

Abstract
In a Dutch pedigree suffering from autosomal dominant nonsyndromic hearing impairment (ADNSHI), linkage was found to the locus for DFNA15, with a two-point logarithm of the odds (LOD) score of 5.1. Sequence analysis of the POU4F3 gene that is involved in DFNA15 revealed the presence of a missense mutation (c.865C>T), segregating with the deafness in this family. The mutation is predicted to result in the substitution of a phenylalanine residue for a leucine residue (p.L289F) in the POU homeodomain of the transcription factor POU4F3. Mutation analysis of the POU4F3 gene in 30 patients suffering from dominantly inherited hearing impairment revealed a second novel missense mutation (c.668T>C), resulting in the substitution of a proline for a leucine residue (p.L223P) within the POU-specific DNA-binding domain of the protein. In a computer model describing the structure of the two DNA-binding domains, the alterations are predicted to affect the tertiary structure of these domains. Transient transfection studies showed that whereas the wild-type POU4F3 is located almost exclusively in the nucleus, part of the mutant proteins was also present in the cytoplasm. In addition, both mutant proteins showed greatly reduced capability for binding to DNA as well as transcriptionally activating reporter gene expression. Together, our results describe the identification of the first missense mutations in POU4F3 causing DFNA15. Furthermore, mutations in this gene do not seem to be a rare cause of hearing impairment in the Dutch population, and the POU4F3 gene may thus be suitable for implementation in diagnostic testing.
Keywords: DFNA15, POU4F3, nonsyndromic hearing impairment, DNA-binding domain, POU-specific domain, homeodomain
INTRODUCTION
Autosomal dominant nonsyndromic hearing impairment (ADNSHI) is very heterogeneous, with respect to both the causative genes as well as the clinical manifestations. Clinically, there is great variability regarding age of onset, the degree of hearing loss and the affected frequencies, and the presence or absence of vestibular defects [Huygen et al., 2007]. Genetically, various genes are known that play a role in the pathology of ADNSHI. Functional characterization of the proteins encoded by these genes will result in a better understanding of the molecular processes involved in normal hearing, and is therefore of vital importance to develop therapeutic strategies.
To date, 51 loci for ADNSHI have been reported, and for 19 loci the causative gene has been identified (Hereditary Hearing Loss Homepage; http://webhost.ua.ac.be/hhh). The proteins encoded by these genes include motor proteins (myosin Ia, -IIa, -IIc, -VI, and -VIIa), cytoskeletal proteins (γ-actin, diaphanous-1), the endoplasmic reticulum (ER)-membrane protein wolframin, the transmembrane channel-like protein TMC1, components of the extracellular matrix (cochlin, the collagen XI α2 chain, α-tectorin), gap junction proteins (connexin 26 and connexin 30), ion channels (KCQN4), and regulators of transcription (TFCP2L3, EYA4, and POU4F3) (reviewed by Petit [2006]). Besides the function of these genes, their expression pattern within the inner ear, both spatial and temporal, and thus their presumed time and site of action is also highly variable.
The prevalence of deafness-causing mutations greatly differs among these genes. Various mutations, in for instance the TECTA and COCH genes, frequently cause DFNA8/12 [Alloisio et al., 1999; Balciuniene et al., 1999; Moreno-Pelayo et al., 2001; Plantinga et al., 2006; Verhoeven et al., 1998] and DFNA9 [Collin et al., 2006; De Kok et al., 1999; Fransen et al., 1999; Manolis et al., 1996; Robertson et al., 1997, 1998; Street et al., 2005], respectively. In contrast, a mutation in the POU4F3 gene (DFNA15) has thus far been described in only one family, in which an 8-bp deletion in the region encoding the POU homeobox DNA-binding domain of this transcription factor results in a reduced capability of binding to its target DNA [Vahava et al., 1998; Weiss et al., 2003].
Here, we report on a large Dutch family with autosomal dominant hearing loss in which the genetic defect mapped to the DFNA15 locus (MIM 602459). Mutation analysis of the POU4F3 gene (MIM 602460) resulted in the identification of the first missense mutation in this gene causing hearing impairment. Subsequent screening of 30 index patients from small families revealed one other novel missense mutation in POU4F3. Furthermore, we provide evidence that these mutations affect the functional properties of the POU4F3 protein.
MATERIALS AND METHODS
Subjects
A large Dutch family (Family W05−549) suffering from progressive hearing loss was ascertained. A total of 61 individuals participated in this study. Medical history was taken from all participants, paying attention to hearing impairment and vestibular symptoms. Furthermore, concomitant disease, use of medication, and any other possible cause of acquired deafness were ruled out. After microotoscopic examination, pure tone audiometry was performed. Clinically affected individuals also underwent speech audiometry. In addition, 30 index patients suffering from nonsyndromic hearing loss with a putative autosomal dominant pattern of inheritance and 100 ethnically-matched controls participated in this study. Written informed consent was obtained from all individuals and this study was approved by the local ethics committee (Commissie Mensgebonden Onderzoek Regio Arnhem Nijmegen 9504−0521).
Linkage Analysis
Genomic DNA from all participating individuals was extracted from peripheral blood lymphocytes according to standard protocols [Miller et al., 1988]. Microsatellite markers flanking known DFNA loci were genotyped in part of the family under standard PCR conditions and analyzed with the GeneMapper program (Applied Biosystems, Foster City, CA). In the region with an allele segregation that was indicative for linkage in this family, two-point logarithm of the odds (LOD) scores for the markers D5S436 and D5S2090 were calculated with the SuperLink v1.4 program in the easyLINKAGE software package [Hoffmann and Lindner, 2005]. Markers were selected from the University of California, Santa Cruz (UCSC) human genome database (www.genome.ucsc.edu).
Mutation Analysis
All exons and intron–exon boundaries of the POU4F3 gene were amplified using standard PCR conditions. To amplify exon 1, forward primer 5′-GCAGGCTGCTTGTAAGATGAG-3′ and reverse primer 5′-AGACAGCGGCGATTGTTC-3′ were used, whereas for exon 2, two PCR products were amplified using forward primer 5′-CTCGGTTGCTTGAAAATGTG-3′ and reverse primer 5′-GGGGATCTTGAGATTAGCC-3′, and forward primer 5′-AGCTGG AAGCCTTCGCC-3′ and reverse primer 5′-GGAAAGTCTGTGGCTTCGG-3′, respectively. Sequence analysis was performed with the ABI PRISM Big Dye Terminator Cycle Sequencing V2.0 Ready Reaction kit and the ABI PRISM 3730 DNA analyzer (Applied Biosystems). To determine the presence of the p.L289F mutation in Family W05−549, and in ethnically-matched controls, the PCR product of exon 2 was digested with XhoI, whose recognition site is disrupted in the mutant allele, and analyzed on a 1.4% agarose gel. The presence of the p.L223P mutation in control individuals was detected in a comparable manner by BlpI digestion, of which the recognition site is also disrupted by the mutation. Mutation nomenclature follows the journal guidelines (www.hgvs.org/mutnomen) with the numbering based on +1 as the A of the ATG translation initiation codon in the reference sequence. The initiation codon is codon 1.
Molecular Modeling
The POU-specific and the POU homeodomain of POU4F3 show high sequence similarity with the corresponding domains in POU2F1/Oct-1 [Klemm et al., 1994]. The crystal structure of POU2F1/Oct-1 was used as a template to build a model of these domains in the human POU4F3 protein. Modeling was performed with the WHAT IF software [Vriend, 1990] using standard parameter settings and protocols as described previously [Chinea et al., 1995; De Filippis et al., 1994]. This modeling is straightforward because the percentage sequence identity is high (53%) and all key residues are conserved between the template and the model. The effects of both the p.L223P and the p.L289F mutations were analyzed with the WHAT IF mutant structure prediction module. A detailed description of the molecular modeling analysis is presented at http://swift.cmbi.ru.nl/gv/service/pou4f3.
Antibodies
Primary monoclonal mouse anti-hemagglutinin (HA) antibodies and rabbit anti-actin antibodies were derived from Sigma (St. Louis, MO). Secondary antibodies for immunocytochemistry and western blot analysis were acquired from Molecular Probes-Invitrogen (Breda, the Netherlands), Rockland (Gilbertsville, PA), and Jackson ImmunoResearch Laboratories (West Grove, PA).
Generation of Plasmids
A cDNA molecule encoding the full-length wild-type POU4F3 protein was amplified from Marathon Ready human fetal brain cDNA (Clontech, Mountain View, CA), using a nested PCR approach. The first PCR was performed with the primers 5′-AAGCCTGATTCCATGTCACC-3′ and 5′-ATAACCATCCCCCGAACC-3′, followed by a nested PCR reaction using the primers 5′-aaaaagcaggcttcATGGCCATGAACTCCAAGCA-3′ and 5′-agaaagctgggtgTCAGTGGACAGCCGAATACT-3′ that contain the attB1 and attB2 sites necessary for Gateway cloning. By using this Gateway technology (Invitrogen, Breda, the Netherlands), cDNAs encoding human full-length POU4F3 were cloned in the pDONR201 vector as previously described [Kantardzhieva et al., 2005]. To introduce the two missense mutations identified in this study, site-directed mutagenesis was performed with the QuickChange site-directed mutagenesis kit, according to the manufacturers protocol (Stratagene, La Jolla, CA) using the primers 5′-CCGGCGTGGGCTCGCCGAGCCAAAGCACCAT-3′ and 5′-ATGGTGCTTTGGCTCGGCGAGCCCACGCCGG-3′ for the p.L223P mutant, whereas for the p.L289F mutant, primers 5′-CCGGAGAAGCGTTCATTCGAGGCCTATTTCG-3′ and 5′-CGAAATAGGCCTCGAATGAACGCTTCTCCGG-3′ were used. Subsequently, the desired fragments were cloned into the HA-tagged destination vector (pcDNA3-HA/DEST; Invitrogen), again as described previously [Kantardzhieva et al., 2005].
Transient Transfection in COS-1 Cells
HA-tagged wild-type and mutant POU4F3 were expressed by using the mammalian expression vector pcDNA3-HA/DEST, that contains a cytomegalovirus (CMV) promoter. COS-1 cells were transiently transfected by using Effectene (Qiagen, Hilden, Germany), according to the manufacturer's instructions. At 24 hr after transfection, cells were washed with phosphate-buffered saline (PBS) and subsequently prepared for either immunolocalization studies or western blot analysis.
Subcellular Localization of Wild-Type and Mutant POU4F3 in COS-1 Cells
To determine the subcellular localization of the HA-tagged wild-type and mutant POU4F3, transiently transfected cells were fixed with 4% paraformaldehyde, incubated with primary anti-HA and secondary goat-anti-mouse Alexa680-labelled antibodies and mounted with Vectashield containing DAPI (Vector Laboratories Inc., Burlingame, CA). Images were taken with an Axioplan2 Imaging fluorescence microscope equipped with a DC350FX camera (Zeiss, Sliedrecht, the Netherlands) and processed using Adobe Photoshop (Adobe Systems, San Jose, CA). To determine the number of cells expressing POU4F3 outside the nucleus, slides were blinded and 100 cells of each transfected mutant were scored; each construct was transfected in duplicate.
Western Blot Analysis
To determine the size of the HA-tagged wild-type and mutant POU4F3 proteins, transfected cells were lysed on ice in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Triton X-100 supplemented with complete protease inhibitor cocktail [Roche, Woerden, the Netherlands]). Supernatants were cleared by centrifugation for 10 min at 11,000 g at 4°C, separated on a 4% to 12% SDS-PAGE gel and transferred onto nitrocellulose filters (Hybond; Amersham Biosciences, Buckinghamshire, United Kingdom). HA-tagged POU4F3 proteins were detected with either primary anti-HA and secondary IRDye800 goat-anti-mouse immunoglobulin G (IgG), or primary anti-β-actin and secondary Alexa680 goat-anti-rabbit IgG antibodies. Signals were analyzed using the Odyssey Infrared Imaging System (LI-COR, Lincoln, NE).
Electrophoretic Mobility Shift Assay (EMSA)
The wild-type mouse Pou4f3 expression plasmid was described previously [Liu et al., 2000]. To generate the p.L223P and p.L289F mutations, murine Pou4f3 cDNA was mutated by PCR mediated site-directed mutagenesis and then subcloned into the pcDNA3 expression vector (Invitrogen). Wild-type and mutant Pou4f3 proteins were produced from the expression plasmids by the coupled TNT transcription/translation system (Promega, Madison, WI), in the presence of [35S]methionine. Similar protein yields were confirmed by running SDS-PAGE and autoradiography. To generate the probe, DNA oligonucleotides containing a consensus Pou4f3 binding site were end-radiolabeled with ATP-γ32P and T4 polynucleotide kinase. Binding reactions were carried out at room temperature for 20 to 30 min in a final volume of 30 μl containing 10 mM HEPES (pH 7.5), 50 mM KCl, 1 mM EDTA, 0.1% Triton-X 100, 5% glycerol, 0.1 mM dithiothreitol (DTT), 0.1 mM phenylmethanesulphonylfluoride (PMSF), 1 μg poly(dI-dC), 5 × 105 cpm of labeled probe, and 5 μl of each desired protein lysate. Competition was performed by adding to the reactions an 500-fold excess amount of unlabelled oligonucleotides. Free and bound probes were resolved in a 5% nondenaturing polyacrylamide gel. The specific oligonucleotide used for probe and competition contained the consensus Pou4f binding site: 5′-CACGCATAATTAATCGC-3′ [Gruber et al., 1997; Liu et al., 2000].
Luciferase Assay
pcDNA3 expression plasmids containing the coding sequence of wild type, p.L223P, or p.L289F Pou4f3 were cotransfected with Prox or Prox3 luciferase reporter plasmids [Trieu et al., 1999] into 293T cells using the Lipofectamine reagent following the manufacturer's instructions (Invitrogen). The pRL-TK Renilla luciferase reporter plasmid was cotransfected for internal control of cell transfection efficiency. Firefly and Renilla luciferase activities were measured 48 hr after transfection using luciferase assay kits according to the manufacturer's protocols (Promega). All values of firefly luciferase activities were normalized with those of the corresponding Renilla luciferase activities derived from the control plasmid. Experiments were performed in triplicate and repeated twice with similar results.
Statistical Analysis
Statistical significance was determined by performing a Student's two-tailed t-test using the GraphPad Prism software package (GraphPad Software Inc., San Diego, CA).
Gene Accession Numbers
Human POU4F3 mRNA: GenBank BC104923.1; Mouse Pou4f3 mRNA: RefSeq NM_131278.
RESULTS
Clinical Characterization
Members of a large Dutch family (Family W05−549) suffered from autosomal dominant hearing loss (Fig. 1A). Clinical assessment of all participating family members revealed 32 affected individuals. Audiometric profiles in this family show a large variability in terms of onset age, level of progression, and shape of the audiogram. Figure 1B shows serial audiograms of two of the affected family members (age range 38−53 and 30−45 years, respectively) and illustrates this variability. In Patient 1 (Fig. 1B, left panel), subjective onset age of hearing impairment was reported to be approximately 13 years of age, whereas in Patient 2 (Fig. 1B, right panel), this was reported to be approximately 20 years. Over the displayed course of 15 years, hearing impairment in Patient 1 showed hardly any progression in the mid frequencies and little progression in the lower and higher frequencies. The last visit audiogram at the age of 53 years shows a gently downsloping configuration with a binaural mean pure tone average at 0.5, 1, and 2 kHz (PTA0.5,1,2 kHz) of 63 dB. Patient 22 did show progression of hearing impairment in the displayed period of 15 years with highest progression rates occurring in the mid and high frequencies (3.5−4.5 dB/year). The last visit audiogram shows a residual hearing with binaural mean PTA0.5,1,2 kHz of 110 dB. A more detailed description of the clinical characteristics of the hearing loss in this family is presented in a separate article [Pauw et al., in press].
FIGURE 1.

A: Pedigree of Family W05−549. Two individuals for which audiograms are shown (B) are indicated by 1 and 2, respectively. Two phenocopies that were affected but did not carry the mutation are indicated by PC. B: Serial audiograms of two of the affected members from Family W05−549 (left panel 1, right panel 2). Shown are binaural mean thresholds in decibels (dB) for each frequency in kilohertz (kHz).The legend on the right shows the age in years (y) at which the audiometric tests were performed.
Linkage Analysis
Since the clinical evaluation revealed no obvious indication for the genetic cause of the hearing impairment in Family W05−549, microsatellite markers flanking known DFNA loci were analyzed in part of the family (see Supplementary Fig. S1; available online at http://www.interscience.wiley.com/jpages/1059−7794/suppmat). For the markers D5S436 and D5S2090, which flank the DFNA15 locus, linkage was found with a maximum two-point LOD score of 5.1 for marker D5S436 (Supplementary Fig. S1).
Mutation Analysis of the POU4F3 Gene
Thus far, DFNA15 has been described to be causative for the hearing impairment in only one family (originally designated Family H), with an 8-bp deletion in the POU4F3 gene as the causative mutation [Vahava et al., 1998]. To determine whether mutations in POU4F3 are responsible for the hearing loss in Family W05−549, all exons and intron–exon boundaries of this gene were sequenced for one affected individual. In exon 2, a missense mutation (c.865C>T) was identified, predicted to result in the substitution of a phenylalanine for a leucine (p.L289F) (Supplementary Fig. S2A). This mutation disrupts a XhoI restriction site, enabling XhoI restriction analysis to show that the mutation segregates with the hearing impairment in this family (data not shown), except for two affected individuals that did not carry the mutation. Therefore, these individuals are considered to be phenocopies. The mutation was not present in 100 ethnically-matched control individuals. To determine whether POU4F3 mutations are a more common cause of hearing impairment in the Netherlands, the gene was sequenced in 30 additional unrelated patients with various types of hearing loss. Interestingly, in one patient, another novel missense mutation was identified (c.668T>C; Supplementary Fig. S2B) predicted to result in the substitution of a proline for a leucine (p.L223P). The mutation was not present in 100 ethnically-matched controls. The patient with the mutation suffers from moderate to severe hearing loss at the age of 40 years, affecting mainly high frequencies. The occurrence of hearing loss in her family is currently under investigation.
Molecular Modeling of the Wild-Type and Mutant POU4F3 Proteins
The human POU4F3 gene encodes a protein of 338 amino acids that functions as a transcription factor [Gerrero et al., 1993; Ninkina et al., 1993]. The POU4F3 protein is comprised of two highly conserved DNA-binding domains, the POU-specific domain with four α-helices (amino acids [aa] 179−256), and the POU homeodomain (aa 274−333) with three α-helices. These two domains are separated by a linker sequence. Within the POU homeodomain, two nuclear localization signals (NLS) are predicted to be present, one monopartite (aa 274−278) and one bipartite (aa 314−331) NLS [Weiss et al., 2003]. A schematic overview of the primary structure of the POU4F3 protein is presented in Figure 2A.
FIGURE 2.
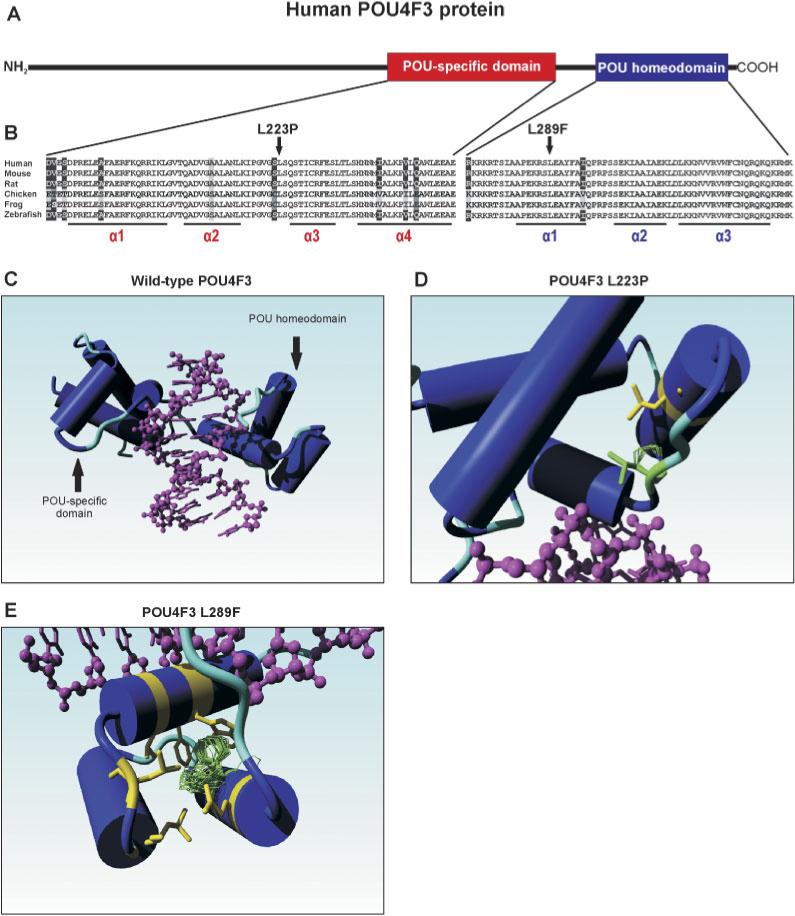
A: Schematic representation of the human POU4F3 protein.The POU-specific and the POU homeodomain are depicted in red and blue, respectively. B: Sequence comparison of the POU-specific and the POU homeodomain of several vertebrate POU4F3 proteins. Identical residues in all sequences are black on a white background, whereas amino acids that are identical in at least four species are gray on a black background. Conserved changes are black on a light gray background, whereas similar amino acids are depicted in white on a black background. The two leucine residues (L223 and L289) that were found to be substituted by a proline and phenylalanine in the two families with hearing impairment are indicated by an arrow. The positions of the various α-helices in the two DNA-binding domains are underlined. Accession numbers of the POU4F3 protein sequences: human: Q15319; mouse Q63955; rat XP 344676; chicken NP 990090; frog AAG17008; and zebrafish NP571353. C: Molecular model of the human POU4F3 protein.The part of the wild-type POU4F3 protein containing the DNA-binding domains is depicted, with the various α-helices represented as cylinders. The target DNA is indicated in purple. D: Graphic representation of the predicted effect of the p.L223P mutation. The leucine residue at position 223 is replaced by a proline (depicted in green), resulting in a clash with leucine residue 215 (in yellow) that is present in the second α-helix of the POU-specific domain. E: Graphic representation of the predicted effect of the p.L289F mutation. The leucine residue 289 is replaced by a phenylalanine. Two possible rotamer orientations of the aromatic side chain are depicted in green. In one case (upper orientation), the phenylalanine side chain clashes with the aromatic ring of a highly conserved tryptophan residue (W321) in the third α-helix of the POU homeodomain, whereas the other (less favored) orientation clashes mainly with the side-chains of two adjacent leucine residues. All residues that may somehow clash with the phenylalanine at position 289 are depicted in yellow. Images as presented in panels C–E were made using the YASARA NOVA program [Krieger et al., 2002]. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
The missense mutation found in the single patient (p.L223P) replaces the leucine residue located just before the third α-helix within the POU-specific domain of POU4F3, and is conserved in all vertebrate POU4F3 proteins (Fig. 2B, left panel). The p.L289F mutation found in Family W05−549 substitutes a phenylalanine for a leucine residue in the middle of the first α-helix of the POU homeodomain. Also, this leucine residue is conserved in all vertebrate POU4F3 proteins (Fig. 2B, right panel), and even in all other human members of the POU family of transcription factors (data not shown).
To address the effect of the two missense mutations on the tertiary structure of the POU4F3 protein, a molecular model of the POU4F3 protein was built based on the known crystal structure of the POU2F1/Oct-1 protein [Klemm et al., 1994]. An overview of the protein part that contains both the POU-specific and the POU homeodomain, and their orientation toward the target DNA (indicated in purple) is presented in Figure 2C. The leucine residue at position 223 (L223) is part of a small hydrophobic domain. The substitution of this leucine by a proline results in an atomic clash between this proline residue and the leucine residue at position 215 (L215, depicted in yellow) in the POU-specific domain of the protein (Fig. 2D). As a result, a local structural change will occur. Since the residues around L223 are involved in DNA contacts, this mutation will probably reduce the strength of the interaction with the target DNA. In addition, L223 is involved in an intricate net of (energetically favorable) hydrophobic interactions (not shown in Fig. 2D). A proline residue is smaller than a leucine residue, so a proline at position 223 will not completely participate in this hydrophobic cluster. As a result, this whole domain will become less stable, probably leading to a faster turnover, slower folding, misfolding, or structural deformations that will result in reduced DNA binding.
Leucine residue L289 is one of the important core residues that holds the homeodomain together. Mutation of this residue as found in Family W05−549 is likely to be devastating for the ability of this domain to properly fold. De Filippis et al. [1994] have reported how a rotamer search can be used to predict where side-chains of mutated residues could potentially be located. The rotamer cloud in Figure 2E shows two clouds of preferred positions for the phenylalanine side chain. The most preferred conformation of the phenylalanine clashes with a very important and highly conserved tryptophan residue in the third helix of the homeodomain (W321). The other, less populated and thus less likely, rotamer distribution clashes less severely, but still is too close to a series of residues (mainly two leucines near the C-terminal end of helix 2 of this domain, L311 and L313). Both favored rotamers of the phenylalanine are thus predicted to severely affect the tertiary structure of the homeodomain and thus will probably also interfere with binding of POU4F3 to its target DNA.
Transient Transfection of Wild-Type and Mutant POU4F3 Constructs
To determine the effect of the two missense mutations on the subcellular localization of POU4F3, COS-1 cells were transiently transfected with constructs encoding either the wild-type or the mutant protein fused to an HA-tag C-terminally. Immunofluorescence analysis revealed that, whereas the wild-type POU4F3 is located almost exclusively in the cell nuclei, the two mutant POU4F3 proteins were present both in the cytoplasm and in the nucleus, in part of the transfected cells (Fig. 3A). For both mutants, the number of cells expressing POU4F3 in the cytoplasm was higher compared to that for cells expressing the wild-type protein (Fig. 3B). Western blot analysis of lysates of transfected cells using an anti-HA antibody showed that the wild-type and mutant POU4F3 proteins were expressed as a single protein of the correct molecular weight (Fig. 3C), indicating that the staining observed in the immunocytochemical analysis is specifically derived from the tagged POU4F3 proteins. Based on the intensity of the POU4F3 bands, there was no indication that the stability of the mutant POU4F3 differs from that of the wild-type protein.
FIGURE 3.
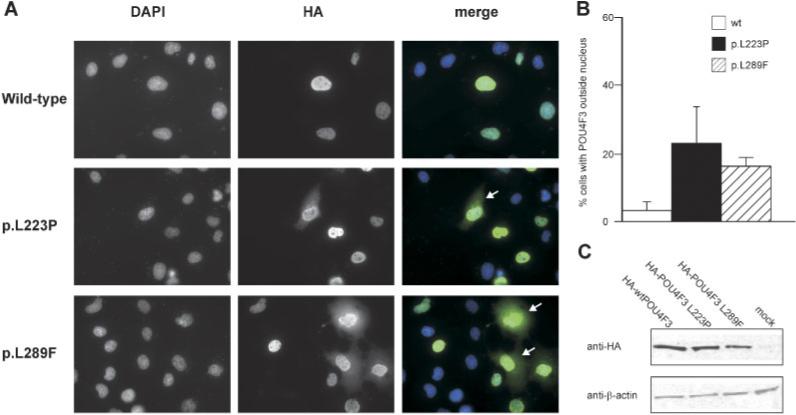
A: Immunofluorescence analysis of transiently transfected HA-tagged wild-type and mutant POU4F3 proteins. Representative examples are shown, with for the two mutant POU4F3 proteins a number of cells expressing POU4F3 outside the cell nuclei indicated by a white arrow. Images are presented as DAPI (staining cell nuclei), anti-HA (detecting the transfected HA-tagged POU4F3 proteins), and merged pictures (DAPI in blue, HA-tagged POU4F3 in green). B: Quantification of the number of transfected cells expressing HA-tagged POU4F3 outside the nucleus. Slides were blinded and for each transfected construct in duplicate, 100 cells were evaluated. C: Western blot analysis of lysates of transfected cells, stained with an anti-HA antibody. Beta-actin levels are used for comparison. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
POU4F3 Missense Mutations Disrupt DNA Binding and Transcriptional Activities
The molecular modeling suggested that both p.L223P and p.L289F mutations alter the 3D structure of the POU4F3 DNA binding domain (Fig. 2) and hence disturb its DNA-binding affinity. To further address this, the missense mutations were introduced in the coding sequence of the mouse Pou4f3. Subsequently, the ability of the two mutant Pou4f3 proteins to bind to a Pou4f consensus binding site (ATAATTAAT; [Gruber et al., 1997]) were investigated by an EMSA. As shown in Figure 4A, in vitro translated wild-type Pou4f3 strongly binds to the consensus site to form a specific protein-DNA complex that could be abrogated by excess amount of unlabelled consensus sites. In contrast, hardly any binding of both the Pou4f3 p.L223P and p.L289F mutant proteins to the consensus site was detected, demonstrating a severe effect of these two mutations on DNA binding activity. In addition, the mutants neither appeared to significantly compete nor otherwise interfere with DNA binding of the wild-type protein.
FIGURE 4.
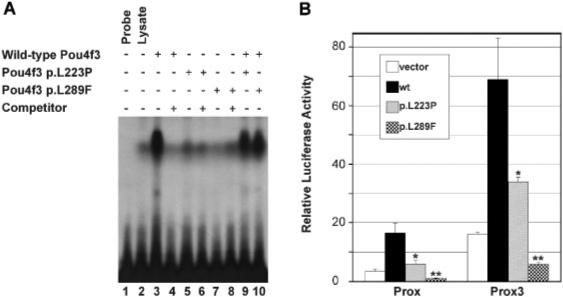
A: EMSA using a labeled Pou4f3 binding site as probe. Reactions contained untreated lysate (lane 2), in vitro translated wild-type Pou4f3 (lanes 3, 4, 9, and 10), and/or translated mutant L223P (lanes 5, 6, and 9) or L289F (lanes 7, 8, and 10) in the presence (lanes 4, 6, and 8) or absence of a 500-fold excess amount of cold specific competitor oligonucleotides. Wild-type Pou4f3 formed a strong protein-DNA complex that could be abolished by excess amounts of cold competitor, whereas p.L223P and p.L289F exhibited no detectable DNA binding activity and did not appear to significantly interfere with DNA binding of the wild-type protein. B: Relative luciferase activities following cotransfection of wild-type and mutant Pou4f3 expression constructs with the Prox or Prox3 luciferase reporter plasmids. Cotransfection was conducted in 293T cells and luciferase activities were measured 48 hr after transfection. Transfection efficiency was controlled by measuring Renilla luciferase activities following cotransfection with the pRL-TK Renilla luciferase reporter plasmid. Results represent means ±SD of triplicate assays in a single experiment. Statistical significance: *P<0.05; **P<0.001.
The disability of the Pou4f3 p.L223P and p.L289F mutants to bind their target DNA will probably result in a failure to activate the target gene transcription. To confirm this, the transcription activation properties of the Pou4f3 proteins were determined in 293T cells using the previously described Prox and Prox3 luciferase reporter constructs [Trieu et al., 1999]. Prox contains the proximal Pou4f1 autoregulatory enhancer that can be activated by Pou4f factors and Prox3 harbors three copies of a Pou4f recognition site identified in the Pou4f1 enhancer [Trieu et al., 1999]. In transient transfection assays, transfection of wild-type Pou4f3 resulted in a five-fold increase in luciferase activity from both the Prox and Prox3 promoters. In contrast, p.L223P activated transcription from these promoters by only about two-fold and p.L289F did not exhibit any transactivation activity at all from these promoters (Fig. 4B). In conclusion, the DFNA15-causing mutations severely affect the ability of the transcription factor to bind DNA, consistent with their deficiency to activate reporter gene expression. Again, these deleterious effects seem to be more pronounced for the p.L289F mutation in the homeobox domain than for the p.L223P mutation within the POU-specific domain of POU4F3.
DISCUSSION
In the present study, we describe the identification of two novel missense mutations in the POU4F3 gene underlying DFNA15. Thus far, only one mutation in this gene had been described, an 8-bp deletion resulting in a frameshift and a truncated protein [Vahava et al., 1998]. The first missense mutation (c.865C>T; p.L289F) segregates with hearing impairment in a large pedigree that clinically presented with a high phenotypic variation, both with respect to the affected frequencies as well as the degree of progression. Subsequent sequence analysis of the POU4F3 gene in 30 additional patients revealed a second novel missense mutation (c.668T>C; p.L223P) in one patient. The clinical variability found in Family W05−549 is in accordance with that observed in the original DFNA15 family H, with regard to the configuration of the audiogram, the age of onset, and the severity and progression of the hearing loss [Avraham, 2000; Frydman et al., 2000; Gottfried et al., 2002]. Although mutations in POU4F3 that cause hearing impairment had thus far been described in only one single family, our results indicate that mutations in this gene do not seem to be a very rare cause of ADNSHI in the Dutch population.
POU4F3 is part of the POU superfamily of transcription factors that in humans consists of 14 family members, all characterized by the presence of two DNA-binding domains, a POU-specific domain and a POU homeodomain [Wegner et al., 1993]. Together, these domains specifically bind an octamer of target DNA with a consensus sequence [Gruber et al., 1997]. The various POU proteins are involved in tissue-specific gene regulation. More specifically, the human class IV POU domain transcription factors POU4F1, POU4F2, and POU4F3 are essential for cell maturation and survival in specific cell-types: POU4F1 in motor and sensory neural cells, POU4F2 in retinal ganglion cells, and POU4F3 in cochlear and vestibular hair cells [Erkman et al., 1996; Xiang et al., 1997a, 1997b; McEvilly et al., 1996]. In mice, targeted disruption of the Pou4f3/Brn-3c gene results in defective inner ear function as a result of complete absence of sensory hair cells. Transient transfection studies have shown that Brn-3c is capable of activating the BDNF and NT-3 promoters in inner ear derived sensory epithelial cell lines [Clough et al., 2004]. Furthermore, expression profiling of inner ears derived from wild-type and Brn-3c−/− mice revealed the zinc-finger transcription factor growth factor independence (Gfi1) as an in vivo target gene that is transcriptionally activated by POU4F3. With respect to hearing deficits, Gfi1−/− mice show a phenotype comparable to that of Brn-3c−/− mice [Hertzano et al., 2004], indicating that Gfi1 is a downstream target of POU4F3 in the inner ear. In addition, the LIM domain transcription factor gene Lhx3 was recently identified as a POU4F3 target that is downregulated in the auditory but not in the vestibular hair cells of Brn-3c−/− mice [Hertzano et al., 2007]. Together, it is hypothesized that reduced expression levels of for instance the aforementioned genes contribute to the molecular mechanisms underlying the hearing loss in Brn-3c−/− mice and thus possibly in humans, although additional yet unidentified in vivo targets may contribute to these processes as well.
The 8-bp deletion that was detected in Family H is predicted to result in a frameshift and premature termination of the POU4F3 protein [Vahava et al., 1998]. This mutant protein lacks part of the POU homeodomain, including one of the two NLSs [Weiss et al., 2003]. Molecular modeling of two missense mutations described in this study (p.L223P and p.L289F) predicted these mutations to affect the tertiary structure of the DNA-binding domains, and thus to decrease binding to target DNA. This was confirmed by electromobility shift assays, in which proteins with either of the substitutions showed reduced binding to Pou4f binding consensus sequences. Both mutant proteins did not interfere with binding of the wild-type Pou4f3 to the consensus DNA, indicating that the mutants do not exert a dominant-negative effect on the wild-type protein. In addition, the mutant proteins were less capable of activating reporter genes than the wild-type protein. Finally, both mutants partially mislocalized outside the nucleus of transiently transfected COS-1 cells. A similar set of experiments has previously been performed for a mutant POU4F3 protein resulting from the 8-bp deletion that was identified in the first DFNA15-family. Like the two missense mutations described in this study, that mutant also showed a decreased binding to consensus DNA, reduced capability to activate reporter gene expression, and partial mislocalization in the cytoplasm [Weiss et al., 2003]. Together, these functional studies strongly indicate that both missense mutations as well as the frameshift mutation affect proper functioning of the POU4F3 protein. Based on these results, haploinsufficiency seems to be the most-likely mechanism underlying the hearing impairment in DFNA15-patients, although heterozygous Brn-3c+/− mice show no obvious hearing problems. However, it cannot be excluded that the pathological mechanism underlying the hearing loss is a gain-of-function, in which the mutant POU4F3 proteins for instance acquire an increased affinity for other target genes. Due to the high degree of variation regarding age of onset and progression of the hearing impairment even within a family, it is hard to draw a clear genotype–phenotype correlation. In addition, there is only one patient carrying the p.L223P mutation.
Besides hearing deficits, Brn-3c−/− mice also suffer from vestibular defects [Xiang et al., 1997a]. In contrast, in the affected individuals from the Jewish Family H [Vahava et al., 1998], no obvious vestibular defects were observed.. In Family W05−549, an extensive analysis of vestibular function was carried out recently, showing reduced vestibular function in some of the family members that also had an impaired hearing ability. Apparently, there can be compensatory factors, either genetic or environmental, involved in the process of maintaining balance in humans. A detailed description of the vestibular function in Family W05−549 will be presented in a separate work (van Drunen FJW, Pauw RJ, Collin RWJ, Kremer H, Huygen PLM, Cremers CWRJ, unpublished results).
In conclusion, we describe the identification of two novel missense mutations in the POU4F3 gene, in two Dutch families suffering from nonsyndromic autosomal dominant hearing impairment DFNA15. These mutations are predicted to affect the tertiary structure of the DNA-binding domains of this transcription factor, thereby altering the affinity for its target DNA. This is further supported by a reduced capability to transcriptionally activate reporter constructs of both mutant proteins compared to the wild-type POU4F3. While mutations in POU4F3 that cause hearing loss had thus far been described in only one family, our results indicate that POU4F3 mutations are not rare in the Dutch hearing-impaired population. Since the POU4F3 gene is comprised of only two exons, it may therefore be a suitable candidate for implementation in routine diagnostic testing.
ACKNOWLEDGMENTS
We acknowledge all the individuals who participated in this study; we thank S. van der Velde-Visser and C. Beumer for technical assistance, and Dr. E. Turner for luciferase constructs. This study was financially supported by the National Institutes of Health (EY12020 and EY015777 to M.X.).
Grant sponsor: Heinsius Houbolt Foundation; Grant sponsor: European Commission FP6 Integrated Project EUROHEAR; Grant number: LSHG-CT-20054 -512063; Grant sponsor: National Institutes of Health; Grant numbers: EY12020; and EY015777.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
The Supplementary Material referred to in this article can be accessed at http://www.interscience.wiley.com/jpages/1059-7794/suppmat.
Supplementary Material
REFERENCES
- Alloisio N, Morle L, Bozon M, Godet J, Verhoeven K, Van Camp G, Plauchu H, Muller P, Collet L, Lina-Granade G. Mutation in the zonadhesin-like domain of alpha-tectorin associated with autosomal dominant non-syndromic hearing loss. Eur J Hum Genet. 1999;7:255–258. doi: 10.1038/sj.ejhg.5200273. [DOI] [PubMed] [Google Scholar]
- Avraham KB. DFNA15. Adv Otorhinolaryngol. 2000;56:107–115. doi: 10.1159/000059091. [DOI] [PubMed] [Google Scholar]
- Balciuniene J, Dahl N, Jalonen P, Verhoeven K, Van Camp G, Borg E, Pettersson U, Jazin EE. Alpha-tectorin involvement in hearing disabilities: one gene—two phenotypes. Hum Genet. 1999;105:211–216. doi: 10.1007/s004390051091. [DOI] [PubMed] [Google Scholar]
- Chinea G, Padron G, Hooft RW, Sander C, Vriend G. The use of position-specific rotamers in model building by homology. Proteins. 1995;23:415–421. doi: 10.1002/prot.340230315. [DOI] [PubMed] [Google Scholar]
- Clough RL, Sud R, Davis-Silberman N, Hertzano R, Avraham KB, Holley M, Dawson SJ. Brn-3c (POU4F3) regulates BDNF and NT-3 promoter activity. Biochem Biophys Res Commun. 2004;324:372–381. doi: 10.1016/j.bbrc.2004.09.074. [DOI] [PubMed] [Google Scholar]
- Collin RW, Pauw RJ, Schoots J, Huygen PL, Hoefsloot LH, Cremers CW, Kremer H. Identification of a novel COCH mutation, G87W, causing autosomal dominant hearing impairment (DFNA9). Am J Med Genet A. 2006;140:1791–1794. doi: 10.1002/ajmg.a.31354. [DOI] [PubMed] [Google Scholar]
- De Filippis V, Sander C, Vriend G. Predicting local structural changes that result from point mutations. Protein Eng. 1994;7:1203–1208. doi: 10.1093/protein/7.10.1203. [DOI] [PubMed] [Google Scholar]
- De Kok YJ, Bom SJ, Brunt TM, Kemperman MH, van Beusekom E, van der Velde-Visser SD, Robertson NG, Morton CC, Huygen PL, Verhagen WI, Brunner HG, Cremers CW, Cremers FP. A Pro51Ser mutation in the COCH gene is associated with late onset autosomal dominant progressive sensorineural hearing loss with vestibular defects. Hum Mol Genet. 1999;8:361–366. doi: 10.1093/hmg/8.2.361. [DOI] [PubMed] [Google Scholar]
- Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O'Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature. 1996;381:603–606. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- Fransen E, Verstreken M, Verhagen WI, Wuyts FL, Huygen PL, D'Haese P, Robertson NG, Morton CC, McGuirt WT, Smith RJ, Declau F, Van de Heyning PH, Van Camp G. High prevalence of symptoms of Meniere's disease in three families with a mutation in the COCH gene. Hum Mol Genet. 1999;8:1425–1429. doi: 10.1093/hmg/8.8.1425. [DOI] [PubMed] [Google Scholar]
- Frydman M, Vreugde S, Nageris BI, Weiss S, Vahava O, Avraham KB. Clinical characterization of genetic hearing loss caused by a mutation in the POU4F3 transcription factor. Arch Otolaryngol Head Neck Surg. 2000;126:633–637. doi: 10.1001/archotol.126.5.633. [DOI] [PubMed] [Google Scholar]
- Gerrero MR, McEvilly RJ, Turner E, Lin CR, O'Connell S, Jenne KJ, Hobbs MV, Rosenfeld MG. Brn-3.0: a POU-domain protein expressed in the sensory, immune, and endocrine systems that functions on elements distinct from known octamer motifs. Proc Natl Acad Sci USA. 1993;90:10841–10845. doi: 10.1073/pnas.90.22.10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried I, Huygen PL, Avraham KB. The clinical presentation of DFNA15/POU4F3. Adv Otorhinolaryngol. 2002;61:92–97. doi: 10.1159/000066819. [DOI] [PubMed] [Google Scholar]
- Gruber CA, Rhee JM, Gleiberman A, Turner EE. POU domain factors of the Brn-3 class recognize functional DNA elements which are distinctive, symmetrical, and highly conserved in evolution. Mol Cell Biol. 1997;17:2391–2400. doi: 10.1128/mcb.17.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzano R, Montcouquiol M, Rashi-Elkeles S, Elkon R, Yucel R, Frankel WN, Rechavi G, Moroy T, Friedman TB, Kelley MW, Avraham KB. Transcription profiling of inner ears from Pou4f3(ddl/ddl) identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum Mol Genet. 2004;13:2143–2153. doi: 10.1093/hmg/ddh218. [DOI] [PubMed] [Google Scholar]
- Hertzano R, Dror AA, Montcouquiol M, Ahmed ZM, Ellsworth B, Camper S, Friedman TB, Kelley MW, Avraham KB. Lhx3, a LIM domain transcription factor, is regulated by Pou4f3 in the auditory but not in the vestibular system. Eur J Neurosci. 2007;25:999–1005. doi: 10.1111/j.1460-9568.2007.05332.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann K, Lindner TH. easyLINKAGE-Plus—automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- Huygen PL, Pauw RJ, Cremers CW. Audiometric profiles associated with genetic non-syndromal hearing impairment: a review and phenotype analysis. In: Martini Alessandro., editor. Genes, Hearing and Deafness. Informa Healthcare; London: 2007. pp. 185–204. [Google Scholar]
- Kantardzhieva A, Gosens I, Alexeeva S, Punte IM, Versteeg I, Krieger E, Neefjes-Mol CA, den Hollander AI, Letteboer SJ, Klooster J, Cremers FP, Roepman R, Wijnholds J. MPP5 recruits MPP4 to the CRB1 complex in photoreceptors. Invest Ophthalmol Vis Sci. 2005;46:2192–2201. doi: 10.1167/iovs.04-1417. [DOI] [PubMed] [Google Scholar]
- Klemm JD, Rould MA, Aurora R, Herr W, Pabo CO. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell. 1994;77:21–32. doi: 10.1016/0092-8674(94)90231-3. [DOI] [PubMed] [Google Scholar]
- Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- Liu W, Khare SL, Liang X, Peters MA, Liu X, Cepko CL, Xiang M. All Brn3 genes can promote retinal ganglion cell differentiation in the chick. Development. 2000;127:3237–3247. doi: 10.1242/dev.127.15.3237. [DOI] [PubMed] [Google Scholar]
- Manolis EN, Yandavi N, Nadol JB, Jr, Eavey RD, McKenna M, Rosenbaum S, Khetarpal U, Halpin C, Merchant SN, Duyk GM, MacRae C, Seidman CE, Seidman JG. A gene for non-syndromic autosomal dominant progressive postlingual sensorineural hearing loss maps to chromosome 14q12−13. Hum Mol Genet. 1996;5:1047–1050. doi: 10.1093/hmg/5.7.1047. [DOI] [PubMed] [Google Scholar]
- McEvilly RJ, Erkman L, Luo L, Sawchenko PE, Ryan AF, Rosenfeld MG. Requirement for Brn-3.0 in differentiation and survival of sensory and motor neurons. Nature. 1996;384:574–577. doi: 10.1038/384574a0. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Pelayo MA, del Castillo I, Villamar M, Romero L, Hernandez-Calvin FJ, Herraiz C, Barbera R, Navas C, Moreno F. A cysteine substitution in the zona pellucida domain of alpha-tectorin results in autosomal dominant, postlingual, progressive, mid frequency hearing loss in a Spanish family. J Med Genet. 2001;38:E13. doi: 10.1136/jmg.38.5.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkina NN, Stevens GE, Wood JN, Richardson WD. A novel Brn3-like POU transcription factor expressed in subsets of rat sensory and spinal cord neurons. Nucleic Acids Res. 1993;21:3175–3182. doi: 10.1093/nar/21.14.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauw RJ, van Drunen FJW, Collin RWJ, Huygen PLM, Kremer H, Cremers CWRJ. Audiometric characterization of a Dutch DFNA15 family with a novel mutation in POU4F3, p.L289F. Arch Otorhinolaryngol Head Neck Surg. (in press) [Google Scholar]
- Petit C. From deafness genes to hearing mechanisms: harmony and counterpoint. Trends Mol Med. 2006;12:57–64. doi: 10.1016/j.molmed.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Plantinga RF, de Brouwer AP, Huygen PL, Kunst HP, Kremer H, Cremers CW. A novel TECTA mutation in a Dutch DFNA8/12 family confirms genotype-phenotype correlation. J Assoc Res Otolaryngol. 2006;7:173–181. doi: 10.1007/s10162-006-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson NG, Skvorak AB, Yin Y, Weremowicz S, Johnson KR, Kovatch KA, Battey JF, Bieber FR, Morton CC. Mapping and characterization of a novel cochlear gene in human and in mouse: a positional candidate gene for a deafness disorder, DFNA9. Genomics. 1997;46:345–354. doi: 10.1006/geno.1997.5067. [DOI] [PubMed] [Google Scholar]
- Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, Nadol JB, Jr, Miyamoto RT, Linthicum FH, Jr, Lubianca Neto JF, Hudspeth AJ, Seidman CE, Morton CC, Seidman JG. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet. 1998;20:299–303. doi: 10.1038/3118. [DOI] [PubMed] [Google Scholar]
- Street VA, Kallman JC, Robertson NG, Kuo SF, Morton CC, Phillips JO. A novel DFNA9 mutation in the vWFA2 domain of COCH alters a conserved cysteine residue and intrachain disulfide bond formation resulting in progressive hearing loss and site-specific vestibular and central oculomotor dysfunction. Am J Med Genet A. 2005;139:86–95. doi: 10.1002/ajmg.a.30980. [DOI] [PubMed] [Google Scholar]
- Trieu M, Rhee JM, Fedtsova N, Turner EE. Autoregulatory sequences are revealed by complex stability screening of the mouse brn-3.0 locus. J Neurosci. 1999;19:6549–6558. doi: 10.1523/JNEUROSCI.19-15-06549.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, Morrow JE, Lee MK, Skvorak AB, Morton CC, Blumenfeld A, Frydman M, Friedman TB, King MC, Avraham KB. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science. 1998;279:1950–1954. doi: 10.1126/science.279.5358.1950. [DOI] [PubMed] [Google Scholar]
- Verhoeven K, Van Laer L, Kirschhofer K, Legan PK, Hughes DC, Schatteman I, Verstreken M, Van Hauwe P, Coucke P, Chen A, Smith RJ, Somers T, Offeciers FE, Van de Heyning P, Richardson GP, Wachtler F, Kimberling WJ, Willems PJ, Govaerts PJ, Van Camp G. Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat Genet. 1998;19:60–62. doi: 10.1038/ng0598-60. [DOI] [PubMed] [Google Scholar]
- Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–6,29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- Wegner M, Drolet DW, Rosenfeld MG. POU-domain proteins: structure and function of developmental regulators. Curr Opin Cell Biol. 1993;5:488–498. doi: 10.1016/0955-0674(93)90015-i. [DOI] [PubMed] [Google Scholar]
- Weiss S, Gottfried I, Mayrose I, Khare SL, Xiang M, Dawson SJ, Avraham KB. The DFNA15 deafness mutation affects POU4F3 protein stability, localization, and transcriptional activity. Mol Cell Biol. 2003;23:7957–7964. doi: 10.1128/MCB.23.22.7957-7964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Gan L, Li D, Chen ZY, Zhou L, O'Malley BW, Jr, Klein W, Nathans J. Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc Natl Acad Sci USA. 1997a;94:9445–9450. doi: 10.1073/pnas.94.17.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Gan L, Li D, Zhou L, Chen ZY, Wagner D, O'Malley BW, Jr, Klein W, Nathans J. Role of the Brn-3 family of POU-domain genes in the development of the auditory/vestibular, somatosensory, and visual systems. Cold Spring Harb Symp Quant Biol. 1997b;62:325–336. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.