement of the supernatant. Supernatants were retained for caspase experiments.
Glucose consumption rate
Cells were cultured in 5, 15, 20, or 25 mmol/l glucose. At the time points indicated in results, an aliquot of medium was taken from each plate, and the glucose concentration remaining in the medium was measured spectrophotometrically using a hexokinase glucose-6-phosphate dehydrogenase enzymatic assay kit from Sigma-Aldrich. Glucose consumption was explained by the generalized reaction of A → B, where A is extracellular glucose and B is consumed glucose. This reaction is described by the following equations: 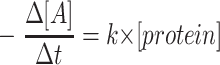 for zero order kinetics,
for zero order kinetics, 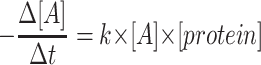 for first order kinetics and
for first order kinetics and 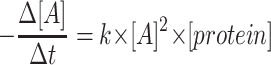 for second order kinetics, where [A] is media glucose concentration, [protein] is total cellular protein, Δt is time, and k is the rate constant (17). To evaluate the order of kinetics of glucose consumption reaction
for second order kinetics, where [A] is media glucose concentration, [protein] is total cellular protein, Δt is time, and k is the rate constant (17). To evaluate the order of kinetics of glucose consumption reaction 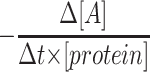 was plotted against [A]. Numerical value of rate constant was subsequently determined by Nonlinear Least Squares Regression at http://statpages.org/nonlin.html.
was plotted against [A]. Numerical value of rate constant was subsequently determined by Nonlinear Least Squares Regression at http://statpages.org/nonlin.html.
Detection of ROS in HRECs
Fluorescent ROS detection.
5-(and-6)-Chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) ROS indicator dye (Invitrogen) was used. HRECs were plated in a flat, clear bottom, 96-well plate at 10,000 cells/well and treated with varying glucose concentrations as described in results. The cells were preloaded with 2 μmol/l CM-H2DCFDA for 30 min followed by fluorescence measurement in a microplate reader (Molecular Devices, Downing, PA). A well containing no cells was subtracted as a background. The cells from a sister well for each experiment were trypsinized and counted, and the results were presented as fluorescence per cell.
Spin-trap electron paramagnetic resonance.
For electron paramagnetic resonance (EPR) measurements, HRECs were cultured in matrix-coated Teflon tubes (1-mm internal diameter) in 5, 15, or 25 mmol/l glucose for the times indicated in results. Several spin traps with different principles of detection were used. First, two commercially available spin traps, 5, 5-dimethylpyrroline-N-oxide (DMPO) from Dojindo Laboratories (Kumamoto, Japan) and α-phenyl-tert-butylnitrone (PBN) from Sigma-Aldrich were used. The EPR silent DMPO or PBN reacts with intracellular radicals to produce stable EPR-detectable DMPO or PBN radicals (see Fig. 3, top and middle). The increase in the corresponding spin adduct intensity corresponds to the amount of free radicals in the cell.
FIG. 3.

Measurements of oxidative stress in high glucose–treated HRECs using spin trap EPR do not show production of oxidative radicals. Spin-trap EPR was used to confirm the failure of high glucose to increase endogenous ROS production by HRECs. HRECs were cultured in matrix-coated Teflon tubes. In each experiment, the cells were incubated for 1–72 h in 5 (⋄), 15 (□), or 25 (▵) mmol/l glucose, and production of radicals was measured. Twenty-four–hour time point is shown; DMPO (top), PBN (middle) or TEMPO-AM (bottom) was used. The principle of detection is shown on the left, representative EPR traces in the middle, and quantitated data on the right. Cells stimulated with 0.5 mmol/l H2O2 in the presence of iron to induce Fenton's reaction served as positive controls (○). Teflon tubes without cells were used as negative controls (♦). Top and middle: Traces A for DMPO and PBN are from 25 mmol/l glucose-stimulated cells. No cells control and 5 and 15 mmol/l glucose traces looked similar to 25 mmol/l glucose trace. Traces B for DMPO and PBN are from the positive control. In DMPO and PBN experiments, exposure to 15 and 25 mmol/l glucose did not cause detectable radical production; no cells control (♦), 5 (⋄), 15 (□), or 25 (▵) mmol/l glucose graphs are super-imposable in the top and middle panels. Only when the cells were stimulated with H2O2 was radical production observed (○). Bottom: TEMPO-AM is more sensitive than DMPO and PBN and is hydrolyzed inside the cell, retained in the cytoplasm, and then reduced to the corresponding hydroxylamine, leading to the loss of paramagnetism. Loss of EPR signal corresponds to the amount of intracellular free radicals. HRECs were incubated for 1–96 h in the varying glucose concentrations, and then TEMPO-AM was added. HRECs exposed to 5 (⋄), 15 (□), or 25 (▵) mmol/l glucose for 24 h all demonstrated a low level of radicals, and there was no difference in radical production between different glucose concentrations. Trace A for TEMPO-AM is from no cell control with no loss of paramagnetism; trace B is from 25 mmol/l glucose; 5 and 15 mmol/l glucose traces looked similar to 25 mmol/l glucose; trace C is from positive control with radical production being detected as a loss of paramagnetism. Results of five independent experiments (mean ± SD) are shown in the graphs.
We also synthesized (according to Inoguchi et al. [18]) cell permeable spin trap 4-acetoxymethylcarboxylate-2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO-AM). The EPR-detectable TEMPO-AM is hydrolyzed inside the cell, retained in the cytoplasm, and then reduced to the corresponding hydroxylamine, leading to the loss of paramagnetism (see Fig. 3, bottom). Thus, the loss of EPR signal corresponds to the amount of intracellular free radicals. EPR spectra were measured on a Bruker ESP 300E spectrometer equipped with a Bruker ER4111VT variable-temperature accessory (Bruker, Rheinstetten, Germany) using EPR cavity (4108 TMH). Data were analyzed using software supplied by Bruker (ESP 1600 data system).
Mitochondrial superoxide production.
HRECs (1 × 104 cells) were grown on coverslips and incubated in treatment medium as described above. At times indicated, the superoxide-specific MitoTracker Red solution was added to the medium at a final concentration of 500 μmol/l, and cells were further incubated in the dark for 20 min at 37°C/5% CO2. The medium was removed; cells were washed twice with PBS (2 ml), fixed in 4% paraformaldehyde for 10 min at room temperature, and washed twice with PBS (2 ml). Coverslips were mounted on slides using an anti-fading fluorescent mounting medium containing DAPI to stain for nuclei (Vector Shield). Digital Images were captured in 12 bits on a Leica DMI 6000 B inverted microscope using a Retiga EXI camera (Q-Imaging, Vancouver, British Columbia, Canada) at ×40 magnification (MitoTrackerRed, excitation 580 nm, emission 600 nm; DAPI, excitation 359, emission 461). Image analysis was performed using Metamorph Imaging Software (Molecular Devices). Red intensity (arbitrary fluorescence units equivalent to mitochondrial superoxide production) was quantified, integrated, and normalized to the integrated DAPI intensity (equivalent to cell number). Results were represented as the ratio of arbitrary fluorescence units to integrated DAPI intensity.
SDS-PAGE and Western blot analysis
SDS-PAGE and Western blot analyses were performed as previously described (19). Primary antibodies dilution was for c-FLIP, 1:800; phospho-IκBα, 1:500; IκBα, 1:1,000; phosphor-p44/42, 1:1,000; phosphor-SAPK/JNK, 1:1,000; phosphor-p38, 1:500; and 4G10, 1:1,000. Secondary antibodies dilutions were 1:3,000. Western blots were quantitated by scanning densitometry using ImageJ software ver. 1.29 (available by ftp at zippy.nimh.nih.gov/ or at http://rsb.info.nih.gov/nih-image; developed by Wayne Rasband [National Institutes of Health, Bethesda, MD]), and expression of protein of interest was normalized to actin.
Electrophoretic mobility gel shift assay
The double-stranded oligonucleotide containing the NF-κB binding sequence derived from human vascular cell adhesion molecule-1 (VCAM-1) promoter were designed and synthesized as follows: 5′ TGCCCTGGGTTTCCCCTTGAAGGGATTTCCCTC3′ and 3′GACCCAAAGGGGAACTTCCCTAAAGGGAGGCGG5′. The oligonucleotides were annealed and labeled in the presence of [32P]dCTP with a random primer kit from Invitrogen, according to the manufacturer's protocol. For binding reactions, nuclear extracts (6 μg) were incubated in 25-μl total reaction volume with 32P-labeled NF-κB oligonucleotides for 20 min at room temperature. DNA-protein complexes were resolved on 6% nondenaturing polyacrylamide gels, and the bands were examined by autoradiography. Incubation of the nuclear extracts with excess cold NF-κB oligonucleotides was used to confirm the specificity of binding activity.
Caspase activity assay
Caspase activities were measured using fluorogenic caspase substrate (DEVD-AFC for caspase-3 and IETD-AFC for caspase-8) as described previously (20,21). Caspase activity was calculated against an AFC standard curve and expressed as picomoles AFC per milligram protein per minute.
Annexin V staining for apoptosis
Annexin V staining was done according to the manufacturer's instruction as previously described (16). HRECs staining positive for Annexin V were counted and expressed as a percentage.
Cytokine measurement by ELISA
IL-1β and TNF-α concentrations in the medium were determined using an IL-1β or TNF-α ELISA kit (Pierce/Endogen) according to the manufacturer's instructions as previously described (21).
Statistical analysis
Data are expressed as the mean ± SD. Factorial ANOVA with post hoc Tukey's test (calculated using http://faculty.vassar.edu/lowry/VassarStats.html) was used for comparing continuous data obtained from independent samples, and Kruskal-Wallis with post hoc Dunn's test was used for comparing Western blot and imaging data. Significance was established at P < 0.05.
RESULTS
Glucose consumption by HRECs is not affected by glucose concentration, unlike HRPEs and HMCs.
We determined the dependency of glucose consumption by HRECs versus glucose concentration in the media. Plot of 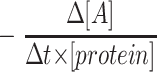 versus [A] is presented in Fig. 1. Nonlinear least squares regression analysis demonstrated that the dependency can be best fitted by the equation Y = 175.09 ± 2.17 nmol glucose · mg−1 protein · h−1 with correlation coefficiency of 0.927, describing a zero-order kinetics reaction (17). Thus, the rate of glucose consumption in HRECs was not dependent on glucose concentration in the media. We then compared the amount of glucose consumed in 24 h by HRECs, HRPEs, and HMCs cultured in 5 and 25 mmol/l glucose. As presented in the inset of Fig. 1, HRECs consumed extremely low amounts of glucose compared with the other retinal cell types examined. In contrast to HRPEs and HMCs, there was no increase in glucose consumption in HRECs cultured in 25 mmol/l glucose. The increase in consumption by HRPEs and HMCs was glucose specific because the osmotic control mannitol did not increase consumption (data not shown).
versus [A] is presented in Fig. 1. Nonlinear least squares regression analysis demonstrated that the dependency can be best fitted by the equation Y = 175.09 ± 2.17 nmol glucose · mg−1 protein · h−1 with correlation coefficiency of 0.927, describing a zero-order kinetics reaction (17). Thus, the rate of glucose consumption in HRECs was not dependent on glucose concentration in the media. We then compared the amount of glucose consumed in 24 h by HRECs, HRPEs, and HMCs cultured in 5 and 25 mmol/l glucose. As presented in the inset of Fig. 1, HRECs consumed extremely low amounts of glucose compared with the other retinal cell types examined. In contrast to HRPEs and HMCs, there was no increase in glucose consumption in HRECs cultured in 25 mmol/l glucose. The increase in consumption by HRPEs and HMCs was glucose specific because the osmotic control mannitol did not increase consumption (data not shown).
FIG. 1.

Rate of glucose consumption in endothelial cells is not dependent on glucose concentration in media. Confluent plates of HRECs were cultured in 5 mmol/l (♦), 15 mmol/l (▴), 20 mmol/l (•), or 25 mmol/l (▪) glucose for 2, 4, 8, 16, 32, and 64 h. HRECs, HRPEs, and HMCs were cultured in 5 mmol/l (white bars) or 25 mmol/l (black bars) glucose for 24 h (inset). Glucose consumption was determined as a rate of decrease in glucose concentration in the media normalized to total cellular protein and plotted against glucose concentration in the media. Nonlinear least squares regression analysis demonstrated that the dependency can be best fitted by the equation Y = 175.09 ± 2.17 nmol glucose · mg−1 protein · h−1 with correlation coefficiency of 0.927, describing a zero-order kinetics reaction. As presented in the inset, HRECs consumed extremely low amounts of glucose compared with HRPEs and HMCs, and there was no increase in glucose consumption in HRECs cultured in 25 mmol/l glucose unlike in the HRPE and HMC cultures. Results represent means ± SD; n = 3 for HRPEs, 4 for HMCs, and 6 for HRECs; *P < 0.05.
High glucose conditions do not induce ROS production in HRECs.
Using the CM-H2DCFDA fluorescent ROS indicator, we were able to observe weak baseline generation of endogenous ROS for cells incubated in normal (5 mmol/l) glucose. However, no increase in endogenous ROS was observed when cells were exposed to intermediate (15 mmol/l) and high (25 mmol/l) glucose for up to 96 h (Fig. 2A). We examined the effect of fluctuating glucose between 5 and 25 mmol/l every 12 h on ROS production because this would more closely represent what can be observed in diabetic patients. As shown in Fig. 2B, there was no difference in ROS production between cultures maintained only at 5 mmol/l and cultures that were exposed to fluctuating glucose concentrations of 5 mmol/l for 12 h and then 25 mmol/l glucose for the next 12 h. This fluctuating pattern was continued for 96 h. Only by 96 h was a change observed, but this was not due to an increase in ROS in the fluctuating group but rather to a decrease in ROS production in 5 mmol/l group. We also monitored ROS generation using MitoTrackerRed, a dye that accumulates in the mitochondria and becomes fluorescent upon reaction with superoxide radicals. No change in endogenous ROS production was observed with high glucose treatment of HRECs in either short-term (up to 144 h) or long-term (4 weeks) exposure to high glucose (Fig. 2C).
FIG. 2.

Measurements of oxidative stress in high glucose–treated HREC fluorescent ROS indicators do not show production of oxidative radicals. CM-H2DCFDA fluorescent ROS indicator measurements were performed on HRECs treated with increasing glucose concentrations (5, 15, and 25 mmol/l) (n = 3) (A) or on HRECs treated with fluctuating glucose concentrations (B). The fluorescence intensity was normalized to cell number in normal 5 mmol/l glucose (white bars) or fluctuating 5–25 mmol/l glucose every 12 h for the times indicated (black bars). Results of nine independent experiments (means ± SD) are shown. *P < 0.01 compared with 5 mmol/l glucose at 1, 24, and 72 h (white bars). #P < 0.01 25 mmol/l glucose compared with 5 mmol/l glucose at 96 h. C: HRECs were incubated in medium containing 5 or 25 mmol/l glucose for times indicated. Mitochondrial superoxide production was measured as described in research design and methods using MitoTrackerRed, a dye that accumulates in the mitochondria and becomes fluorescent upon reaction with superoxide radicals. Results are presented as the mean ± SD (MitoTracker Red/DAPI intensity; n = 6). At time points indicated, representative fluorescent images (×40 objectives) of MitoTracker Red and DAPI overlay are shown on the left.
To confirm the failure of high glucose to increase endogenous ROS production by HRECs, we developed a more sensitive spin-trap EPR approach. In each experiment, the cells were incubated for 1–72 h in 5, 15, or 25 mmol/l glucose and DMPO- or PBN-radical production was measured. Cells stimulated with 0.5 mmol/l H2O2 in the presence of iron to induce Fenton's reaction served as a positive control. Exposure to 15 and 25 mmol/l glucose did not cause detectable radical production (no cells control, 5, 15, and 25 mmol/l glucose graphs are super-imposable; Fig. 3, top and middle). Although only 24-h data are shown, the rest of the time points produced similar results. Only when the cells were stimulated with H2O2 was radical production observed.
We then used the even more sensitive EPR-detectable TEMPO-AM spin trap. TEMPO-AM is hydrolyzed inside the cell, retained in the cytoplasm, and then reduced to the corresponding hydroxylamine, leading to the loss of paramagnetism (Fig. 3, bottom). Thus, the loss of EPR signal corresponds to the amount of intracellular free radicals. HRECs were incubated for 1–96 h in the varying glucose concentrations, and then TEMPO-AM was added. HRECs exposed to 5, 15, or 25 mmol/l glucose for 24 h demonstrated a low level of radicals, and there was no difference in radical production between different glucose concentrations. Incubation of the cells with glucose over all time points produced similar results (data not shown). When the cells were stimulated with H2O2, radical production was detected as a loss of paramagnetism. As expected, Teflon tubes without cells show no loss of paramagnetism (Fig. 3, bottom, ♦).
High glucose does not affect MAPK signaling in HRECs.
Because hyperglycemia is the key metabolic mediator associated with diabetes, we examined the effect of high glucose on all major MAPK pathways in HRECs. Exposure to high glucose (25 mmol/l) for 10 min to 96 h did not affect phosphorylation status of extracellular signal–related kinase (ERK)1/2, p38, or JNK MAPKs (Fig. 4A and B). The 24-, 72-, and 96-h time points for 25 mmol/l glucose are shown; shorter time points, intermediate (15 mmol/l) glucose concentration, and l-glucose osmotic control produced similar results (results not shown).
FIG. 4.

High glucose does not lead to MAPK phosphorylation and NF-κB activation in HRECs. HRECs were incubated in either 5 or 25 mmol/l glucose for designated time periods. Immunoblot analyses using anti–phospho-ERK1/2, anti–phospho p38, and anti–phospho JNK antibodies was performed. Equal amounts of protein were added to each lane as confirmed by actin levels. Exposure to high glucose (25 mmol/l) for 24, 72, and 96 h did not affect phosphorylation status of ERK1/2, p38, or JNK MAPKs. Representative results from at least three independent experiments are shown in the left (A). Densitometry analysis of Western blots (normalized to actin, n = 3) are presented as mean ± SD in B. C: NF-κB EMSA gel shift assays were performed on HRECs treated with 5 or 25 mmol/l glucose for times indicated. IL-1β (1 ng/ml) treatments served as positive controls. Representative image of NF-κB gel shift is show in C; densitometry analysis of three independent EMSA assays is shown in D.
To determine the effect of high glucose on activation of NF-κB pathway, an electrophoretic mobility shift assay (EMSA) was performed in HRECs after high glucose (25 mmol/l) exposure for 48 h and 4 weeks. A double-stranded DNA probe containing the specific NF-κB binding site from human VCAM-1 promoter was used to study the activation and binding of NF-κB to the promoters of adhesion molecules. As shown in Fig. 4C, high glucose exposure did not induce NF-κB binding to the VCAM-1 promoter. In contrast to high glucose, stimulation with 1 ng/ml IL-1β for 30 min induced a robust 3.5 increase in NF-κB binding to the VCAM-1 promoter.
High glucose induced only low-level activation of proapoptotic caspases.
Apoptosis of retinal endothelial cells has been demonstrated in vivo (22) and ultimately results in formation of acellular capillaries. Several proapoptotic initiator caspases, such as capase-2, -8, and -9, are known to trigger apoptotic events. Therefore, we examined high glucose–induced activation of these proapoptotic initiator caspases. Caspase-8 was the only initiator caspase that became activated, and its activity increased slightly by 38 ± 4.3% at 96 h and 40 ± 1.9% by 144 h (Fig. 5A). Western blot analysis confirmed activation of capase-8 indicated by cleavage of caspase-8 zymogen (55 kDa) (Fig. 5A). Interestingly, the initiator caspase-9, a caspase associated with mitochondria-mediated apoptosis, was not activated (data not shown). To further confirm that mitochondria-mediated apoptosis was not induced by high glucose, we examined protein levels of the proapoptotic bcl2 family member bax and the anti-apoptotic bcl2. Protein levels of both bcl2 family members were unchanged in high glucose–treated HRECs compared with control (data not shown).
FIG. 5.

High glucose induces slight activation of proapoptotic caspase signaling pathways in short-term but not in long-term treatments. A: HRECs were incubated in medium containing 5 or 25 mmol/l glucose. Activities of caspase-8, the only initiator caspase that became activated, are presented as means ± SD (n = 8) with *P < 0.05 significantly different from normal glucose. Western blot analyses were done to confirm activation of caspase-8 by determining cleavage of caspase-8 protein over time in high glucose (25 mmol/l)–treated HRECs compared with control cells (55-kDa full-length caspase-8 protein, 43-kDa cleaved fragment). This Western blot is representative of eight independent experiments. B: In HRECs, the c-FLIPL (or α-isoform) (∼55 kDa) is the dominant isoform of this family of caspase-8 signaling regulator and apoptosis inhibitor. Western blot analysis showed that HRECs have high levels of c-FLIPL protein (55 kDa), and high glucose (25 mmol/l) treatment induces loss of c-FLIPL. This Western blot is representative of three independent experiments. Densitometry analysis of the Western blots is presented as the mean ± SD of the c-FlipL–to–actin ratio (n = 3) with *P < 0.05 significantly different from normal glucose. C: Caspase-3 activity showed a slight but significant increase over time by 15 ± 0.5 and 18 ± 0.5% at 96 and 144 h, respectively, compared with control. At 4 weeks of high glucose treatment, no significant changes in caspase-3 activity were detectable. Activities of caspase-3 are presented as means ± SD (n = 8) with *P < 0.05 significantly different from normal glucose.
A major regulator of caspase-8 signaling and inhibitor of apoptosis is the family of c-FLIP proteins. In HRECs, the c-FLIPL (or α-isoform) (∼55 kDa) is the dominant isoform. Western blot analysis showed that HRECs have high levels of c-FLIPL protein (Fig. 5B). Decrease of pro-survival c-FLIPL was evident at 96 h of high glucose exposure, a time point at which caspase-8 activation occurs, suggesting that c-FLIPL needs to be deactivated before caspase-8 activation can occur.
Next, we characterized the downstream effect of high glucose on the activation of executioner caspases, such as caspase-3, to confirm that the weak activation of caspase-8 led to the execution of cell death of HRECs. Caspase-3 activity showed a slight but significant increase over time by 15 ± 0.5 and by 18 ± 0.5% at 96 and 144 h, respectively, compared with control (Fig. 5C, left). At 144 h, high glucose induced 9.3 ± 1.1% cell death in HRECs compared with control cells (4.2 ± 2.1%) as measured by Annexin V staining (n = 5). Interestingly, no significant increase in caspase-3 activity was observed in HRECs exposed to 25 mmol/l glucose for 4 weeks compared with control, indicating that HRECs are highly capable of adapting to high glucose conditions (Fig. 5C, right).
High glucose does not activate proinflammatory caspase-1 or lead to production and release of proinflammatory cytokines by HRECs.
We next investigated whether elevated glucose increased the production and release of proinflammatory cytokines. Incubation of HRECs for 24 up to 144 h in high glucose did not induce proinflammatory caspase-1 activation, the enzyme required to generate mature IL-1β. Caspase-1 activity in HRECs exposed to normal glucose conditions ranged from 40.1 ± 0.8 (24 h) up to 42.9 ± 0.9 pmol AFC · mg−1 · min−1 (144 h) over time. Caspase-1 activity in HRECs treated in high glucose ranged from 39.4 ± 1.0 pmol (24 h) up to 43.4 ± 1.1 pmol AFC · mg−1 · min−1 (144 h) over time. In addition, HRECs did not produce and release the proinflammatory cytokines IL-1β and TNF-α as measured by ELISA when incubated in high glucose for 4 up to 96 h (data not shown).
High glucose induces IL-1β in HRPEs and HMCs.
To determine whether high glucose induces inflammatory mediators by neighboring cells, we measured IL-1β production in HRPEs and HMCs exposed to high glucose. In sharp contrast to endothelial cells, exposure of HRPEs and HMCs to high glucose for 4 h induced marked increase in IL-1β production. For HRPEs, IL-1β production showed an ∼10-fold increase from 5.4 ± 4.2 pg · ml−1 · mg−1 protein in 5 mmol/l glucose to 50.1 ± 12.1 in 25 mmol/l glucose (P < 0.05). For HMCs, IL-1β production increased >10-fold from 20.4 ± 8.5 in 5 mmol/l glucose to 246.6 ± 36.3 pg · ml−1 · mg−1 protein in 25 mmol/l glucose (P < 0.05).
IL-1β increases glucose consumption and subsequent mitochondrial superoxide production in HRECs.
In contrast to high glucose (shown in Fig. 1), exposure of HRECs cultured in normal 5 mmol/l glucose to 1 ng/ml IL-1β for 6 h induced a 2.38-fold increase in glucose consumption (Fig. 6A). In addition, the same treatment induced a threefold increase in mitochondrial oxidative stress (Fig. 6B) compared with normal and high glucose treatment (Fig. 2B).
FIG. 6.

IL-1β increases glucose consumption and subsequent mitochondrial superoxide production in HRECs. A: HRECs were incubated in medium containing 5 mmol/l glucose (white bar) or 5 mmol/l glucose + 1 ng/ml IL-1β (black bar) for 6 h, and glucose consumption was measured as described in research design and methods. Results are presented as the mean ± SD; n = 5, *P < 0.05. B: HRECs were incubated in medium containing 5 mmol/l glucose or 5 mmol/l glucose + 1 ng/ml IL-1 β for 6 h. Mitochondrial superoxide production was measured as described in research design and methods. Results are presented as the mean ± SD (MitoTracker Red/DAPI intensity; n = 6) with *P < 0.05 significantly different from normal glucose. Representative fluorescent images (×40 objectives) of MitoTracker Red and DAPI overlay are shown on the left.
Cytokines induce MAPK phosphorylation and IκBα phosphorylation and degradation in HRECs.
Because high glucose conditions did not activate or stimulate HRECs in any of the assays we tested thus far, we next examined the direct effect of TNF-α and IL-1β on HRECs. HRECs were exposed to 5 ng/ml TNF-α or 1 ng/ml IL-1β for 0–30 min. Both cytokines induced marked ERK1/2 and JNK phosphorylation (Fig. 7A).
FIG. 7.

Cytokines, but not high glucose conditions, induce MAPK phosphorylation, tyrosine phosphorylation, and IκBα phosphorylation and degradation in HRECs. A: Since high glucose conditions did not activate or stimulate HRECs, the direct effects of TNF-α and IL-1β on HRECs were tested. HRECs were serum starved overnight and stimulated with 5 ng/ml TNF-α or 1 ng/ml IL-1β for different time periods as indicated. The activation of ERK and JNK signaling pathways was assessed by immunoblot analyses using anti–phospho-ERK1/2 and anti–phospho JNK antibodies as indicated. Equal amounts of protein were added to each lane as confirmed by actin levels. Both cytokines induced marked ERK1/2 and JNK phosphorylation. Representative results from at least three independent experiments are shown on the left and quantified and presented as means ± SD on the right. *P < 0.01 compared with control. B: Tyrosine phosphorylation in HRECs was assessed by immunoblot using anti-phospho-tyrosine antibody after treatment with high glucose for 0.5–78 h with or without IL-1β (1 ng/ml). Stimulation for 10 min with either 0.5 mmol/l H2O2 (positive control) or 1 ng/ml IL-1β led to a marked increase in tyrosine-phosphorylated proteins. Twenty-four–hour exposure data are shown; similar results were obtained for 0.5- to 78-h time points. C: The effects of high glucose (top) conditions, TNF-α, and IL-1β (bottom image) on IκBα phosphorylation and degradation, the first step in NF-κB pathway activation, were examined in HRECs. High glucose had no effect, whereas 5 ng/ml TNF-α or 1 ng/ml IL-1β for 0–30 min led to a dramatic increase in IκBα phosphorylation and degradation. Representative results from at least three independent experiments are shown on the left and quantified and presented as means ± SD on the right. *P < 0.01 compared with control.
IL-1β, but not high glucose, induces tyrosine phosphorylation in HRECs.
In addition to MAPK protein, tyrosine kinases play an important role in activation of inflammatory and apoptotic signaling cascades. The effect of high glucose and cytokine exposure on tyrosine phosphorylation in HRECs was next examined. Stimulation with 0.5 mmol/l H2O2 for 10 min was used as a positive control. HRECs were treated with high glucose for 0.5–78 h with or without 1 ng/ml IL-1β. IL-1β was added for the last 10 min before harvesting the cells. As expected, stimulation with 0.5 mmol/l H2O2 for 10 min as well as 1 ng/ml IL-1β led to a marked increase in tyrosine-phosphorylated proteins in HRECs (Fig. 7B). However, exposure to high glucose for 0.5–78 h did not change the tyrosine phosphorylation status of HRECs (24-h data shown, other time points produced similar results).
Cytokines, but not high glucose conditions, induce NF-κB pathway in HRECs.
Activation of the NF-κB pathway initiates an inflammatory response in retinal endothelium (23). Thus, the effect of high glucose conditions, TNF-α, and IL-1β on the first step in NF-κB pathway activation, namely IκBα phosphorylation and degradation, was examined. High glucose had no effect, whereas 5 ng/ml TNF-α or 1 ng/ml IL-1β for 0–30 min led to a dramatic increase in IκBα phosphorylation and degradation (Fig. 7C).
Activation of caspases in HRECs by cytokines.
As demonstrated above, HRECs are very responsive to cytokine exposure. IL-1β (1 ng/ml) under normoglycemic conditions activated caspase-8 (Fig. 8A) and dramatically decreased protein levels of c-FLIPL after 12-h treatment, as shown by Western blot analysis (Fig. 8B, right lane). In comparison, c-FLIPL levels were only moderately decreased after 96 h of high glucose incubation (Fig. 8B, left lane). In contrast to high glucose treatment, cytokine treatment induced the activities of a variety of caspases, such as caspase-2 (31.3 ± 7.1 pmol AFC · mg−1 · min−1), caspase-6 (35.9 ± 5.9 pmol AFC · mg−1 · min−1), and caspase-9 (13.0 ± 10.3 pmol AFC · mg−1 · min−1), compared with control (17.0 ± 12.4, 24.0 ± 8.7, and 0.8 ± 6.4 pmol AFC · mg−1 · min−1, respectively), indicating the activation of a mitochondria-mediated apoptotic pathway, which is further supported by the presence of mitochondrial superoxide production as shown in Fig. 6B.
FIG. 8.

IL-1β induces activation of caspase-8 and -3, and loss of protective c-FLIPL in HRECs. A: HRECs were incubated in medium containing 5 mmol/l glucose or 1 ng/ml IL-1β for 12 h. Activities of caspase-8 were measured and presented as means ± SD (n = 6) with *P < 0.05 significantly different from normal glucose. B: IL-1β strongly decreased protein levels of pro-survival c-FLIPL within 12 h compared with 96 h incubation in 25 mmol/l glucose and control as shown by Western blot analysis in HRECs. The Western blot is representative of three independent experiments. Densitometry analysis of the Western blots is presented as the mean ± SD of the c-FlipL–to–actin ratio (n = 3) with *P < 0.05 significantly different from normal glucose. C: HRECs were treated in 5 or 25 mmol/l glucose in the presence or absence of IL-1β (1 ng/ml) for 12 h, and caspase-3 activities were measured (left). High glucose did not influence caspase-3 activity in a synergistic manner. Caspase-3 activities are presented as means ± SD (n = 6) with *P < 0.05 significantly different from normal glucose. HRECs were cultured alone or in co-culture with HMCs in 5 or 25 mmol/l glucose for 48 h as described in research design and [scap]methods (right). Caspase-3 activities of HRECs either cultured alone or in HREC/HMC co-culture were measured and presented as means ± SD (n = 6) with *P < 0.05 compared with HRECs treated in 5 mmol/l glucose.
As expected, 1 ng/ml IL-1β effectively increased HREC caspase-3 activity by 234 ± 13.2% compared with control (Fig. 8C, left) within 12 h. To test the influence of high glucose on the effect of cytokine treatment on caspase-3 activity, HRECs were treated with 1 ng/ml IL-1β in high glucose conditions. High glucose did not alter the effect of cytokine treatment alone on caspase-3 activation (Fig. 8C, left), indicating that high glucose has neither priming nor protective effect. To test whether other retinal cell types known to release cytokines after high glucose treatment, such as HMCs, have an effect on HREC viability, HRECs were co-cultured with HMCs in 5 and 25 mmol/l glucose for 48 h. Caspase-3 activities of HRECs cultured alone or co-cultured with HMCs in different glucose conditions were measured. Only co-culture of HRECs with HMCs in high glucose led to a significant increase in caspase-3 activity (53.1 ± 4.5 pmol AFC · mg−1 · min−1) in HRECs (Fig. 8C, right), indicating that high glucose–stimulated HMCs are causing a detrimental effect on HRECs.
DISCUSSION
Our in vitro results suggest that HRECs respond to cytokines rather than high glucose for ROS generation, induction of inflammatory pathways, and apoptotic changes. Our studies suggest that in vivo diabetes–related endothelial injury in the retina may be primarily due to glucose-induced cytokine release by neighboring cells rather than a direct effect of high glucose on endothelial cells themselves.
A number of studies have demonstrated that glucose-induced oxidative stress and cytokine production in the retina are associated with diabetic retinopathy (7,24–27). However, before the current study, it was not clear whether endothelial dysfunction is a primary effect of direct glucose toxicity in retinal endothelial cells or a secondary effect mediated by cytokines. In this study, we systematically compared the effects of elevated glucose levels to the effects of two key cytokines on various parameters associated with endothelial cell toxicity using HRECs as a model.
Our first key finding demonstrated that HRECs had a very low level of glucose consumption compared with other retinal cell types, HRPEs and HMCs, from the same donors. Moreover, the rate of glucose consumption by HRECs was independent of glucose concentration in the media. There are a number of parameters that can affect glucose toxicity in different cell types. Among them are expression and regulation of different types of glucose transporters and hexokinase isoforms, the rate of glycolysis versus oxidative phosphorylation, antioxidant protection level, and differential response to insulin. As we and others described before, the predominant glucose transporter in HRECs is GLUT1 with minor GLUT3 expression level (15,28,29). The expression level of these transporters was not controlled by high glucose in HRECs in our studies; moreover, the rate of glucose consumption observed in this study (175.09 nmol · mg protein−1 · h−1) was below the Vmax for glucose uptake observed by us in high glucose in HRECs (56.00 pmol · mg protein−1 · s−1 = 201.60 nmol · mg protein−1 · h−1) (15). Taken together, these data suggest that glucose consumption in HRECs is not likely to be limited by glucose uptake rate but rather by further glucose metabolism steps.
If HRECs do not increase glucose consumption under high glucose conditions, there is no reason to expect glucose-induced radical production by these cells. We performed repeated tests using H2DCFDA ROS indicator dye, MitoTracker Red mitochondrial O2·-specific probe, and three different spin traps for EPR measurements: DMPO, PBN, and TEMPO-AM, all of which were negative for high glucose–induced radical production by HRECs.
Similarly, high glucose had no effect on MAPK protein phosphorylation signaling cascades, namely ERK1/2, p38, and JNK phosphorylation, on tyrosine phosphorylation or on activation of NF-κB pathway. High glucose also did not induce caspase activation or apoptosis for the first 96 h of glucose exposure. Only after 96–144 h of exposure did we detect a modest induction of caspases and a minor amount of apoptosis. Only caspase-8 and caspase-3 were activated in HRECs by elevated glucose levels, indicating a type I caspase signaling pathway, which requires upstream caspase-8 activation followed by direct downstream activation of caspase-3. Caspases and other proapoptotic proteins involved in the mitochondrial death pathway were unaffected by high glucose. This is consistent with the findings that ROS formation and mitochondrial O2· production were not increased in high glucose because mitochondria-mediated apoptosis requires the production of O2· within the mitochondria itself to release cytochrome c from the mitochondrial membrane (30). A major regulator of caspase-8 activation is the protein c-FLIP. c-FLIPs are well-known inhibitors of cell death. To date, three isoforms of the protein have been identified, c-FLIPL, c-FLIPS, and c-FLIPR. All isoforms are structurally similar to caspase-8 but do not have a catalytically active site serving as an alternative substrate for caspase-8 (31). High glucose–induced caspase-8 activation only occurred after c-FLIPL levels declined, indicating that apoptosis observed in HRECs was associated with the reduction of c-FLIPL. Interestingly, although caspase-3 was slightly but significantly activated in HRECs by high glucose at 144 h, this activation of caspase-3 seems to be transient. Long-term exposure of HRECs in high glucose up to 4 weeks did not lead to increased caspase-3 activity or cell death, indicating that HRECs are very capable of adapting to high glucose conditions.
Overall, we found that HRECs were remarkably unresponsive to intermediate, high, and fluctuating glucose conditions in various assays associated with endothelial cell toxicity used in this study.
A prominent feature of diabetic retinopathy is increased levels of proinflammatory cytokines in the retina, and several inflammatory pathways are activated at the early stage of diabetic retinopathy. Diabetes leads to the activation of caspase-1, the enzyme responsible for the production of the proinflammatory cytokines IL-1β and IL-18 in the retinas of diabetic animals and diabetic patients (20,21,27). IL-1β can induce IL-6, TNF-α, and itself (32). IL-1β and TNF-α have many overlapping physiological functions. Both have also been associated with the induction of oxidative stress and the mitochondrial death pathway (33). Cytokines, such as TNF-α, its soluble receptor, and IL-1β, are increased in serum of diabetic patients, and increased TNF-α levels have been associated with the development of diabetes complications (34–36). Furthermore, increases in TNF-α and IL-1β levels have been shown in vitreous fluid of diabetic patients and in retinas of streptozotocin-induced diabetic rats and mice (26,35,36).
Endothelial cells are extremely susceptible to cytokines, such as IL-1β, TNF-α, and IFN-γ, which induce production of endothelial cell–derived cytokines (e.g., IL-8, MCP-1, and RANTES), recruitment of leukocytes to the cell surface, and cell death (37,38). They are also very responsive to cytokine stimulation, leading to ICAM-1 and VCAM-1 upregulation, as we have previously shown (19,23). In the current study, we demonstrated that high glucose did not induce caspase-1 activation, the enzyme responsible for IL-1β production, and did not lead to the production and release of IL-1β and TNF-α by HRECs. In contrast, we have shown that IL-1β induced more than a twofold increase in glucose consumption and mitochondrial O2· generation in HRECs, whereas glucose alone did not. We also show that IL-1β is a potent inducer of caspase-3 activity and apoptosis in HRECs. After 12 h of IL-1β stimulation, we detected a 234% increase in caspase-3 activity compared with control, an activation never reached with high glucose alone at any given time point in these cells. In contrast to high glucose treatment, IL-1β led to the activation of caspases involved in the mitochondrial type II death pathway and was preceded by the formation of mitochondrial oxidative stress required for mitochondrial-mediated apoptosis. Anti-apoptotic c-FLIPL levels were dramatically decreased by IL-1β, indicating that HRECs might be very well protected against hyperglycemia but seem to be less able to protect themselves against proinflammatory stress.
In addition to retinal endothelial cells, almost all other cell types in the retina are known to be adversely affected in diabetes. These include pericytes, microglia, astrocytes, HMCs, and HRPEs cells, as well as retinal neural element, such as ganglion cells, bipolar cells, and photoreceptors (rev. in 39). Any of these cells could be the actual source of the glucose-induced cytokine production. In this study, we used two examples, HRPEs and HMCs, to demonstrate the principle of potential involvement of other retinal cell types in glucose-induced toxicity. In agreement with other studies, our results demonstrate that HMCs and HRPEs produce proinflammatory cytokines, such as IL-1β, in response to elevated glucose (21,40). HRPEs and HMCs are known to produce growth factors and neuroactive peptides (41) that maintain endothelial health; however, pathologically high levels of growth factors and cytokines can exist during diabetes and during retinal ischemia (42–44). Because HMCs are known to encircle the microvasculature, protecting the integrity of the microvasculature, the likelihood of cytokines produced by HMCs reaching endothelial cells is very possible (45). Our study indicates that high glucose–induced stimulation of HMCs might have detrimental effects on the vasculature. But again, most other retinal cell types surrounding the vasculature can possibly affect endothelial cell viability in the similar fashion.
Although glucose-induced ROS expression by endothelial cells has been reported by others (18,46–48), outcomes are highly dependent on experimental conditions and the vascular bed and the species of origin of the endothelial cells (49). Perhaps more important is that cell types typically contaminating retinal endothelial cultures, such as HMCs, HRPEs, pericytes, microglia, and astrocytes, may be the actual source of the glucose-induced cytokine production measured previously in retinal endothelial cell cultures. HRPEs and HMCs exposed to high glucose produced 2.5 and 12.3 ng/ml IL-1β, respectively, which is 2.5- to 12.3-fold higher than the 1 ng/ml IL-1β required for stimulation of endothelial cells. Thus, cytokines secreted by these cells could in turn stimulate endothelial cell ROS production, apoptosis, activation of MAPK signaling cascades, tyrosine phosphorylation, NF-κB pathway activation, and adhesion molecules expression. We have extensively published studies using primary HREC cultures and only HRECs of 99% or higher purity, as were used in the current study.
In conclusion, our in vitro studies show that high glucose conditions do not increase endogenous ROS generation by HRECs but rather that these cells respond to cytokines that upregulate ROS. Furthermore, our in vitro studies also suggest that in vivo retinal endothelial injury resulting in capillary drop-out and the retinal pathology associated with diabetic retinopathy may be due primarily to glucose-induced cytokine release by neighboring cells rather than the direct effect of high glucose on endothelial cells.
Acknowledgments
J.V.B. has received Juvenile Diabetes Research Foundation Grant 2-2005-97, National Institutes of Health grants DK-065014 and EY-016077, and MEAS Grant MICL02163. S.M. has received Juvenile Diabetes Research Foundation Grant 2-2006-255, American Diabetes Association Grant 7-06-RA-95, and National Institutes of Health Grants EY-014380 and EY-017206. M.B.G. has received Juvenile Diabetes Research Foundation Grant 4-2000-847 and National Institutes of Health Grants EY-012601 and EY007739.
We thank Weiqin Chen, Madalina Opreanu, Jason A. Vincent, E. Chepchumba Yego, and P. Spoerri for technical support, John McCracken for help with EPR measurements, Denis A. Proshlyakov for help with data analysis, and Michael E. Boulton for careful review of this manuscript.
Published ahead of print at http://diabetes.diabetesjournals.org on 16 April 2008.
J.V.B. and S.M. contributed equally to this study.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Baynes JW: Role of oxidative stress in development of complications in diabetes. Diabetes 40 :405 –412,1991 [DOI] [PubMed] [Google Scholar]
- 2.Koya D, King GL: Protein kinase C activation and the development of diabetic complications. Diabetes 47 :859 –866,1998 [DOI] [PubMed] [Google Scholar]
- 3.Vinores SA, Campochiaro PA, Williams EH, May EE, Green WR, Sorenson RL: Aldose reductase expression in human diabetic retina and retinal pigment epithelium. Diabetes 37 :1658 –1664,1988 [DOI] [PubMed] [Google Scholar]
- 4.Brownlee M: Lilly Lecture 1993: Glycation and diabetic complications. Diabetes 43 :836 –841,1994 [DOI] [PubMed] [Google Scholar]
- 5.Tilton RG, Kawamura T, Chang KC, Ido Y, Bjercke RJ, Stephan CC, Brock TA, Williamson JR: Vascular dysfunction induced by elevated glucose levels in rats is mediated by vascular endothelial growth factor. J Clin Invest 99 :2192 –2202,1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M: Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404 :787 –790,2000 [DOI] [PubMed] [Google Scholar]
- 7.Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH, Liou GI, Caldwell RW: Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets 6 :511 –524,2005 [DOI] [PubMed] [Google Scholar]
- 8.Lee HC, Wei YH: Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood) 232 :592 –606,2007 [PubMed] [Google Scholar]
- 9.Nishikawa T, Araki E: Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal 9 :343 –353,2007 [DOI] [PubMed] [Google Scholar]
- 10.Lassegue B, Sorescu D, Szocs K, Yin QQ, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK: Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88 :888 –894,2001 [DOI] [PubMed] [Google Scholar]
- 11.Ellis EA, Grant MB, Murray FT, Wachowski MB, Guberski DL, Kubilis PS, Lutty GA: Increased NADH oxidase activity in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med 24 :111 –120,1998 [DOI] [PubMed] [Google Scholar]
- 12.Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, Deleo FR, Quinn MT: Role of NF-{kappa}B in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-{alpha}. J Leukoc Biol 82 :729 –741,2007 [DOI] [PubMed] [Google Scholar]
- 13.Lefer DJ, Nakanishi K, Vinten-Johansen J: Endothelial and myocardial cell protection by a cysteine-containing nitric oxide donor after myocardial ischemia and reperfusion. J Cardiovasc Pharmacol 22 (Suppl. 7):S34 –S43,1993 [PubMed] [Google Scholar]
- 14.Powell ED, Field RA: Diabetic retinopathy and rheumatoid arthritis. Lancet 41 :17 –18,1964 [DOI] [PubMed] [Google Scholar]
- 15.Busik JV, Olson LK, Grant MB, Henry DN: Glucose-induced activation of glucose uptake in cells from the inner and outer blood-retinal barrier. Invest Ophthalmol Vis Sci 43 :2356 –2363,2002 [PubMed] [Google Scholar]
- 16.Kusner LL, Sarthy VP, Mohr S: Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase: a role in high glucose-induced apoptosis in retinal Muller cells. Invest Ophthalmol Vis Sci 45 :1553 –1561,2004 [PubMed] [Google Scholar]
- 17.Keusch P: Chemical Kinetics: Rate Laws, Arrhenius Equation—Experiments [article online],2003. . Available from http://www.uniregensburg.de/Fakultaeten/nat_Fak_IV/Organische_Chemie/Didaktik/Keusch/kinetics.htm. Accessed 5 March 2008
- 18.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H: High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49 :1939 –1945,2000 [DOI] [PubMed] [Google Scholar]
- 19.Chen W, Jump DB, Grant MB, Esselman WJ, Busik JV: Dyslipidemia, but not hyperglycemia, induces inflammatory adhesion molecules in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci 44 :5016 –5022,2003 [DOI] [PubMed] [Google Scholar]
- 20.Mohr S, Xi X, Tang J, Kern TS: Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes 51 :1172 –1179,2002 [DOI] [PubMed] [Google Scholar]
- 21.Vincent JA, Mohr S: Inhibition of caspase-1/interleukin-1β signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 56 :224 –230,2007 [DOI] [PubMed] [Google Scholar]
- 22.Mizutani M, Kern TS, Lorenzi M: Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest 97 :2883 –2890,1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Esselman WJ, Jump DB, Busik JV: Anti-inflammatory effect of docosahexaenoic acid on cytokine-induced adhesion molecule expression in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci 46 :4342 –4347,2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Y, Smith MA, Miller CM, Kern TS: Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J Neurochem 80 :771 –779,2002 [DOI] [PubMed] [Google Scholar]
- 25.Kanwar M, Chan PS, Kern TS, Kowluru RA: Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci 48 :3805 –3811,2007 [DOI] [PubMed] [Google Scholar]
- 26.Kowluru RA, Odenbach S: Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol 88 :1343 –1347,2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohr S: Potential new strategies to prevent the development of diabetic retinopathy. Expert Opin Investig Drugs 13 :189 –198,2004 [DOI] [PubMed] [Google Scholar]
- 28.Kumagai AK, Glasgow BJ, Pardridge WM: GLUT1 glucose transporter expression in the diabetic and nondiabetic human eye. Invest Ophthalmol Vis Sci 35 :2887 –2894,1994 [PubMed] [Google Scholar]
- 29.Knott RM, Robertson M, Forrester JV: Regulation of glucose transporter (GLUT 3) and aldose reductase mRNA inbovine retinal endothelial cells and retinal pericytes in high glucose and high galactose culture. Diabetologia 36 :808 –812,1993 [DOI] [PubMed] [Google Scholar]
- 30.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S: Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A 99 :1259 –1263,2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krueger A, Baumann S, Krammer PH, Kirchhoff S: FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol 21 :8247 –8254,2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sparacio SM, Zhang Y, Vilcek J, Benveniste EN: Cytokine regulation of interleukin-6 gene expression in astrocytes involves activation of an NF-kappa B-like nuclear protein. J Neuroimmunol 39 :231 –242,1992 [DOI] [PubMed] [Google Scholar]
- 33.Goossens V, Grooten J, De Vos K, Fiers W: Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A 92 :8115 –8119,1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S: Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye 20 :1366 –1369,2006 [DOI] [PubMed] [Google Scholar]
- 35.Limb GA, Soomro H, Janikoun S, Hollifield RD, Shilling J: Evidence for control of tumour necrosis factor-alpha (TNF-alpha) activity by TNF receptors in patients with proliferative diabetic retinopathy. Clin Exp Immunol 115 :409 –414,1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doganay S, Evereklioglu C, Er H, Turkoz Y, Sevinc A, Mehmet N, Savli H: Comparison of serum NO, TNF-alpha, IL-1beta, sIL-2R, IL-6 and IL-8 levels with grades of retinopathy in patients with diabetes mellitus. Eye 16 :163 –170,2002 [DOI] [PubMed] [Google Scholar]
- 37.Mantovani A, Bussolino F, Introna M: Cytokine regulation of endothelial cell function: from molecular level to the bedside. Immunol Today 18 :231 –240,1997 [DOI] [PubMed] [Google Scholar]
- 38.Kowluru RA, Odenbach S: Role of interleukin-1beta in the development of retinopathy in rats: effect of antioxidants. Invest Ophthalmol Vis Sci 45 :4161 –4166,2004 [DOI] [PubMed] [Google Scholar]
- 39.Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, Kester M, Kimball SR, Krady JK, LaNoue KF, Norbury CC, Quinn PG, Sandirasegarane L, Simpson IA: Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 55 :2401 –2411,2006 [DOI] [PubMed] [Google Scholar]
- 40.Krady JK, Basu A, Allen CM, Xu Y, LaNoue KF, Gardner TW, Levison SW: Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 54 :1559 –1565,2005 [DOI] [PubMed] [Google Scholar]
- 41.Carrasco E, Hernandez C, Miralles A, Huguet P, Farres J, Simo R: Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care 30 :2902 –2908,2007 [DOI] [PubMed] [Google Scholar]
- 42.Bensaoula T, Ottlecz A: Biochemical and ultrastructural studies in the neural retina and retinal pigment epithelium of STZ-diabetic rats: effect of captopril. J Ocul Pharmacol Ther 17 :573 –586,2001 [DOI] [PubMed] [Google Scholar]
- 43.Handa JT, Verzijl N, Matsunaga H, Aotaki-Keen A, Lutty GA, te Koppele JM, Miyata T, Hjelmeland LM: Increase in the advanced glycation end product pentosidine in Bruch's membrane with age. Invest Ophthalmol Vis Sci 40 :775 –779,1999 [PubMed] [Google Scholar]
- 44.Miyamoto N, de Kozak Y, Jeanny JC, Glotin A, Mascarelli F, Massin P, BenEzra D, Behar-Cohen F: Placental growth factor-1 and epithelial haemato-retinal barrier breakdown: potential implication in the pathogenesis of diabetic retinopathy. Diabetologia 50 :461 –470,2007 [DOI] [PubMed] [Google Scholar]
- 45.Distler C, Dreher Z: Glia cells of the monkey retina: II. Muller cells. Vision Res 36 :2381 –2394,1996 [DOI] [PubMed] [Google Scholar]
- 46.Ulker S, McMaster D, McKeown PP, Bayraktutan U: Antioxidant vitamins C and E ameliorate hyperglycaemia-induced oxidative stress in coronary endothelial cells. Diabetes Obes Metab 6 :442 –451,2004 [DOI] [PubMed] [Google Scholar]
- 47.Dragomir E, Manduteanu I, Voinea M, Costache G, Manea A, Simionescu M: Aspirin rectifies calcium homeostasis, decreases reactive oxygen species, and increases NO production in high glucose-exposed human endothelial cells. J Diabetes Complications 18 :289 –299,2004 [DOI] [PubMed] [Google Scholar]
- 48.Pricci F, Leto G, Amadio L, Iacobini C, Cordone S, Catalano S, Zicari A, Sorcini M, Di Mario U, Pugliese G: Oxidative stress in diabetes-induced endothelial dysfunction involvement of nitric oxide and protein kinase C. Free Radic Biol Med 35 :683 –694,2003 [DOI] [PubMed] [Google Scholar]
- 49.Du Y, Sarthy VP, Kern TS: Interaction between NO and COX pathways in retinal cells exposed to elevated glucose and retina of diabetic rats. Am J Physiol Regul Integr Comp Physiol 287 :R735 –R741,2004 [DOI] [PubMed] [Google Scholar]