

Abstract
The discovery in mice of a new lineage of CD4+ effector T helper (Th) cells that selectively produce IL-17 has provided exciting new insights into immune regulation, host defence, and the pathogenesis of autoimmune and other chronic inflammatory disorders. This population of CD4+ Th cells, which has been termed 'Th17', indeed plays an apparently critical role in the pathogenesis of some murine models of autoimmunity. Interestingly, murine Th17 cells share a common origin with Foxp3+ T regulatory cells, because both populations are produced in response to transforming growth factor-β, but they develop into Th17 cells only when IL-6 is simultaneously produced. Initial studies in humans have confirmed the existence of Th17 cells, but they have shown that the origin of these cells in humans differs from that in mice, with IL-1β and IL-23 being the major cytokines responsible for their development. Moreover, the presence in the circulation and in various tissues of Th cells that can produce both IL-17 and interferon-γ, as well as the flexibility of human Th17 clones to produce interferon-γ in addition to IL-17 in response to IL-12, suggests that there may be a developmental relationship between Th17 and Th1 cells, at least in humans. Resolving this issue has great implications in tems of establishing the respective pathogenic roles of Th1 and Th17 cells in autoimmune disorders. In contrast, it is unlikely that Th17 cells contribute to the pathogenesis of human allergic IgE-mediated disorders, because IL-4 and IL-25 (a powerful inducer of IL-4) are both potent inhibitors of Th17 cell development.
Introduction
The adaptive effector CD4+ T helper (Th)-mediated immune response is highly heterogeneous, based on the development of distinct subsets that are characterized by various profiles of cytokine production. Initially, two polarized forms of Th effectors, namely type 1 (Th1) and type 2 (Th2), were identified in both mice and humans [1,2]. Th1 cells produce interferon (IFN)-γ and their primary role is to protect against intracellular microbes; in contrast, Th2 cells produce IL-4, IL-5, IL-9 and IL-13 and are involved in protection against gastrointestinal nematodes, but they are also responsible for allergic disorders [3,4].
Th1 and Th2 cells develop via activation of various transcription factors, the most important being signal transducer and activator of transcription (STAT)-4 and T box expressed in T cells (T-bet) for Th1 cells, and STAT-6 and GATA-binding protein (GATA)-3 for Th2 cells [5]. T-bet binds the promoter of IFN-γ whereas GATA-3 drives epigenetic changes in the Th2 cytokine cluster (IL-4, IL-5 and IL-13), thus giving rise to the development of Th1 and Th2 cells, respectively [6]. The mechanisms responsible for the polarization of the naïve Th cells toward the Th1 or Th2 profile of cytokine production have not yet been completely clarified. However, early production of IFN-γ, IFN-α, or IL-12 by cells of the innate immune system drives Th1 differentiation, whereas early production of IL-4, in the absence of IL-12, drives Th2 differentiation [3,4]. Natural killer cells are the major source of IFN-γ, whereas plasmocytoid dendritic cells (DCs) are the major source of IFN-α [6]. The most powerful Th1-polarizing cytokine is IL-12 [7], which is produced by myeloid DCs after triggering of many of their Toll-like receptors by pathogen products. However, the expression by DCs of various ligands for the Notch receptors present on the naïve Th cells appears also to be involved in the differentiation process. The prevalent expression on DCs of Jagged favours Th2 polarization, even independently of IL-4 production, whereas expression of Delta ligand triggers Th1 polarization [8]. This latter finding has recently been confirmed, including in humans. Immature myeloid DCs express Jagged-1, which triggers a Th2-polarizing programme in CD4+ T cells, whereas stimulation of Toll-like receptors on DCs upregulates the Delta-4 ligand, which triggers in the same cells an opposite, Th1-polarizing programme [9]. In addition, at least in mice, early IL-4 production by naïve Th cells can also be induced by IL-25, a cytokine that is produced not only by Th2 cells but also by an unidentified cell type found in the gut of worm-infested mice [10] or by lung epithelial cells [11]. A third type of Th cell that can produce both Th1 and Th2 cytokines, namely type 0 (Th0), has also been described [12].
During the past few years, a novel family of CD4+ Th cells was detected, which is essentially characterized by IL-17 production and was therefore named 'Th17' [13-19]. Th17 cells exist in both mice and humans, but their phenotypic and functional features, as well as the mechanisms responsible for their development in the two species, appear to be different. In this review I describe the main characteristics of human as compared with murine Th17 cells, and I discuss their possible roles in protection against infectious agents and in immunopathology.
Discovery of murine Th17 cells and their origin
Although the existence of IL-17 as a product of activated CD4+ T cells has been known for more than 10 years, only recently was the existence of Th17 cells as a distinct subset recognized [13-19]. The breakthrough leading to the discovery of the Th17 lineage came from murine models of autoimmunity. Experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA) have historically been associated with unchecked Th1 responses, largely based on studies in which disease development was ablated by treatment with neutralizing antibodies specific for IL-12p40 or gene-targeted mice deficient in the p40 subunit of IL-12.
The link with IL-12 in these diseases was called into question by the discovery that a new IL-12 family member, namely IL-23, shares with IL-12 the p40 subunit, the heterodimer of IL-12 being composed of p40 and p35, and that of IL-23 being composed of p40 and p19. Therefore, an elegant series of studies was conducted that showed that EAE and CIA did not develop in mice deficient in IL-23p19 subunit, whereas they could develop in those deficient in IL-12p35 subunit [13,14]. This suggests that IL-23, but not IL-12, is critically linked to autoimmunity, at least in these models. Moreover, a positive correlation was established between the availability of IL-23 and IL-17 producing effector T cells and disease development, and a negative correlation was established between IL-12 and IFN-γ producing Th1 cells and disease development [20]. Subsequent studies demonstrated that although IL-12 polarized cells (prototypic Th1 cells) expressed genes associated with cytotoxicity, such as those encoding IFN-γ, Fas ligand and granzymes, IL-23 polarized cells expressed genes associated with chronic inflammation, such as IL-17, IL-17F, IL-6, tumour necrosis factor-α and proinflammatory chemokines. Based on these findings, a new role for Th17 cells in immunopathology and the distinct origin of Th1 and Th17 cells under differential IL-12 or IL-23 conditioning was proposed [16]. According to this model, early differentiation of Th1 and Th17 cells from naïve CD4+ T-cell precursors was shared, and thus Th1 and Th17 diverged contingent upon selective availability of IL-12 and IL-23 acting on a common 'Th1 precursor' or 'pre-Th1 intermediate' that co-expressed both IL-12 and IL-23 receptors [21,22].
More recently, however, a completely different model of murine Th17 development has been described. Although IL-23 appeared to be required for Th17-mediated immunopathology, different reports indicated that IL-23 was not critical for Th17 commitment, but only appeared to be required to amplify and/or stabilize the Th17 phenotype [17]. More importantly, three different groups independently demonstrated that transforming growth factor (TGF)-β was required for initiation and that IL-6 was a critical co-factor for Th17 differentiation (Figure 1). IL-1β and tumour necrosis factor-α were also found to amplify the Th17 response induced by TGF-β and IL-6, but they could not substitute for either of these cytokines [23-25]. More recently, it was shown that IL-21, a cytokine produced by Th17 cells themselves, provides an additional autocrine amplificatory signal (Figure 1) [26,27]. Of note, the Th17 polarizing cytokine TGF-β was already known for its ability to promote the development of Foxp3+ T regulatory (Treg) cells. However, expression of IL-17 or Foxp3 was restricted to separate subsets, so that TGF-β driven Th17 and Treg development from naïve precursors appeared to be mutually exclusive. Importantly, in the presence of IL-6, TGF-β induced development of Treg cells was blocked, whereas blockade of IL-6 permitted development of Foxp3+ Treg cells, suggesting that IL-6 inhibited Treg development while enhancing Th17 development induced by TGF-β [17,24,25]. In a recent study, however, an IL-6 independent pathway of murine Th17 differentiation has been discovered [28].
Figure 1.
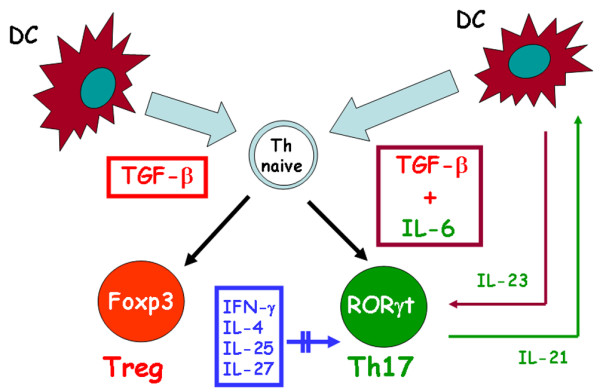
Pathway of murine Th17 differentiation. IL-17 producing CD4+ T helper (Th17) cells originate in response to transforming growth factor (TGF)-β and IL-6 produced by dencritic cells (DCs), whereas TGF-β alone in the absence of IL-6 promotes differentiation of the naïve T helper (Th) cell into a Foxp3+ T regulatory (Treg) cell. IL-23 produced by DCs allows the expansion and/or survival of Th17 cells. Th17 cells themselves produce IL-21, which provides an autocrine amplification loop. IFN-γ, IL-4, IL-25 and IL-27 play an inhibitory role in the development of murine Th17 cells. Double parallel lines across the arrow mean inhibitory effect. ROR, orphan retinoid nuclear receptor.
Properties of murine Th17 cells and their role in protection and immunopathology
The master regulator that directs the differentiation program of Th17 cells is the orphan retinoid nuclear receptor (ROR)γt, whereas neither GATA-3 nor T-bet are required for this function [29]. More recently, it was found that Th17 cells express high levels of another related nuclear receptor, namely RORα, induced by TGF-β and IL-6, which is dependent on STAT-3 [30].
The major functions of cytokines produced by Th17 cells is to chemoattract different cell types through induction of other cytokines and chemokines. Both IL-17 (or IL-17A) and IL-17F act on a broad range of cell types to induce the expression of cytokines (such as IL-6, granulocyte/macrophage colony-stimulatory factor and granulocyte colony-stimulatory factor) and chemokines (such as IL-8, CXC chemokine ligand 1 and 10, and CC chemokine ligand 20), as well as metallo-proteinases (Figure 2). Therefore, both IL-17 and IL-17F are key cytokines for the recruitment, activation and migration of neutrophils. Th17 cells also produce IL-21, which is a powerful B-cell differentiating factor [31], but it also plays an important autocrine amplifying role on Th17 responses [26,27] (see above). Th17 cells also produce IL-22, which is a member of the IL-10 family that has been found to be strongly upregulated during chronic inflammatory disorders and can induce acantosis and dermal inflammation [32].
Figure 2.
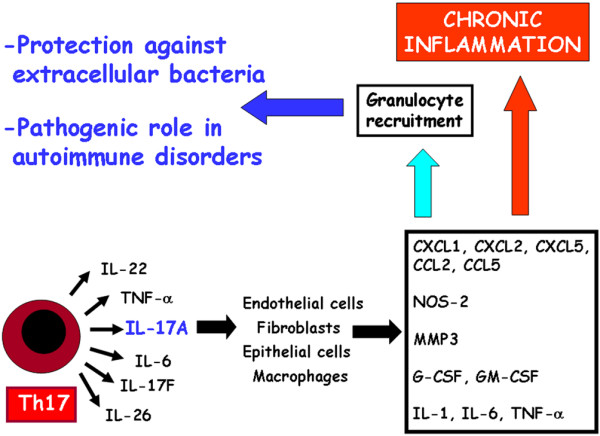
Main activities attributed to Th17 cells. IL-17 producing CD4+ T helper (Th17) cells produce several cytokines, the most important being IL-17 or IL-17A, which activated multiple cell types to produce proinflammatory cytokines, chemokines, nitric oxide synthase (NOS)-2, metallo-proteinases (matrix metalloproteinase [MMP]3) and colony-stimulating factor. This results in granulocyte recruitment, which plays an important role in protection against extracellular bacteria but also in macrophage recruitment and establishment of chronic inflammation. CCL, CC chemokine ligand; CXCL, CXC chemokine ligand; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte/macrophage colony-stimulating factor; Th, T helper; TNF, tumour necrosis factor.
However, in other tissues, such as liver, IL-22 has been shown to counteract the destructive nature of the inflammatory response, thus playing a protective role [33]. Finally, Th17 cells produce IL-26 [34], whose exact role in the Th17 response is not yet established (Figure 2). Murine Th17 cells are subjected to strict control by several cytokines (Figure 1). The development of these cells is indeed inhibited under Th1 or Th2 polarizing conditions, which means that IL-12, IFN-γ and IL-4 play negative regulatory roles in the development of Th17 cells [6]. Even IL-2 has a negative effect on the Th17 differentiation [35]. More recently, IL-25 and IL-27 were also found to inhibit Th17 cells [36,37]. Of note, IL-25 is an inducer of IL-4 [9,10] whereas IL-27 is a potent inducer of IL-10 in CD4+T cells [38,39] and allows the development of Th1 cells [40].
Because of their functional properties, Th17 cells have been suggested to play an important role in responses against extracellular Gram-negative bacteria and fungi, in which granulocyte infiltration is highly protective (Figure 2). Accordingly, preferential IL-17 production by T cells has been found during infection with Klebsiella pneumoniae, Bacteroides fragilis, Citrobacter rodentium, Escherichia coli, Borrelia burgdoferi and fungal species, whereas IL-17 appears to play a modest role (if any) in protecting against intracellular mycobacteria [41]. Notably, however, high fungal burden was found to counter-regulate IL-12 production and to induce production of both IL-23 and IL-17, which subverted the inflammatory program of neutrophils, thus resulting in severe tissue inflammatory pathology rather than protection [42].
In addition to infections, Th17 cells play an important role in the induction and propagation of autoimmunity in various animal models (Figure 2). IL-17 deficient mice or mice treated with an IL-17 receptor antagonist are resistant to development of CIA and develop EAE with delayed onset and reduced severity [16,43]. Furthermore, administration of an IL-17 blocking antibody in mice immunized with a myelin antigen prevents chemokine expression in the brain and the subsequent development of EAE [44]. These data support the idea that IL-17 is involved in the pathogenesis of several autoimmune diseases in mice and possibly also in humans. In this context, the presence, and sometimes the prevalence, of Th1 cells in the inflammatory tissues of murine autoimmune disorders has been interpreted as a protective, rather than proinflammatory, mechanism, based on the following observations: IFN-γ or IFN-γ receptor deficient mice are still susceptible to EAE and CIA [45,46]; and IFN-γ inhibits development of Th17 cells [6]. However, other authors do not agree with this conclusion. First, T-bet has been found also to be required for optimal IL-17 production in the presence of IL-23 [47]. Second, therapeutic administration of small interfering RNA specific for T-bet significantly improved the clinical course of established EAE by limiting the differentiation of autoreactive Th1 cells and inhibiting pathogenic Th17 cells through regulation of IL-23 receptor [48]. Third, in Helicobacter induced colitis, IFN-γ but not IL-17 was the crucial T cell effector cytokine when Treg cells were absent [49]. Fourth, using T cell specific TGF-β deficient mice, the major pathogenic population generated during the establishment of colitis was Th1 cells [50]. Fifth, even in mice with EAE, acquisition of pathogenic function by effector Th17 cells was found to be mediated by IL-23 rather than by TGF-β and IL-6 [51]. Finally, an impressive series of previous observations clearly demonstrated a pathogenic, rather than protective, role of IFN-γ in various murine models of autoimmune disorders [52-60]. Hence, even in murine models, several lines of evidence suggest that Th1 cells can contribute to the inflammatory process rather than simply protecting tissues from Th17-driven inflammation.
Phenotypic and functional features of human Th17 cells
Recent systemic studies have been performed to identify Th17 cells in humans and to characterize their phenotype and functions. Two independent studies have demonstrated the existence of CD4+ memory T cells producing IL-17 after polyclonal stimulation in human peripheral blood and in gut from healthy individuals or patients with Crohn's disease [61,62]. Both studies revealed the presence in these cells of RORγt, IL-23 receptor and the CC chemokine receptor 6, whereas they lacked CXC chemokine receptor 3, a chemokine receptor that is usually espressed by Th1 cells. Moreover, one of the studies identified possible specificity for Candida albicans hyphae of T cells producing IL-17. In the other study various functional features of human Th17 cells were assessed, including at the clonal level [62]. Human Th17 cells exhibited poor proliferative capacity and cytotoxic potential; could induce the production by B lymphocytes of IgG, IgM and IgA, but not IgE; and appeared to be less susceptible than Th1 or Th2 clones to the suppressive activity of an autologous Foxp3+ Treg clone. Moreover, some differences between the two studies were also observed. For example, we identified the existence of a remarkable number of either double positive (IFN-γ+IL-17+) freshly derived cells or T-cell clones (that we named 'Th17/Th1'), a finding that has also been reported, but not sufficiently stressed, in mouse studies. In contrast, in our study clones producing both IL-17 and IL-4 were not observed [62]. More importantly, we found that both classic Th17 and Th17/Th1 clones consistently exhibited not only expression of RORγt but also that of T-bet, at both mRNA and protein levels [62]. The incubation of Th17 clones with IL-12 allowed these cells to produce IFN-γ in addition to IL-17, and this effect was associated with reduced RORγt and increased T-bet expression. Notably, the IL-12 mediated effects were partially inhibited in the presence of IL-23, suggesting the existence of a flexibility between Th17 and Th1 cells and a possible developmental relationship between the two cell types [62].
Induction and regulation of Th17 cells in humans
More recent studies, although partially contradictory, have clearly shown that the induction of Th17 cells in humans is completely different from that in mice. Acosta-Rodriguez and coworkers [63] reported an essential role for IL-1β, in addition to IL-6, but no activity in response to TGF-β in promoting differentiation of naïve CD4+ T cells into Th17 cells. Chen and colleagues [64] found that both IL-6 and TGF-β upregulated RORγt expression, but they did not induce Th17 differentiation in human naïve T cells. Conversely, IL-23 promoted the generation of human Th17 cells but was also an important inducer of other pro-inflammatory cytokines. Wilson and coworkers [65] found that either IL-1β or IL-23 alone was sufficient to induce development of IL-17 producing cells. Finally, van Beelen and colleagues [66] demonstrated that human Th17 cells could be derived only by memory, and not by naïve, T cells, and this effect was due to the nucleotide oligomerization domain 2 ligand muramylpeptide, which enhanced IL-23 and IL-1 production by DCs.
Thus, in contrast to the findings reported in mice, all of these studies agreed that TGF-β was not essential for or inhibited the development of human Th17 cells [63-66]. However, these studies were performed using CD45RA+ T cells purified from peripheral blood of adults. Hence, the possibility remains that the procedure of purification of CD45RA+ T cells could have resulted in contamination of the culture with memory CD4+ T cells, thus affecting the results [67]. To circumvent this problem, we investigated the mechanisms responsible for the differentiation of human Th17 cells using purified CD4+ T cells obtained from umbilical cord blood. In our system, either IL-1β or IL-23 upregulated the expression of both IL-23 receptor and RORγt, and IFN-γ, but only the combination of the two cytokines allowed CD4+ T cells to express and produce IL-17. Again, we observed the development not only of Th17 but also of Th17/Th1 cells. Moreover, although the addition of IL-4 to the mixture of IL-1 and IL-23 consistently inhibited the expression of IL-23 receptor, RORγt and IL-17, the addition of IL-12 reduced the expression of IL-17 without affecting Th17/Th1 cells. Moreover, TGF-β did not affect either RORγt or the IL-23 receptor, but virtually abolished T-bet expression, thus reducing Th1 and Th17/Th1 while increasing Th17 cells (unpublished data).
Taken together, these data support the difference in origin of Th17 cells between mice and humans, and once again suggest the existence of a developmental relationship between human Th17 and Th1 cells (Figure 3).
Figure 3.
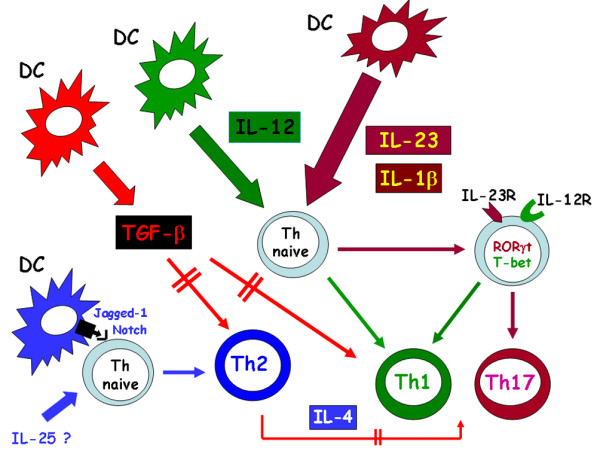
Pathway of human Th17 differentiation. IL-17 producing CD4+ T helper (Th17) cells originate in presence of IL-23 and IL-1β, each of which upregulates orphan retinoid nuclear receptor (ROR)γt, T box expressed in T cells (T-bet), IL-23 receptor (IL-23R) and IL-12 receptor (IL-12R) in the naïve T hlper (Th) cell. When the two cytokines are produced in combination, IL-17 mRNA is also expressed and IL-17 is produced alone or in combination with IFN-γ. The presence of IL-12, in absence of IL-1β and IL-23, shifts the differentiation of the naïve Th cell toward the Th1 phenotype. IL-4, which is produced by the Th naïve cell itself following interaction of its Notch receptors with Jagged-1 expressed on the dendritic cell (DC) and/or by the presence of IL-25 (still unclear in humans), has a potent inhibitory effect on the expression of RORγt, T-bet, IL-23R, IL-12R, IFN-γ, and IL-17. Transforming growth factor (TGF)-β strongly inhibits the development of both Th1 and Th2 cells, whereas it has little or no effect on the development of Th17, thus indirectly favouring their expansion. Double parallel lines across the arrows mean inhibitory effect.
Possible role of Th17 cells in human immunopathology
Because of differences in some properties and in the mechanism of origin between murine and human Th17 cells, it is difficult to identify the role played by this novel member of the CD4+ T cell effector family in the pathogenesis of human disorders; furthermore, this difficulty is exacerbated by the paucity of information currently available.
A greater number of IL-17 mRNA expressing cells were found by using in situ hybridization in cerebrospinal fluid than in peripheral blood from patients with multiple sclerosis [68]. More importantly, human Th17 lymphocytes have been found to promote blood-brain barrier disruption and central nervous system inflammation through CD4+ lymphocyte recruitment [69]. Moreover, both IL-17 and IL-23p19 were found in sera, synovial fluid, and synovial biopsies of most patients with rheumatoid arthritis (RA), whereas both of them were absent in osteoarthritis [70,71]. IL-17 has also been detected in the sera and diseased tissues of patients with systemic lupus erythematosus [72] or systemic sclerosis [73]. Recently, increased expression of CC chemokine ligand 20 (the chemokine able to bind CC chemokine receptor 6 expressing Th17 cells [61,62]) in the inflamed joints of patients with RA has been reported [74]. High IL-17 levels have also been found in the sera and colonic biopsies of patients with Crohn's disease [75,76], in whom IL-17F appears to play an important role [77], as well as in the affected skin of patients with inflammatory skin disorders such as nickel-induced dermatitis, psoriasis and atopic dermatitis [78,79]. However, the great majority of these studies were performed by assessing the presence of mRNA for IL-17 in tissues and/or measuring IL-17 protein in biologic fluids. Moreover, some of these findings were recently challenged. For example, in a recent study the frequency of Th17 cells was significantly decreased in the joints a compared with peripheral blood from the same RA patients, whereas Th1 cells were more abundant in the joints than in peripheral blood [80]. Thus, the role played by Th17 cells in the pathogenesis of human autoimmune disorders, although very probable, is not yet proven. More importantly, the respective roles of Th17 and Th1 cells in inflammatory sites remain unclear; the solution of this problem rests mainly on the demonstration of whether (at least in humans) a developmental relationship between the two cell types does indeed exist.
With regard to the possible pathogenic role of Th17 cells in allergic disorders, it has been claimed that these cells may play a critical role in the granulocyte infiltration that is present in the bronchi of some asthmatic patients [81]. Obviously, because Th17 cells have a particular ability to recruite granulocytes, it cannot be excluded that IL-17 recruited cells can enhance bronchial inflammation in severe asthma complicated by bacterial infections. However, it is highly unlikely that Th17 plays a role in classic IgE-mediated allergic disorders. First, Th17 cells can induce production of IgM, IgG and IgA antibodies, but not IgE antibodies [62]. Second, the presence of IL-4 is among the most effective inhibitory signals for the differentiation of Th17 cells in both mice [6] and humans [63-66] (unpublished data). Finally, we recently examined the phenotype of T cell clones specific for amoxicillin, which were generated from the peripheral blood of a patient who had suffered an amoxicillin-induced anaphylactic shock and exhibited amoxicillin-specific IgE antibodies in his serum. All T cell clones specific for amoxicillin derived from this patient had a classic Th2 profile and none of them was able to produce IL-17 (unpublished data), supporting the view that Th17 cells do not play any role in uncomplicated, IgE-mediated allergic disorders.
Conclusion
The discovery in both mice and humans of a new member of the CD4+effector T-cell family (Th17 cells) has provided exciting and novel insights into the immune mechanisms that are responsible for protection and immunopathology. However, some critical differences appear to exist with regard to the mechanisms that are involved in the differentiation of murine and human Th17 cells, which render it difficult to establish new therapeutic procedures to target these cells or the cytokines that are responsible for their development. In particular, in murine experimental models of autoimmunity Th17 cells share their origin with Treg cells and are pathogenic, whereas Th1 cells appear to play a protective role in response to them. More importantly, that inhibition of Th17 cells favours development of the Treg cell population represents a source of considerable confusion, with respect to possible therapeutic options targeting Th17 cells. In contrast, Th17 cells appear to have a different origin in humans than in mice, and whether classic Th1 cells play a protective role against the pathogenic activity of Th17 cells or contribute alongside them to the pathogenesis of autoimmune disorders remains unclear. If we are to resolve this issue, we must determine whether human Th17 cells are developmentally related to Th1 cells. This represents another impressive example of how murine models, although extremely useful, cannot dogmatically be regarded as optimal models for the development of novel immunotherapeutic strategies in humans.
Abbreviations
CIA = collagen-induced arthritis; DC = dendritic cell; EAE = experimental autoimmune encephalomyelitis; GATA = GATA-binding protein; IFN = interferon; IL = interleukin; RA = rheumatoid arthritis; ROR = orphan retinoid nuclear receptor; STAT = signal transducer and activator of transcription; T-bet = T box expressed in T cells; TGF = transforming growth factor; Th = T helper; Treg = T regulatory.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
The experiments reported in this paper were performed with grants from the Associazione Italiana per la Ricerca sul cancro, the Ministero dell'Istruzione, dell'Università e della Ricerca, the Ministero della Salute and FP6 European Union project INNOCHEM, LSHB-CT 2500-518157.
References
- Mosmann TR, Cherwinski C, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clones. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- Del Prete GF, De Carli M, Mastromauro C, Biagiotti R, Macchia D, Falagiani , Ricci M, Romagnani S. Purified protein derivative of Mycobacterium tuberculosis and excretory/secretori antigen(s) of Toxocara canis expand in vitro human T cells with stable and opposite (type 1 T helper or type 2 T helper) profile of cytokine production. J Clin Invest. 1991;88:346–350. doi: 10.1172/JCI115300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- Romagnani S. The Th1/Th2 paradigm. Immunol Today. 1997;18:263–266. doi: 10.1016/S0167-5699(97)80019-9. [DOI] [PubMed] [Google Scholar]
- Rengarajan J, Szabo SJ, Glimcher LH. Transcriptional regulation of Th1/Th2 polarization. Immunol Today. 2000;21:479–483. doi: 10.1016/S0167-5699(00)01712-6. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17 and effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsen F, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–526. doi: 10.1016/S0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- Liotta F, Frosali F, Querci V, Mantei A, Filì L, Maggi L, Mazzinghi B, Angeli R, Ronconi E, Santarlasci V, Biagioli T, Lasagni L, Ballerini C, Parronchi P, Scheffold A, Cosmi L, Maggi E, Romagnani S, Annunziato F. Human immature myeloid dendritic cells trigger a Th2-polarizing program via Jagged-1/Notch interaction. J Allergy Clin Immunol. 2008 doi: 10.1016/j.jaci.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Fallon PG, Ballantyne SJ, Mangan NE, Barlow H, Davarma A, Hewett DR, McIlgorm A, Jolin HE, McKenzie AN. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203:1105–1116. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angkasekwinal P, Park H, Wang YH, Chang SH, Corry DB, Liu VJ, Zhu Z, Domg C. Interleukin 25 promotes the initiation of proallergic type responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Scherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wickwoski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwuick JD, Cua DJ. Divergent pro-and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1958. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami A. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor β2-deficiet mice. IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelinization in the central nervous system. J Immunol. 2003;170:2153–2160. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kalestein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage J, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Park H, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1069–1070. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin-17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1069–1070. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Kuchroo VK. IL-12-and IL-23-induced T helper cell subsets: birds of the same feather flock together. J Exp Med. 2005;201:169–171. doi: 10.1084/jem.20042279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGF-beta in the context of an inflammatory cytokine milieu supports differentiation of IL-17-producing T cells. Immunity. 2006;25:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Pasnopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awashi A, Jager A, Strom TB, Oukksa M, Kuchroo VK. IL-21 initiates an alternative pathway to onduce pro-inflammatory Th17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin-17-producing T helper cells. Proc Natl Acad Sci USA. 2007;104:12099–12104. doi: 10.1073/pnas.0705268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Mackenzie BS, Zhou L, Tadokoro CE, Lepelley A, LKafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatopry IL-17+ T helper cells. Cell. 2006;126:1121–1131. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Yang XO, Pappau BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by organ nuclear receptors RORα and RORγ. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, de Waal Malefyt R, Tangye SG. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol. 2007;179:8180–8190. doi: 10.4049/jimmunol.179.12.8180. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a Th17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AL, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647–659. doi: 10.1016/j.immuni.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberts E. IL17/Th17 targeting: On the road to prevent chronic destructive arthritis? Cytokine. 2007;41:84–91. doi: 10.1016/j.cyto.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Laurence A, Tato CT, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Kleinsheck MA, Owyang A, Jooyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, Kastelein RA, Cua DJ. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, Gran B, Zhang G-X, Rostami A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:3268–3275. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin-27 in generating interleukin-10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, Yu S, Saris CL, Bran B, Ciric B, Rostami A. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin-27-stimulated T cells. Nat Immunol. 2007;8:1372–1379. doi: 10.1038/ni1540. [DOI] [PubMed] [Google Scholar]
- Chen Q, Ghilardi N, Baker T, Xie M-H, Gurner A, Grewal IS, de Sauvage FJ. Development of Th1-type immune responses requires the type I cytokine receptor TCCR. Nature 2000. 2007;407:916–920. doi: 10.1038/35038103. [DOI] [PubMed] [Google Scholar]
- Aujla S, Dubin PJ, Kolls JK. Th17 cells and mucosal host defence. Semin Immunol. 2007;19:377–382. doi: 10.1016/j.smim.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, Belladonna ML, Vacca C, Conte C, Mosci P, Bistoni F, Puccetti P, Kastelein RA, Kopf M, Romani L. IL-23 and the Th17 pathway promote inflammation and impari antifungal immune resistance. Eur J Immunol. 2007;37:2695–2706. doi: 10.1002/eji.200737409. [DOI] [PubMed] [Google Scholar]
- Koenders MI, Lubberts E, Oppers-Walgren B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Joosten LA, van den Berg WB. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167:141–149. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyttenhove C, Sommereyns C, Thèate I, Mivhiels T, Van Snick J. Anti-IL-17 autovaccine prevents clinical and histological manifestations of experimental autoimmune enecephalomyelitis. Ann N Y Acad Sci. 2007;1110:330–336. doi: 10.1196/annals.1423.035. [DOI] [PubMed] [Google Scholar]
- Ferber IA, Brocce S, Taylor Edwards C, Ridgway W, Dinisco C, Steinman L, Dal ton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- Kageyama Y, Koide Y, Yoshida A, Uchijima M, Arai T, Miyamoto S, Ozeki T, Hiyoshi M, Kushida K, Inoue T. Reduced susceptibility to collagen-induced arthritis in mice deficient in IFN-gamma receptor. J Immunol. 1998;161:1542–1548. [PubMed] [Google Scholar]
- Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–2598. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, Lovett-Racke AE. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–1348. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, Mckenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1-and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain Th-cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Xiao BG, Bai XF, van der Meide PH, Orn A, Link H. Mice with IFN-gamma receptor deficiency are less susceptible to experimental autoimmune myasthenia gravis. J Immunol. 1999;162:3775–3781. [PubMed] [Google Scholar]
- Yu S, Medling B, Yagita H, Braley-Mullen H. Characteristics of inflammatory cells in spontaneous autoimmune thyroiditis of NOD.H-2h4 mice. J Autoimmun. 2001;16:37–45. doi: 10.1006/jaut.2000.0458. [DOI] [PubMed] [Google Scholar]
- Haas C, Ryffel B, Le Hir M. IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J Immunol. 1997;158:5484–5491. [PubMed] [Google Scholar]
- Egwuagu CE, Sztein J, Mahdi RM, Li W, Chao-Chan C, Smith JA, Charukamnoetkanok P, Chepelinsky AB. IFN-gamma increases the severity and accelerates the onset of experimental auto-immune uveitis in transgenic rats. J Immunol. 1999;162:510–517. [PubMed] [Google Scholar]
- Jones LS, Rizzo LV, Agarwal RK, Tarrant TK, Chan CC, Wiggert B, Caspi RR. IFN-gamma-deficient mice develop experimental autoimmune uveitis in the context of a deviant effector response. J Immunol. 1997;158:5997–6005. [PubMed] [Google Scholar]
- Schwarting A, Tesch G, Kinoshita K, Maron R, Weiner HL, Kelley VR. IL-12 drives IFN-gamma-dependent autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1999;163:6884–6891. [PubMed] [Google Scholar]
- Schwarting A, Wada T, Kinoshita K, Tesch G, Kelley VR. IFN-gamma receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1998;161:494–503. [PubMed] [Google Scholar]
- Wang B, Andre I, Gonzalez A, Katz JD, Aguet M, Benoist C, Mathis D. Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:13844–13489. doi: 10.1073/pnas.94.25.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan A, Grusby MJ, Kaplan CD, O'Neill SK, Eibel H, Koreny T, Czipri M, Mikecz K, Zhang J. IL-4 and IL-12 regulate proteoglycan-induced arthritis through Stat-dependent mechanisms. J Immunol. 2002;169:3345–3352. doi: 10.4049/jimmunol.169.6.3345. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Filì L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Napoletani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NJ, Boniface K, Chan JR, McKenzie B, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron J-C, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat SAJ, Kapsenberg ML, de Jong EC. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:1–10. doi: 10.1016/j.immuni.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Laurence A, O'Shea JJ. Th17 differentiation: of mice and men. Nat Immunol. 2007;9:903–905. doi: 10.1038/ni0907-903. [DOI] [PubMed] [Google Scholar]
- Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- Kebir H, Kteymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human Th17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honorati MC, Meliconi R, Pulsatelli L, Cane S, Frizziero L, Facchini A. High in vivo expression of interleukin-17 receptor in synovial endothelial cells and chondrocytes from arthritis patients. Rheumatology. 2001;40:522–527. doi: 10.1093/rheumatology/40.5.522. [DOI] [PubMed] [Google Scholar]
- Wong CK, Ho CY, Li EK, Lam CW. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus. 2000;9:589–593. doi: 10.1191/096120300678828703. [DOI] [PubMed] [Google Scholar]
- Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, Takabayashi K, Iwamoto I. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. 2000;43:2455–2463. doi: 10.1002/1529-0131(200011)43:11<2455::AID-ANR12>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Naomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204:2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen OH, Kirman I, Rudiger N, Hendel J, Vainer B. Upregulation of interleukin-12 and -17 in active inflammatory bowel disease. Scand J Gastroenterol. 2003;38:180–185. doi: 10.1080/00365520310000672. [DOI] [PubMed] [Google Scholar]
- Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiderer J, Elben I, Diegelmann J, Glas J, Stallhofer J, Tillack C, Pfennig S, Jurgens M, Schmechel S, Konrad A, Goke B, Ochsenkuhn T, Muller-Myhsok B, Lohse P, Brands S. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn's disease and analysis of the IL-17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis. 2008;14:437–445. doi: 10.1002/ibd.20339. [DOI] [PubMed] [Google Scholar]
- Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinmocytes: synergistic effects with IFN-gamma and TNF-alpha. J Immunol. 1999;162:494–502. [PubMed] [Google Scholar]
- van Beleen AJ, Teunissen MBM, Kapsenberg ML, de Jong EC. Interleukin-17 in inflammatory skin disorders. Curr Opin Allergy Clin Immunol. 2007;7:374–381. doi: 10.1097/ACI.0b013e3282ef869e. [DOI] [PubMed] [Google Scholar]
- Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, Iwamoto Y, Yoshikai Y. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. 2007 doi: 10.1136/ard.2007.080341. [DOI] [PubMed] [Google Scholar]
- Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, Ceuppens JL. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocyte influx? Respir Res. 2006;7:135–143. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]