

Abstract
Proteolytic cleavage of component C3 to C3b is a central step in the activation of complement. Whereas C3 is largely biologically inactive, C3b is directly involved in various complement activities. While the recently described crystal structures of C3 and C3b provide a molecular basis of complement activation, they do not reflect the dynamic changes that occur in solution. In addition, the available C3b structures diverge in some important aspects. Here we have utilized hydrogen/deuterium exchange coupled with mass spectrometry (HDX-MS) to investigate relative changes in the solution-phase structures of C3 and C3b. By combining two forms of mass spectrometry we could maximize the primary sequence coverage of C3b and demonstrate the feasibility of this method for large plasma proteins. While the majority of the 82 peptides that could be followed over time showed only minor alterations in HDX, we observed clear changes in solvent accessibility for 16 peptides, primarily in the α-chain (α’NT, MG6-8, CUB, TED, C345C domains). Most of these peptides could be directly linked to the structural transitions visible in the crystal structures and revealed additional information about the probability of the structural variants of C3b. In addition, a discontinuous cluster of seven peptides in the MG3, MG6, LNK and α’NT domains showed a decreased accessibility after activation to C3b. Although no gross conformational changes are detected in the crystal structure, this area may reflect a structurally flexible region in solution that contributes to C3 activation and function.
Keywords: Complement, Activation, C3, C3b, Hydrogen-deuterium exchange, Mass Spectrometry
1. Introduction
As a central component of innate immunity, the human complement system plays a major role in the recognition and elimination of microbial intruders and other pathogenic cells. Recent research revealed even more important functions for this tuned cascade of soluble and membrane proteins, such as bridging to adaptive immune responses and additional cascades (e.g. coagulation system). Activation of complement component C3 is the point of convergence in the initiation of the complement cascade by the lectin, alternative and classical pathways. This proteolytic conversion leads to the production of the biologically active effector protein C3b and the anaphylatoxin C3a from the biologically inactive protein C3. Covalent attachment of C3b on the surface of foreign cells (i.e. opsonization) induces a variety of terminal complement actions from cell lysis and phagocytosis to the stimulation of downstream immune responses (Markiewski and Lambris, 2007). Since complement activation on host cells has devastating effects and may lead to a number of severe diseases, the cascade activity has to be carefully controlled. The ability to enable and restrict molecular interaction within a single protein template is a central aspect of the control strategy.
The biological activities of C3b are directly related to the dynamic exposure of binding sites that are necessary for protein-protein interactions (Gros et al., 2007). While native C3 has a very limited amount of physiological binding partners, C3b gains the ability to bind a variety of essential proteins, including C5; properdin; factors B, H and I; membrane co-factor protein; decay accelerating factor; and complement receptor 1 (Sahu and Lambris, 2001). It is through these interactions that C3b and its breakdown fragments are capable of propagating the innate immune response and influencing adaptive immunity. It is not surprising, therefore, that increased activation of C3 or impaired regulation of C3b have been attributed to an increasing number of diseases (Ricklin and Lambris, 2007; Thurman, 2006; Volanakis and Frank, 1998), rendering these proteins as potential targets of therapeutic intervention (Ricklin and Lambris, 2007). In addition, the C3-to-C3b transition is also critical for complement evasion of human pathogens (Lambris et al., 2007). For example, the extracellular fibrinogen-binding protein from Staphylococcus aureus has been shown to preferentially bind the native over the activated form of C3, which may be pivotal for its inhibitory activity (Hammel et al., 2007b). Numerous biological and biophysical techniques have been utilized to investigate the structural changes accompanying the increase in function upon activation of C3. Earlier evidence accumulated through neoepitope mapping with antibodies (Alsenz et al., 1990), chemical modification strategies (Isenman et al., 1981), solution scattering (Perkins and Sim, 1986), and electron microscopy (Smith et al., 1984) indicated significant structural rearrangements during this process. These observations have recently been confirmed by the publication of crystal structures for C3 (Janssen et al., 2005) and C3b (Abdul Ajees et al., 2006; Janssen et al., 2006; Wiesmann et al., 2006) as well as detailed electron microscopy studies (Nishida et al., 2006). While these studies offered a first insight into the mechanism by which C3 activation is propagated, they all were taken under static, non-solute conditions. Even more, the available crystal structures for C3b diverge in some important points and are currently matters of scientific debate (Janssen et al., 2007). Given these structural uncertainties, the versatile biological functions of C3b, as well as the dynamic process in which C3 activity is regulated, more detailed information about how this rearrangement takes place in solution is highly sought after.
Hydrogen deuterium exchange (HDX1) coupled with mass spectrometry (MS) has evolved into an indispensable tool for characterizing such dynamic structural changes in solution. HDX takes advantage of the ability of amide backbone hydrogen atoms to exchange with water hydrogens in solution (Busenlehner and Armstrong, 2005; Hoofnagle et al., 2003; Wales and Engen, 2006). When D2O is substituted for H2O in the buffer in which the protein is dissolved, solvent deuterium atoms exchange with backbone hydrogen atoms at a rate influenced by the local structure of the protein. Amide hydrogens that are buried within the protein or are involved in hydrogen bonds exchange more slowly with solvent deuterium atoms than do more accessible hydrogen atoms at the protein surface (Englander and Kallenbach, 1983). By analyzing the rate and/or magnitude of the deuterium incorporation into the protein backbone, one can make inferences about the relative structure of the protein. To monitor the amount of deuterium incorporation, a physical technique such as nuclear magnetic resonance (NMR) or MS is employed (Englander, 2006). The choice of analytical technique depends upon the size of the protein; NMR is best suited for smaller proteins (less than ~30 kDa) while MS allows for the investigation of proteins in excess of 30 kDa. However, low sequence coverage and other experimental parameters render the analysis of larger proteins increasingly difficult for MS as well (Cravello et al., 2003). Characterization of multidomain plasma proteins such as C3 (184 kDa; Fig. 1A) therefore demands an especially high level of instrumental and experimental precision. Although HDX-MS does not provide a three-dimensional structure of the protein, it is capable of reporting structural changes when the protein is studied under varying conditions or in different states (Eyles and Kaltashov, 2004; Hamuro et al., 2003; Schuster et al., 2007). Furthermore, the technique has also been utilized to identify interacting surfaces in protein-protein (Melnyk et al., 2006) and protein-ligand interactions (Hamuro et al., 2006). HDX-MS reflects changes in protein structure that occur in solution but might not be evident from crystallographic structures. As a consequence, the combination of HDX-MS with high-resolution crystal structures can provide an unequaled insight into the solution phase dynamics of proteins.
Fig. 1.
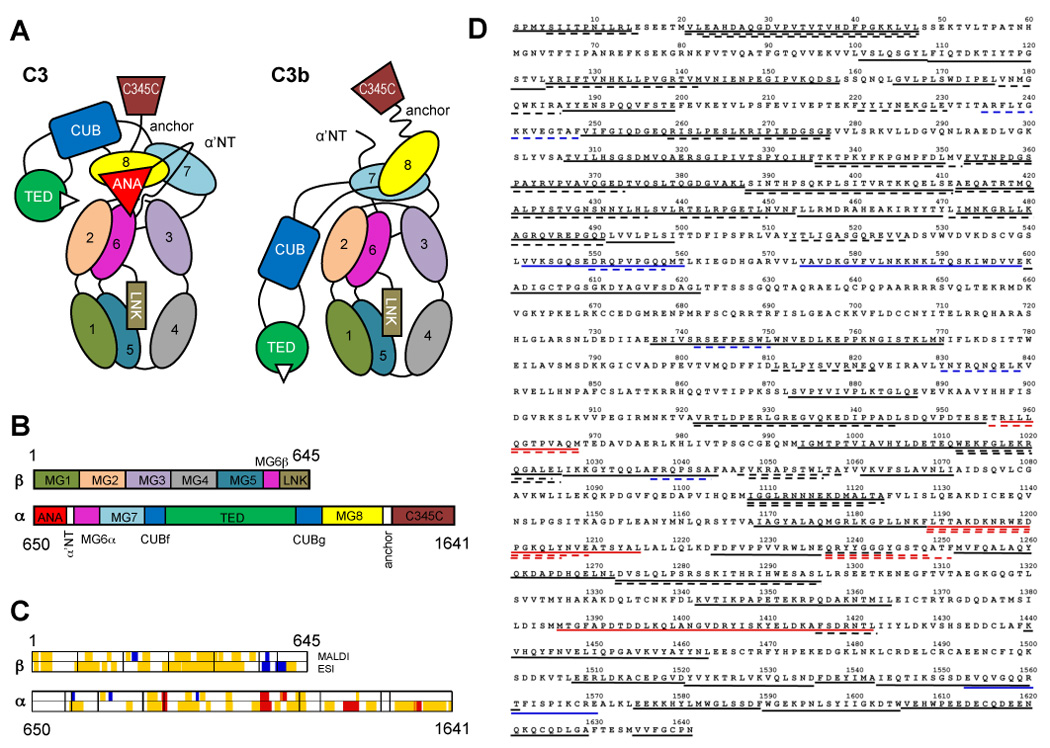
Domain organization of C3/C3b, and sequence coverage by mass spectrometry. (A) Individual domains in C3 and C3b are represented by different colors and labeled with the domain name abbreviation. Numbered domains signify the corresponding macroglobulin domains (MG1–MG8). Panel (B) shows the domain locations on the primary sequence of the C3 α and β-chains. Upon activation, the ANA domain is removed from C3 and allows the α’NT domain to shift from one face of the protein to the other. As a consequence, the MG8, CUB, and TED domains undergo a large relocation in relation to the protein core (MG1-6) and expose the thioester moiety (white triangle) in C3b that is essential for opsonization of foreign surfaces. A set of 82 peptides spanning 61% of the C3 sequence have been utilized for the analysis and are plotted against the domain scheme (C) and the primary sequence of C3 (D). Areas with significantly increased HD exchange in C3b (≥10%; red) were mainly identified in the β-chain, while the α-chain contained more peptides with significant decrease in HD exchange (≤−10%; blue). Yellow and black areas signify peptides with no significant change (>−10% or <10%) in (C) and (D), respectively. Dashed and solid lines represent MALDI- and ESI-derived peptides, respectively, in the sequence plot (D).
In a previous study, we have successfully used HDX-MS to investigate the structural changes in C3 that take place during its hydrolysis to C3(H2O) (Schuster et al., 2007; Winters et al., 2005). Here we have utilized this approach to investigate the relative solution structures of C3 and C3b. Two MS techniques were employed, matrix-assisted laser desorption/ionization time of flight (MALDI) and electrospray ionization with ion trap (ESI), to maximize the sequence coverage for these large proteins. When analyzed in the context of the available crystal structures for C3 and C3b, our data reveal that there is a cluster of four discontinuous peptides within the MG3, MG6, LNK, and α’NT domains that exhibit increased deuterium exchange in C3 when compared to C3b. The same regions only showed minimal differences in the two crystallographic structures, suggesting that this area may exhibit conformational flexibility in solution. These data not only indicate that this region plays an important role in the activation process but also demonstrate the applicability of the technology to large plasma proteins.
2. Materials and Methods
2.1. Protein Preparation
C3 was purified from human plasma (University of Pennsylvania Blood Bank) using established methods (Hammer et al., 1981; Katragadda et al., 2006; Sahu et al., 2000), and was precipitated by dialysis against 5 mM MES buffer (pH 6.0) at 4 °C. C3b was obtained from C3 by limited digestion with trypsin as previously described (Janssen et al., 2006) and was purified by gel filtration over a Superdex 200 size-exclusion column, followed by anion exchange chromatography over a Mono-Q column. Isolated C3b was treated with iodoacetamide to prevent dimerization, purified over a Mono-S column and then concentrated using an Ultrafree-MC centrifugal filter device. Protein purity was carefully controlled by SDS-PAGE to exclude contaminations, and aliquots of the proteins were stored at −70 °C. At the time of use, protein samples was thawed on ice, centrifuged at 3000 rpm in a Sorvall Biofuge Fresco (Thermo Scientific, Waltham, MA) at 4 °C and reconstituted in the appropriate buffer.
2.2. HDX-MS using MALDI
Aliquots of C3 or C3b were deuterated by the addition of 6 µl protein solution (4 mg/ml in PBS; 10 mM sodium phosphate, 150 mM sodium chloride, pH 7.4) to 120 µl D2O (99.9%, Cambridge Isotope Laboratories, Andover, MA). After timed intervals at room temperature (10 s, 30 s, 1 min, 2.5 min, 5 min, 10 min, 15 min), 10-µl aliquots of the protein/D2O solution were removed, added to 10 µL of 0.2% trifluoroacetic acid (TFA) and immediately frozen in liquid nitrogen to quench the amide HDX process (final pH=2.5). To control for HDX that occurred after the quenching process, a reference exchange sample was prepared by mixing 2 µl of protein solution with 40 µl 0.2% TFA; 10 µl of this solution was then mixed with 10 µl D2O and immediately frozen in liquid nitrogen.
Prepared samples were analyzed by MALDI MS on the same day. A single frozen aliquot was thawed at room temperature and the solution was mixed with 20 µl of immobilized pepsin (Pierce, Rockford, IL) at pH 2.6 and 0 °C for 5 min. The pepsin was quickly removed by centrifuging the sample for 30 s at 12,000 × g at 4 °C. Immediately following centrifugation, 1 µl of the peptide mixture was mixed with a 1-µl aliquot of matrix solution (20 mg/ml α-cyano-4-hydroxycinnamic acid in 1:1 acetonitrile: 0.1% TFA). 1 µl of this mixture was quickly spotted onto an ice-chilled MALDI target plate, dried under moderate vacuum for 1 min, and then analyzed on a MALDI time-of-flight mass spectrometer (MALDI micro MX; Waters, Milford, MA) at an acceleration voltage of 20 kV. Typically, 40 single-shot mass spectra were summed to give a composite spectrum. Care was taken to ensure a consistent timing between all steps for each aliquot.
Sequence assignment of the pepsin-generated peptides was performed using an MS/MS-derived C3 peptide map of 354 peptides spanning 80% of the native C3 primary structure. This peptide map had been generated in a previous study (Winters et al., 2005) using an ESI-Ion Trap LCQ-Duo mass spectrometer (Thermo Scientific).
All MALDI data are reprocessed using MassLynx software (Waters) and centroid values of the isotopic envelopes were calculated using MagTran (version 1.02, Dr. Zhongqi Zhang, Amgen Inc. (Zhang and Marshall, 1998)) as described previously (Winters et al., 2005). MALDI-based exchange reactions were performed in triplicate. For a given peptide, the average difference in deuteration was calculated by comparing the percentage of deuterium incorporation (measured deuterium incorporation divided by theoretical maximum deuterium incorporation, multiplied by 100) at each time point from the C3 sample with the corresponding time point in C3b. The mean of the resulting percentages across all time points was used.
2.3. Generation of C3 Peptide Map for ESI Experiments
In order to obtain peptide fragments of suitable size for ESI analysis, digestion conditions were optimized using non-deuterated C3. A pepsin column was prepared by immobilizing porcine pepsin on Poros 20 AL medium (Applied Biosystems, Foster City, CA) at 30 mg/ml according to the manufacturer’s instructions (Hamuro et al., 2003). In general, 20 µl of 30 µM C3 was diluted with 30 µl of cold acidic buffer containing urea and tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Pierce) at varying concentrations. 45 µl of this solution was digested over a refrigerated pepsin column and separated by reversed-phase HPLC prior to analysis by ESI MS. The composition of the acidic buffer solution, the proteolysis, and the HPLC gradient were all optimized for analysis of C3. Several aliquots of non-deuterated C3 were processed using optimized conditions, and the resulting peptides were analyzed by ESI in the data-dependent MS/MS mode with dynamic exclusion. SEQUEST software (Thermo Scientific) was used to identify the sequence-selected parent peptide ions dynamically. The tentatively identified peptides were verified by visual confirmation of the parent ion charge state assigned by SEQUEST. These results provided a map of C3 peptides correlated by m/z, peak retention time, and sequence identity.
2.4. HDX-MS using ESI
Samples containing C3 (15.3 mg/ml in PBS, pH 7.4) or C3b (16.7 mg/mL in PBS, pH 7.4) were analyzed independently under the same conditions. A single time-point deuteration reaction was accomplished by mixing 10 µl of protein solution with 10 µl D2O and incubating the mixture at 4 °C for a predetermined time of 15 s, 50 s, 2.5 min, or 8.5 min, at 4 °C. To quench the exchange reaction, 30 µl of a solution containing 8 M urea and 1 M TCEP at pH 3.0 and 4 °C was added to the mixture. 45 µl of the quenched solution was immediately injected to a pepsin column (104-µl bed volume) in 0.05% TFA (200 µl/min) for 3 min, with subsequent collection of proteolytic products by a trap column (Magic C4, Michrom BioResources, Inc.; 4-µL bed volume). The peptide fragments were eluted from the trap column and separated on a C18 column (Magic C18, Michrom BioResources, Inc.) with a linear gradient of 13–40% solvent B over 23 min (solvent A, 0.05% TFA in water; solvent B, 95% acetonitrile, 5% solvent A). Mass spectrometric analyses were carried out with a Thermo Finnigan LCQ mass spectrometer (Thermo Scientific, Waltham, MA) with capillary temperature at 215 °C. These partially deuterated samples were then subjected to the analysis, along with control samples of non-deuterated (run without deuterated buffers) and fully deuterated C3 (incubated in 50 mM TCEP in 50% D2O for 5 h at 60 °C).
The centroids of probe peptide isotopic envelopes were measured using a program developed in-house in collaboration with Sierra Analytics. The deuterium incorporation was calculated and corrected for back-exchange by using previously described methods (Hamuro et al., 2006; Zhang and Smith, 1993). The percent deuterium incorporation at each time point was compared for the corresponding C3- and C3b-derived peptides in order to calculate the average difference in deuteration across all time points (Table 1 and Table 2).
Table 1.
Differential deuteration in the β-chain of C3- and C3b-derived peptides.
| C3 Sequence Number | Chain | Domain | Average Difference in Deuteration a | |
|---|---|---|---|---|
| MALDI | ESI | |||
| 1–14 | β | MG1 | n/a | 0.2% |
| 5–15 | β | MG1 | −5.5% | n/a |
| 21–47 | β | MG1 | 2.1% | −0.5% |
| 23–47 | β | MG1 | −1.1% | n/a |
| 101–108 | β | MG1-2 | n/a | −3.7% |
| 109–124 | β | MG2 | n/a | −6.7% |
| 125–142 | β | MG2 | −2.4% | −2.5% |
| 143–158 | β | MG2 | n/a | 2.4% |
| 165–176 | β | MG2 | n/a | 9.4% |
| 177–186 | β | MG2 | −4.0% | n/a |
| 187–199 | β | MG2 | n/a | −1.7% |
| 221–230 | β | MG3 | −3.1% | n/a |
| 235–248 | β | MG3 | −18.9% | n/a |
| 249–277 | β | MG3 | n/a | −5.4% |
| 259–277 | β | MG3 | −7.1% | n/a |
| 307–335 | β | MG3-4 | n/a | −4.9% |
| 336–352 | β | MG4 | −4.2% | n/a |
| 336–350 | β | MG4 | n/a | 0.6% |
| 353–373 | β | MG4 | −2.7% | n/a |
| 353–387 | β | MG4 | n/a | −2.3% |
| 388–411 | β | MG4 | −1.5% | 2.6% |
| 412–436 | β | MG4-5 | −3.4% | 1.8% |
| 437–453 | β | MG5 | n/a | −2.4% |
| 440–449 | β | MG5 | −3.7% | n/a |
| 454–470 | β | MG5 | n/a | 1.9% |
| 471–491 | β | MG5 | 1.2% | n/a |
| 472–491 | β | MG5 | n/a | 1.9% |
| 492–499 | β | MG5 | n/a | 3.0% |
| 513–526 | β | MG5 | −1.5% | n/a |
| 542–560 | β | MG6β | n/a | −11.1% |
| 550–558 | β | MG6β | −24.1% | n/a |
| 574–599 | β | MG6β-LNK | n/a | −17.7% |
| 600–622 | β | LNK | n/a | 0.3% |
Average percentage change in amide backbone deuterium level after correction for side chain contributions (MALDI) or deuterium back-exchange (ESI). Positive and negative numbers indicate increased and decreased deuterium incorporation in C3b compared to C3, respectively. Changes greater than ±10% in at least one method were considered significant and are highlighted in bold italic type.
Table 2.
Differential deuteration in the α-chain of C3- and C3b-derived peptides.
| C3 Sequence Number | Chain | Domain | Average Difference in Deuteration a | |
|---|---|---|---|---|
| MALDI | ESI | |||
| 737–750 | α | α’NT | n/a | −8.5% |
| 742–750 | α | α’NT-MG6α | −10.9% | n/a |
| 751–770 | α | MG6α | n/a | −1.9% |
| 811–822 | α | MG7 | −1.7% | n/a |
| 830–839 | α | MG7 | −21.3% | n/a |
| 873–887 | α | MG7 | n/a | −6.1% |
| 922–945 | α | CUBg | 0.1% | n/a |
| 922–955 | α | CUBg | n/a | 4.6% |
| 956–968 | α | CUBg-TED | 11.0% | n/a |
| 957–968 | α | CUBg-TED | n/a | 35.4% |
| 994–1026 | α | TED | n/a | −2.3% |
| 1012–1025 | α | TED | −0.9% | n/a |
| 1012–1026 | α | TED | −0.4% | n/a |
| 1027–1044 | α | TED | n/a | 1.8% |
| 1037–1043 | α | TED | −10.4% | n/a |
| 1047–1058 | α | TED | 0.5% | n/a |
| 1048–1056 | α | TED | −3.2% | n/a |
| 1061–1071 | α | TED | n/a | −6.9% |
| 1108–1121 | α | TED | −6.9% | n/a |
| 1108–1123 | α | TED | −9.2% | −9.2% |
| 1169–1188 | α | TED | n/a | 0.8% |
| 1189–1206 | α | TED | 12.9% | n/a |
| 1189–1209 | α | TED | 12.5% | n/a |
| 1189–1215 | α | TED | n/a | 14.6% |
| 1224–1236 | α | TED | n/a | −2.1% |
| 1237–1244 | α | TED | 2.1% | n/a |
| 1237–1248 | α | TED | 18.1% | n/a |
| 1237–1251 | α | TED | 15.0% | n/a |
| 1252–1272 | α | TED-CUBf | n/a | −0.6% |
| 1273–1296 | α | CUBf | −1.6% | 1.3% |
| 1342–1364 | α | MG8 | n/a | 4.6% |
| 1386–1422 | α | MG8 | n/a | 12.1% |
| 1416–1422 | α | MG8 | −6.2% | n/a |
| 1440–1463 | α | MG8 | n/a | −0.4% |
| 1508–1520 | α | C345C | n/a | −5.8% |
| 1521–1535 | α | C345C | n/a | −0.2% |
| 1536–1542 | α | C345C | n/a | 4.2% |
| 1543–1561 | α | C345C | n/a | 3.6% |
| 1553–1570 | α | C345C | n/a | 12.6% |
| 1575–1589 | α | C345C | n/a | −1.8% |
| 1590–1605 | α | C345C | n/a | 6.4% |
| 1606–1629 | α | C345C | n/a | 4.6% |
| 1635–1641 | α | C345C | n/a | 2.0% |
Average percentage change in amide backbone deuterium level after correction for side chain contributions (MALDI) or deuterium back-exchange (ESI). Positive and negative numbers indicate increased and decreased deuterium incorporation in C3b compared to C3, respectively. Changes greater than ±10% in at least one method were considered significant and are highlighted in bold italic type.
2.5. Mapping of identified HDX peptides on the C3/C3b crystal structures
The crystal structure data for C3 (PDB accession code: 2A73 (Janssen et al., 2005)), C3b (PDB: 2HR0 (Abdul Ajees et al., 2006) and 2I07 (Janssen et al., 2006)), and a C3b:CRIg complex (PDB: 2ICF (Wiesmann et al., 2006)) were visualized and aligned in PyMol (version 0.99, DeLano Scientific LLC). The C3b structure 2I07 has been used for all mapping studies of C3b unless indicated otherwise. In accordance with similar studies (Brudler et al., 2006; Hamuro et al., 2006; Horn et al., 2006), changes in deuterium incorporation of more than ±10% were considered to be significant. Peptides that showed an increased (≥10%), decreased (≤−10%), or not significantly altered hydrogen/deuterium exchange were highlighted in red, blue, and yellow, respectively.
3. Results
3.1. MALDI Experiment – Hydrogen/Deuterium Exchange
A set of 35 peptides that are common to C3 and C3b and cover 27% of the primary sequence of C3b have been selected based on their peak quality and intensity (Table 1 and Table 2; Fig. 1C and D). The differential deuterium incorporation for these 35 peptides was calculated across all time points. Peptides that exhibited a significant difference in average deuterium incorporation (>+10% or <−10%) between C3 and C3b included eight peptides from the α-chain (742–750, 830–839, 956–968, 1037–1043, 1189–1206, 1189–1209, 1237–1248, 1237–1251) and two peptides from the β-chain (235–248, 550–558). The remaining 25 peptides had an absolute average difference in deuteration of less than 10% (Table 1 and Table 2).
The results of the MALDI HDX data were mapped onto the crystal structures of C3 and C3b (Janssen et al., 2006; Janssen et al., 2005) (Fig. 2). In the case of four peptides (1189–1206, 1189–1209, 1237–1248, and 1237–1251), the increased deuterium uptake in C3b could be accounted for by the large conformational changes that occurred during C3 activation. However, several other peptides showed a decrease in exchange upon conversion to C3b (peptides 235–248, 550–558, 742–750, 830–839, 1037–1043), which could not be entirely explained by the structural changes evident in the crystal structure models. As a consequence, we performed additional HDX experiments using ESI in order to improve upon sequence coverage and validate our results.
Fig. 2.
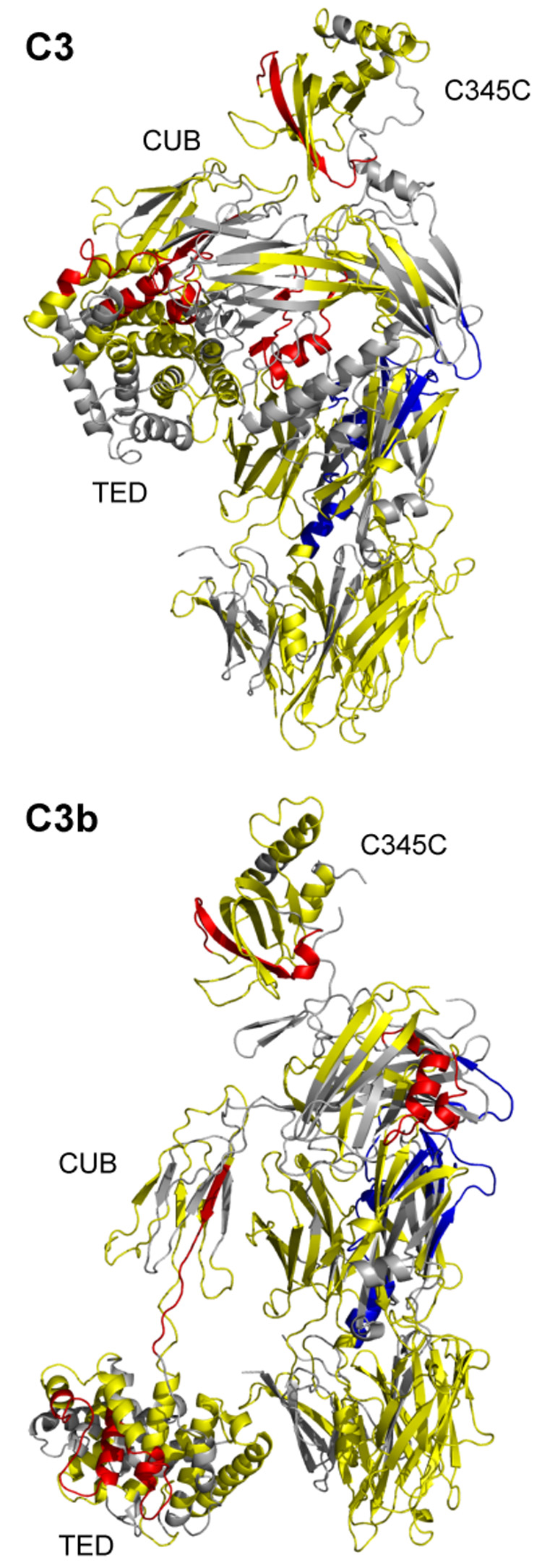
Mapping of differential HDX data on the C3 and C3b crystal structures. Peptides with increased exchange in C3b as compared to C3 are highlighted in red (average difference in deuteration ≥10%); peptides with decreased exchange in C3b as compared to C3 are in blue (≤−10%); peptides with minimal difference in exchange are in yellow (>−10% or <10%). Peptides for which no HDX data are available are represented in gray.
3.2. Generation of C3 Peptide Map for ESI Experiments
We optimized the digestion conditions to produce C3 fragments of suitable size and distribution for ESI analysis. The best results were obtained with acidic buffer containing 8 M urea and 1 M TCEP at pH 3.0, a protein flow of 200 µl/min over the immobilized pepsin column and a linear gradient of 13–40% solvent B over 23 min on the C18 HPLC column. Under these conditions, 155 peptides spanning 68% of the amino acid sequence of C3 were identified. None of those peptides spanned one of the two known glycosylation sites of C3/C3b (Asn-63, Asn-917 (Hirani et al., 1986)).
3.3. ESI Experiment – Hydrogen/Deuterium Exchange
The deuterium incorporation for 47 well-suited peptides from C3 and C3b was followed across all time points (Table 1 and Table 2; Fig. 1C and D). These peptides spanned 55% of the C3b primary sequence and included 26 peptides from the α-chain and 21 peptides from the β-chain. For each peptide, the percent deuterium incorporation was determined at each time point, and the differential change between C3 and C3b was calculated as described above. Six peptides were observed to have an absolute average difference in deuteration of at least 10% (>+10% or <−10%) including four peptides from the α-chain (957–968, 1189–1215, 1386–1422, and 1553–1570; Table 2) and two peptides from the β-chain (542–560, 574–599; Table 1). The remaining peptides exhibited an absolute average difference in deuteration of less than 10%.
3.4. Combining MALDI and ESI HDX Data - Deuterium Incorporation Upon Conversion of C3 to C3b
The combined MALDI- and ESI-based HDX strategies provided for the assessment of 82 peptides common to C3 and C3b, accounting for 61% of the C3b primary sequence (Fig. 1C and D). Comparison of the deuteration profiles showed that nine peptides exhibited increased exchange in C3b when compared to their exchange in C3 (956–968, 957–968, 1189–1206, 1189–1209, 1189–1215, 1237–1248, 1237–1251, 1386–1422 and 1553–1570), and seven peptides exhibited decreased exchange in C3b when compared to their exchange in C3 (235–248, 542–560, 550–558, 574–599, 742–750, 830–839 and 1037–1043). The remaining 66 peptides exhibited a lower difference (<10%) in deuterium incorporation.
The exchange data for all peptides was overlaid onto the C3 and C3b crystal structures (Fig. 2). All peptides that showed increased deuterium exchange in C3b were located within the α-chain (Table 2, Fig. 3). These peptides were distributed among four crystallographic domains: TED, CUB, C345C, and MG8 (Fig. 1B and C). The overlapping peptides 956–968 and 957–968 reside within the TED-CUB interface, which is largely involved in the conformational changes accompanying C3 activation. Examination of the respective crystal structures revealed that this portion of the protein is clearly more solvent-exposed in C3b than in C3, a conclusion with which the HDX data were in full agreement. Two groups of overlapping peptides (1189–1206, 1189–1209, 1189–1215 and 1237–1248, 1237–1251) were in the TED domain, which undergoes a translation of >45 Å upon conversion of C3 to C3b. Also, the peptides in the first of these two groups included a putative binding site for factor H (Herbert et al., 2006; Lambris et al., 1988), a complement regulatory protein with affinity for C3b but not C3, suggesting that the binding site gains solvent accessibility upon structural change. Together, these observations support the increased deuterium exchange observed in the HDX experiments.
Fig. 3.
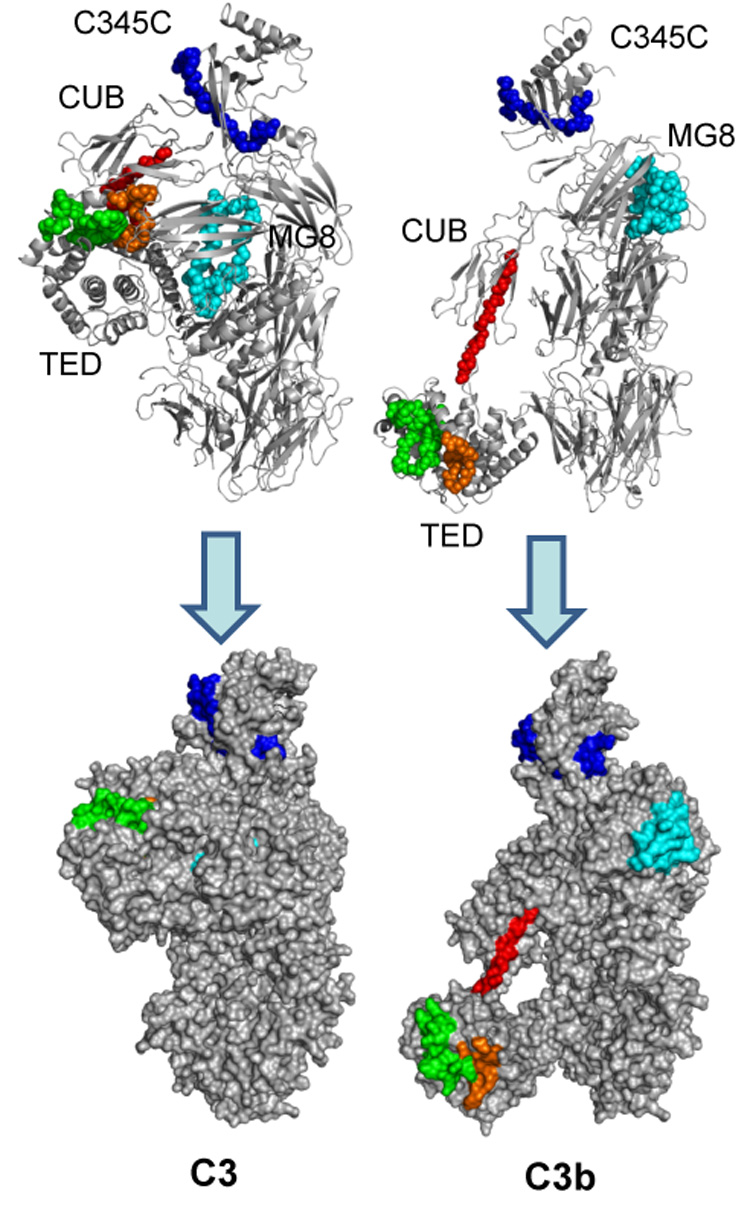
Areas that exhibit increased deuterium exchange upon activation of C3. Peptides with significantly increased HDX in C3b (≥10%) are highlighted in the crystal structures of C3 (left) and C3b (right). The backbones of these peptides are represented as balls in a cartoon representation in the top panels. In the bottom panel, the structures are represented as the calculated solvent-exposed surface. Peptide sequence identification: red = 956–968; green = 1189–1215; orange = 1237–1251; light blue = 1386–1415; blue = 1553–1570. In general, HDX data for these peptides are in agreement with reported changes in protein structure upon conversion of C3 to C3b.
The peptide 1386–1422 is found within the MG8 domain, which undergoes rotation and a 24-Å translation upon conversion of C3 to C3b (Abdul Ajees et al., 2006; Janssen et al., 2006; Wiesmann et al., 2006). This transformation exposes the proposed properdin binding site at residues 1402–1435, which may be reflected by the increased deuterium exchange of peptide 1386–1422 in C3b (Daoudaki et al., 1988). Finally, within the C345C domain, peptide 1553–1570 also exhibited increased exchange in C3b when compared to C3. The C345C domain rotates approximately 30° upon conversion of C3 to C3b and possesses a binding site for factor B, a key protein in the formation of the alternative pathway convertase (Janssen and Gros, 2007). In summary, the observed increases in exchange for these peptides are consistent with the available crystal structures for C3 and C3b.
In contrast to the situation for peptides that exhibited increased deuterium exchange, the HDX data for the peptides that exhibited decreased deuterium exchange in C3b compared to C3 were not consistent with the changes observed in the crystal structures (Fig. 4). These peptides spanned the following domains: MG3 (peptide 235–248), MG6 (542–560, 550–558), the interfaces of MG6-LNK (574–599) and α’NT-MG6 (742–750), MG7 (830–839), and TED (1037–1043) (Fig. 1B and C). With the exception of the TED domain, these domains were found to undergo minimal structural changes upon conversion of C3 to C3b (Janssen et al., 2006). Interestingly, mapping of the peptides that were identified in these domains (235–248, 542–560, 574–599, 742–750 and 830–839) on the crystal structures of C3 and C3b revealed that they form a cluster of loops in close proximity to one another (Fig. 4). When comparing this region between C3 and C3b, only slight changes in the arrangement of these peptides but no frank conformational changes are evident. As a consequence, this area may exhibit local fluctuations in secondary or tertiary structure that are more apparent in solution than in the crystal. At first view, the significance of peptide 1037–1043 seems to be contradictory between experiments. This peptide in the TED domain loses solvent exposure upon conversion from C3 to C3b in the MALDI experiment (−10.4%; Table 2). Structural differences in this area are only visible in the C3b structures by Janssen and Wiesman (Janssen et al., 2006; Wiesmann et al., 2006), whereas this TED area is largely unaltered between C3 and the C3b structure by Abdul Ajees (Abdul Ajees et al., 2006) (Fig. 5A). However, the larger, encompassing peptide 1027–1044 exhibited no significant change in deuterium uptake when analyzed by ESI MS (+1.8%). While this discrepancy may be based on experimental conditions, it may also indicate a statistical compensation due to higher solvent exposure of the first 10 residues. Indeed, comparison of the crystal structures clearly shows that this stretch of amino acids (1027–1036) lies exactly at the contact interface between the TED and MG2 domain and is mainly shielded from the solvent in C3. Upon activation, the same patch becomes solvent-exposed due to the extended positioning of the TED domain in C3b (Fig. 6). This comparison therefore supports both the large relocation of the TED domain and the smaller structural fluctuations visible in two of the three C3b structures.
Fig. 4.
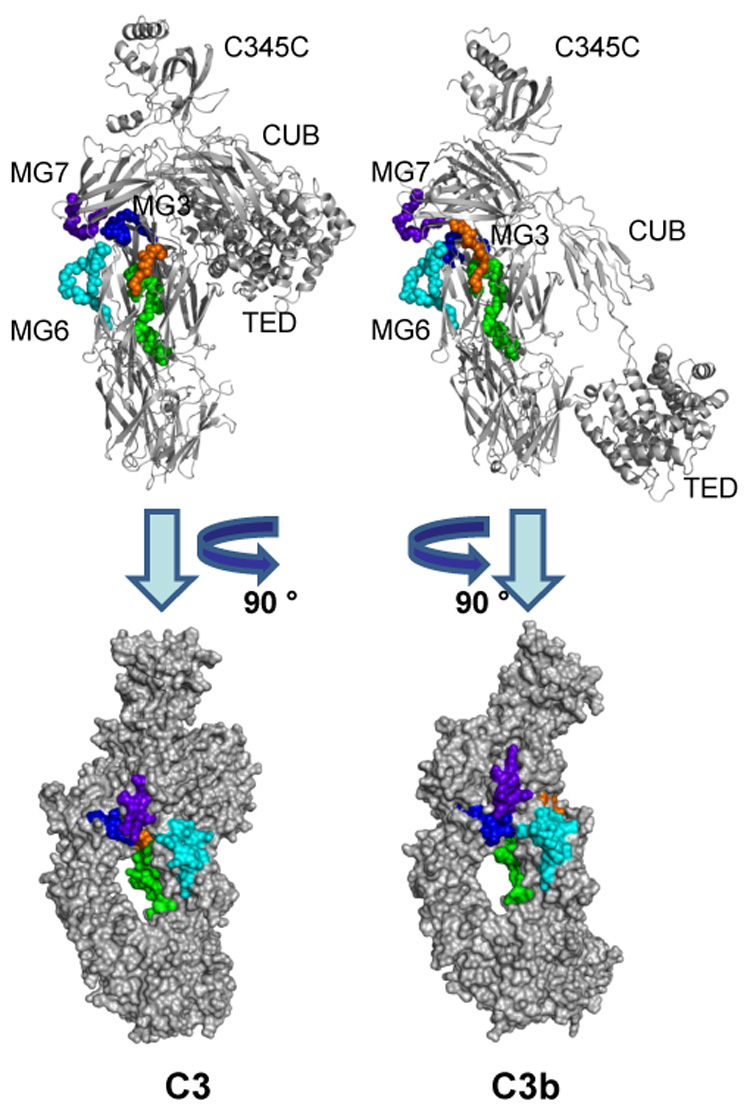
Areas that exhibit decreased deuterium exchange upon activation of C3. Peptides showing a significantly lower HDX (≤−10%) in C3b as compared to C3 are highlighted in the crystal structures of C3 (left) and C3b (right). The backbones of these peptides are represented as balls in a cartoon representation in the top panels to improve clarity. In the bottom panel, the structures have been rotated and represented as the calculated solvent-accessible surface. Peptide sequence identification: blue = 235–248; light blue = 542–560; green = 574–599; orange 742–750; purple = 830–839. While no large structural changes have been observed in the corresponding area of the crystal structures, these peptides form a cluster at the interface of the MG3, MG6, MG7, LNK, and α’NT domains that may show a higher degree of structural flexibility in solution. Peptide 1037–1043, which also showed decreased HDX in C3b, is located on the TED domain and does not contribute to the cluster described above. Its exact location and differential exposition is shown separately in Fig. 6.
Fig. 5.
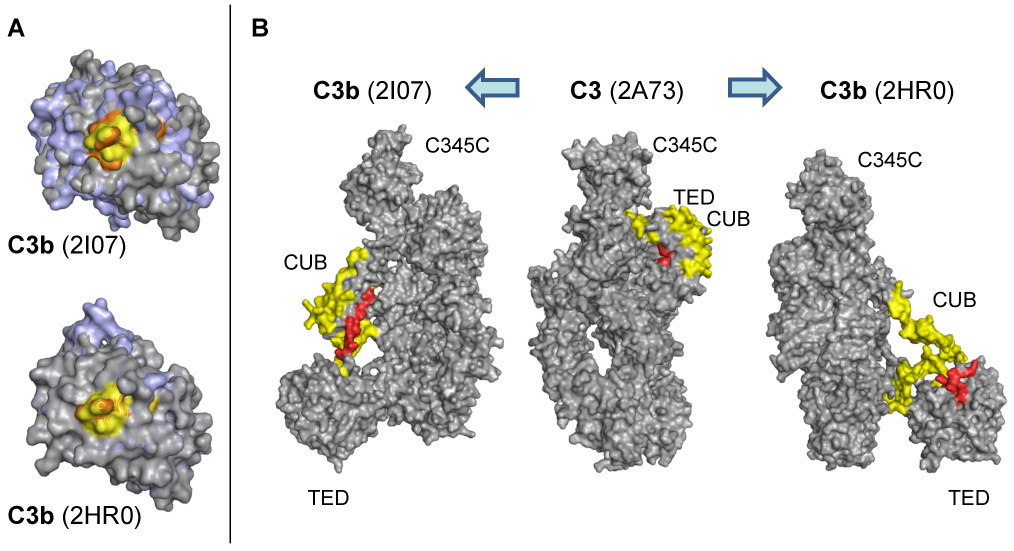
Structural changes in the TED (a) and CUB (b) domains between C3 and the contradicting crystal structures of C3b. (A) Significant conformational changes between the TED domains of C3 (grey) and C3b (pale blue) are only observed for the C3b structure by Janssen et al. (2I07; left) but not for the alternative structure by Abdul Ajees et al. (2HR0, right). In the case of peptide 1037–1044 (yellow and orange in C3 and C3b, respectively), the observed changes in HDX are therefore in closer agreement with structure 2I07. (B) While the CUB domain remains in a compact state in 2I07, the same domain appears rather unfolded and elongated in 2HR0. In the HDX analysis, the majority of the CUB-derived peptides show no significant change in deuteration (yellow), which would again support the 2I07 structure. In agreement with HDX data, peptide 957–968 (red) gets more solvent-exposed in both C3b variants.
Fig. 6.
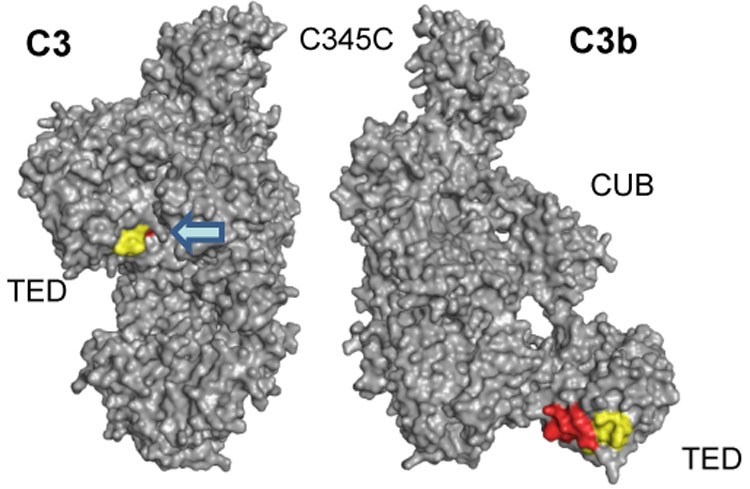
Detailed structural analysis of peptides 1037–1043 (MALDI) and 1027–1044 (ESI). The diverging HDX results for the MALDI-derived peptide 1037–1043 (−10.4%) and the encompassing peptide 1027–1044 (+1.8%) from ESI analysis can be explained by the relocation of TED during C3 activation. While the common peptide stretch 1037–1043 (yellow) is similarly accessible in both C3 and C3b, the N-terminal elongation of the ESI peptide (1027–1036; red) is buried in C3b and gets exposed in C3b. As a consequence, the signal observed in ESI may be the sum of a HDX decrease (as in MALDI) and an HDX increase (for the N-terminus).
3.5. Differential analysis of the available C3b structures
Based on the differences found for peptide 1037–1043 (see above), we extended our analysis and compared the distribution of all identified HDX peptides on the C3b structures by Janssen et al. (Janssen et al., 2006) and Abdul Ajees et al. (Abdul Ajees et al., 2006). The third structure by Wiesmann et al. (co-crystal with the CRIg receptor (Wiesmann et al., 2006)) shows a high degree of similarity with unbound C3b by Janssen and was therefore not evaluated separately. The major differences between the analyzed C3b structures are found in the CUB and TED domains. In the structure by Janssen, the CUB domain is shifted in its position but remains in a highly folded, C3-like state (Fig. 5B). The TED domain undergoes a number of small but significant conformational adaptations and is therefore different between C3 and C3b (Fig. 5A). In the structure by Abdul Ajees, on the other hand, the CUB domain appears in a rather unfolded state, leading to a different elongation and positioning of the TED domain (Fig. 5B). In addition, there are nearly no structural changes within the TED domain between C3 and this structural variant (Fig. 5A). The large unfolding of CUB in the latter structure should lead to a significantly enhanced solvent accessibility of this domain. However, such large changes in solvent exposure could not be confirmed by HDX-MS since most of the peptides in this region showed shifts below 10%. The only peptide with significantly enhanced HDX signal was located at the immediate interface between CUB and TED (957–968, +35.4%). Indeed, this peptide is buried in the backfolded state of TED in native C3 but becomes highly solvent exposed during CUB elongation in both C3b variants. Interestingly, peptide 957–968 is stretched out as part of the CUB arm in the structure by Janssen, whereas it is present in a more folded state as part of TED in case of C3b by Abdul Ajees (Fig. 5B). Together, these data indicate that the C3b crystal structures by Janssen and Wiesmann more closely correspond to the solution-based data we acquired using HDX-MS, especially in the CUB and TED domains.
4. Discussion
Activation and regulation of the human complement system is largely driven by a complex cascade of interactions, enzymatic cleavages, and conformational changes that unveil cryptic binding sites (Gros et al., 2007). C3 and its activated fragment C3b are at the center of this cascade and essential for both the amplification and effector functions of complement. The recent reports of the C3 and C3b crystal structures have provided detailed solid-state structures of these proteins and offered an important insight on their function on a molecular level (Abdul Ajees et al., 2006; Janssen et al., 2006; Janssen et al., 2005; Wiesmann et al., 2006). However, there are significant and fundamental differences between the available structures for C3b. Furthermore, these proteins may exhibit structural fluctuations in solution that are not apparent from their crystal structure. Therefore, we investigated the relative conformations of these two proteins in the liquid state by HDX-MS and mapped the identified peptides on the corresponding structures. Although HDX is incapable of providing an atomic-resolution structure of macromolecules, it can provide insight into protein structural dynamics in solution and supplement crystal data. While the C3b used in this study has been generated by tryptic cleavage of C3, the product is known to closely correspond to the convertase-derived C3b (Bokisch et al., 1969). Our initial experiments using MALDI revealed that many differences in deuterium uptake could be explained by the gross structural changes that are observed upon conversion of C3 to C3b (Gros et al., 2007; Janssen et al., 2006). However, sequence coverage was limited by this technology and some peptides with decreased deuterium uptake in C3b could not be explained by differences in the crystal structures. To validate the results and expand our coverage of the proteins, C3 and C3b were therefore further analyzed by ESI. With this method, the coverage of C3 could be remarkably expanded, and the combination of both techniques provided a rich peptide pool for the investigation of structural changes. In light of the often-faced size restrictions for MS-based proteins analyses (Cravello et al., 2003), the sequence coverage of 61% for the 175-kDa C3b molecule is an important achievement and demonstrates the feasibility of this method for large plasma proteins.
For most areas of the proteins, our combined HDX-MS data were in good agreement with the crystallographic structures of C3 and C3b. Significant increase of deuteration upon activation of C3 to C3b were essentially observed in the CUB, TED, MG8, and C345C domains. These areas are not only known to undergo large conformational changed during the conversion but were also attributed to carry binding sites for a series of complement factors, receptors, and regulators (Gros et al., 2007; Janssen and Gros, 2007). For example, C345C exhibits a putative binding site for factor B (Kolln et al., 2005; Kolln et al., 2004), MG8 was found important for the binding of properdin (Daoudaki et al., 1988), and TED is described to carry several sites for complement receptor 2 (Janssen and Gros, 2007), factor H (Herbert et al., 2006; Jokiranta et al., 2006; Lambris et al., 1988), and bacterial complement evasion proteins (Hammel et al., 2007a; Hammel et al., 2007b). In addition, the CUB domain has been shown to bind factor B (O'Keefe et al., 1988) and its proper structure and orientation is considered important for the assembly of the alternative pathway C3 convertase (Gros et al., 2007). In this respect, our HDX data are highly consistent with the conformational changes upon C3 activation that were described in the crystal structures.
ESI analysis also confirmed our observation that C3 may adopt conformations in solution that result in an increased solvent exposure in a focal area around the MG3, MG6, and MG7 domains when compared to that of C3b. Specifically, discontinuous peptides 235–248, 542–560, 574–599 and 742–750 exhibited increased deuterium exchange in C3 when compared to C3b. The idea that C3 may have greater conformational flexibility in this area is intriguing and may contribute to our understanding of specific functions of C3 and C3b. Indeed, antibody studies have previously indicated that this area of the β-chain is important in factor B binding to C3b, a key step in formation of the alternative pathway convertase (Alsenz et al., 1990). In the alternative pathway of complement activation, the C3 convertase is formed from either C3b or the product of C3 thioester hydrolysis, C3(H2O). While C3b is formed upon cleavage of C3 by the convertase, C3(H2O) forms spontaneously in solution from non-enzymatic C3 thioester hydrolysis. Since C3b and C3(H2O) have binding epitopes and biological functions in common, it is assumed that they also share structural features (Lambris, 1988; Sahu and Lambris, 2001). Indeed, Nishida et al. recently utilized electron microscopy to explore the structural transitions undergone by C3 upon activation (Nishida et al., 2006). They found that conformers of C3 that have been activated by nucleophiles (i.e. C3(N)), including C3(H2O), have a similar general structure and domain arrangement as C3b. However, in order for C3 to form C3(N), the anaphylatoxin (ANA) and α’NT domains of C3 must move from the MG3/MG8 side of C3 to the MG7 side. Structurally, this movement requires that the ANA and α’NT domains must pass through the β-ring formed by domains MG1-MG4, and through the half-ring created by MG5-MG6 (Fig. 1A). Nishida et al. have hypothesized that the MG6 domain in C3 is capable of unfolding to allow this transition to occur. Our observations support this hypothesis: three of the four peptides that exhibited increased exchange in C3 when compared to C3b (542–560, 574–599 and 742–750) were partially or entirely located within the MG6 domain. Thus, it appears that the MG6 domain of C3 is unfolding or adopting a different structure during the timescale of our experiments and in doing so exposes its backbone to the solvent, leading to increased deuterium exchange. Furthermore, our data suggest that peptide 235–248 of the MG3 domain may also exhibit conformational flexibility in C3 and perhaps also play a permissive role in the formation of C3(N).
Our data interpretation is also indirectly supported by the C3b-CRIg co-crystal structure (Wiesmann et al., 2006). CRIg is a complement receptor that is found on the surfaces of resident macrophages and appears to be necessary for the clearance of C3-opsonized pathogens (Helmy et al., 2006). CRIg does not bind to native C3 but interacts with C3b and C3c. Furthermore, binding of CRIg to C3b was found to inhibit the functions of the C3 and C5 convertases of the alternative complement pathway (Katschke et al., 2007). In the co-crystal structures with C3b and C3c, CRIg was localized in the middle of the β-ring, simultaneously interacting with the MG3, MG4, MG5, MG6, and LNK domains. Interestingly, three of the peptides that exhibited decreased deuterium exchange in C3b (235–248, 542–560, 574–599) cover residues that form contacts with CRIg in the co-crystal structure. We therefore speculate that the greater organization of these peptides in C3b influences CRIg’s ability to bind C3b but not C3.
The validity of the structural model of C3b by Abdul Ajees et al. has recently been questioned (Janssen et al., 2007). While all structures agree in an elongation and repositioning of the TED domain, which is responsible for the covalent attachment onto foreign surfaces (opsonization), the exact structural transitions at the CUB/TED interface and the relative position of TED vary considerably. The conservative changes in solvent accessibility in the CUB area, the large exposure of peptide 957–968 and the observed changes in the TED area all seem to better agree with the two structures published by Janssen et al. (Janssen et al., 2006) and Wiesmann et al. (Wiesmann et al., 2006), respectively, than with the C3b structure by Abdul Ajees et al. (Abdul Ajees et al., 2006). In their reply to the recent controversy (Janssen et al., 2007), Abdul Ajees et al. suggest that their structure may represent a different physiological stage in the activation process of C3b, in which CUB is further unfolded compared to the other structure. On the other hand, a rather large gap in the crystal lattice of 2HR0 may also indicate the presence of an impurity caused by a regulator of complement activation, which may have induced additional cleavage (Janssen et al., 2007). While further investigations and additional experiments are required to confirm the true solvent structure of active C3b, our HDX data generally show preferential agreement with the structures published by Janssen et al. and Wiesmann et al.
Finally, the comparison of the current results for C3b with our previous data for C3(H2O) (Winters et al., 2005) provides further support for shared structural elements between these two proteins. In the prior analysis of C3(H2O), a >10% decrease in deuterium exchange (in going from C3 to C3(H2O)) was observed for peptides 235–248, 542–566, 1108–1121 and 1108–1123. Analysis of the relevant peptides for C3b, described herein, also showed a decrease in deuterium exchange of >10% for peptides 235–248 and 550–558 and a smaller decrease for peptides 1108–1121 and 1108–1123. A similar pattern was observed for the peptides that exhibited an increase in deuterium exchange. Most of the peptides that exhibited a >10% increase in deuterium exchange in going from C3 to C3(H2O) (922–945, 956–968, 957–968, 1027–1036, 1189–1206, 1189–1209, 1237–1248 and 1237–1251) also showed a similar change for the conversion to C3b. The two only exceptions were the MALDI-derived peptide 922–945 (no significant change) and the ESI-derived peptide 1029–1044 (minimal increase in deuteration). This high agreement in the deuteration patterns provides more evidence for a structural similarity between these two related proteins and cross-validates the experimental method.
Acknowledgments
We thank Dr. Debbie McClellan for her editorial assistance and Dr. Bert Janssen for his thoughtful review of the manuscript. This work was supported by the National Institutes of Health Grants GM-069736, GM-62134, AI-30040, AI-072106, and AI-068730. M.C.S. received stipend support from an Amgen Rheumatology Fellowship Award.
Abbreviations
- ANA
anaphylatoxin domain
- C3(H2O)
hydrolyzed C3
- C3(N)
nucleophilically activated C3
- CRIg
complement receptor of the immunoglobulin superfamily
- CUB
C1r/C1s, Uegf, Bmp1 domain
- ESI
electrospray ionization
- HDX
hydrogen/deuterium exchange
- LNK
linker domain
- MALDI
matrix-assisted laser desorption/ionization
- MG
macroglobulin domain
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- PDB
protein data bank
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- TED
thioester domain
- TFA
trifluoroacetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdul Ajees A, Gunasekaran K, Volanakis JE, Narayana SV, Kotwal GJ, Murthy HM. The structure of complement C3b provides insights into complement activation and regulation. Nature. 2006;444:221–225. doi: 10.1038/nature05258. [DOI] [PubMed] [Google Scholar]
- Alsenz J, Becherer JD, Nilsson B, Lambris JD. Structural and functional analysis of C3 using monoclonal antibodies. Curr. Top. Microbiol. Immunol. 1990;153:235–248. doi: 10.1007/978-3-642-74977-3_13. [DOI] [PubMed] [Google Scholar]
- Bokisch VA, Muller-Eberhard HJ, Cochrane CG. Isolation of a fragment (C3a) of the third component of human complement containing anaphylatoxin and chemotactic activity and description of an anaphylatoxin inactivator of human serum. J Exp Med. 1969;129:1109–1130. doi: 10.1084/jem.129.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudler R, Gessner CR, Li S, Tyndall S, Getzoff ED, Woods VL., Jr PAS domain allostery and light-induced conformational changes in photoactive yellow protein upon I2 intermediate formation, probed with enhanced hydrogen/deuterium exchange mass spectrometry. J. Mol. Biol. 2006;363:148–160. doi: 10.1016/j.jmb.2006.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busenlehner LS, Armstrong RN. Insights into enzyme structure and dynamics elucidated by amide H/D exchange mass spectrometry. Arch. Biochem. Biophys. 2005;433:34–46. doi: 10.1016/j.abb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Cravello L, Lascoux D, Forest E. Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom. 2003;17:2387–2393. doi: 10.1002/rcm.1207. [DOI] [PubMed] [Google Scholar]
- Daoudaki ME, Becherer JD, Lambris JD. A 34-amino acid peptide of the third component of complement mediates properdin binding. J. Immunol. 1988;140:1577–1580. [PubMed] [Google Scholar]
- Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Soc. Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- Eyles SJ, Kaltashov IA. Methods to study protein dynamics and folding by mass spectrometry. Methods. 2004;34:88–99. doi: 10.1016/j.ymeth.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Gros P, Milder FJ, Janssen BJ. Complement driven by conformational changes. Nat. Rev. Immunol. 2007;8:48–58. doi: 10.1038/nri2231. [DOI] [PubMed] [Google Scholar]
- Hammel M, Sfyroera G, Pyrpassopoulos S, Ricklin D, Ramyar KX, Pop M, Jin Z, Lambris JD, Geisbrecht BV. Characterization of Ehp, a secreted complement inhibitory protein from Staphylococcus aureus. J. Biol. Chem. 2007a;282:30051–30061. doi: 10.1074/jbc.M704247200. [DOI] [PubMed] [Google Scholar]
- Hammel M, Sfyroera G, Ricklin D, Magotti P, Lambris JD, Geisbrecht BV. A structural basis for complement inhibition by Staphylococcus aureus. Nat. Immunol. 2007b;8:430–437. doi: 10.1038/ni1450. [DOI] [PubMed] [Google Scholar]
- Hammer CH, Wirtz GH, Renfer L, Gresham HD, Tack BF. Large scale isolation of functionally active components of the human complement system. J. Biol. Chem. 1981;256:3995–4006. [PubMed] [Google Scholar]
- Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y, Coales SJ, Southern MR, Nemeth-Cawley JF, Stranz DD, Griffin PR. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J. Biomol. Tech. 2003;14:171–182. [PMC free article] [PubMed] [Google Scholar]
- Helmy KY, Katschke KJ, Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, van Lookeren Campagne M. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124:915–927. doi: 10.1016/j.cell.2005.12.039. [DOI] [PubMed] [Google Scholar]
- Herbert AP, Uhrin D, Lyon M, Pangburn MK, Barlow PN. Disease-associated sequence variations congregate in a polyanion recognition patch on human factor H revealed in three-dimensional structure. J. Biol. Chem. 2006;281:16512–16520. doi: 10.1074/jbc.M513611200. [DOI] [PubMed] [Google Scholar]
- Hirani S, Lambris JD, Muller-Eberhard HJ. Structural analysis of the asparagine-linked oligosaccharides of human complement component C3. Biochem. J. 1986;233:613–616. doi: 10.1042/bj2330613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle AN, Resing KA, Ahn NG. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- Horn JR, Kraybill B, Petro EJ, Coales SJ, Morrow JA, Hamuro Y, Kossiakoff AA. The role of protein dynamics in increasing binding affinity for an engineered protein-protein interaction established by H/D exchange mass spectrometry. Biochemistry. 2006;45:8488–8498. doi: 10.1021/bi0604328. [DOI] [PubMed] [Google Scholar]
- Isenman DE, Kells DI, Cooper NR, Muller-Eberhard HJ, Pangburn MK. Nucleophilic modification of human complement protein C3: correlation of conformational changes with acquisition of C3b-like functional properties. Biochemistry. 1981;20:4458–4467. doi: 10.1021/bi00518a034. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444:213–216. doi: 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Gros P. Structural insights into the central complement component C3. Mol. Immunol. 2007;44:3–10. doi: 10.1016/j.molimm.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson-Ekdahl K, Nilsson B, Gros P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature. 2005;437:505–511. doi: 10.1038/nature04005. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Read RJ, Brunger AT, Gros P. Crystallography: crystallographic evidence for deviating C3b structure. Nature. 2007;448:E1–E2. doi: 10.1038/nature06102. discussion E2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokiranta TS, Jaakola VP, Lehtinen MJ, Parepalo M, Meri S, Goldman A. Structure of complement factor H carboxyl-terminus reveals molecular basis of atypical haemolytic uremic syndrome. Embo J. 2006;25:1784–1794. doi: 10.1038/sj.emboj.7601052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katragadda M, Magotti P, Sfyroera G, Lambris JD. Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J. Med. Chem. 2006;49:4616–4622. doi: 10.1021/jm0603419. [DOI] [PubMed] [Google Scholar]
- Katschke KJ, Jr, Helmy KY, Steffek M, Xi H, Yin J, Lee WP, Gribling P, Barck KH, Carano RA, Taylor RE, Rangell L, Diehl L, Hass PE, Wiesmann C, van Lookerenb Campagne M. A novel inhibitor of the alternative pathway of complement reverses inflammation and bone destruction in experimental arthritis. J. Exp. Med. 2007;204:1319–1325. doi: 10.1084/jem.20070432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolln J, Bredehorst R, Spillner E. Engineering of human complement component C3 for catalytic inhibition of complement. Immunol. Lett. 2005;98:49–56. doi: 10.1016/j.imlet.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Kolln J, Spillner E, Andra J, Klensang K, Bredehorst R. Complement inactivation by recombinant human C3 derivatives. J. Immunol. 2004;173:5540–5545. doi: 10.4049/jimmunol.173.9.5540. [DOI] [PubMed] [Google Scholar]
- Lambris JD. The multifunctional role of C3, the third component of complement. Immunol. Today. 1988;9:387–393. doi: 10.1016/0167-5699(88)91240-6. [DOI] [PubMed] [Google Scholar]
- Lambris JD, Avila D, Becherer JD, Muller-Eberhard HJ. A discontinuous factor H binding site in the third component of complement as delineated by synthetic peptides. J. Biol. Chem. 1988;263:12147–12150. [PubMed] [Google Scholar]
- Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat. Rev. Microbiol. 2007;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 2007;171:715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk RA, Hewitt KM, Lacy DB, Lin HC, Gessner CR, Li S, Woods VL, Jr, Collier RJ. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J. Biol. Chem. 2006;281:1630–1635. doi: 10.1074/jbc.M511164200. [DOI] [PubMed] [Google Scholar]
- Nishida N, Walz T, Springer TA. Structural transitions of complement component C3 and its activation products. Proc. Natl. Acad. Sci. U.S.A. 2006;103:19737–19742. doi: 10.1073/pnas.0609791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe MC, Caporale LH, Vogel CW. A novel cleavage product of human complement component C3 with structural and functional properties of cobra venom factor. J. Biol. Chem. 1988;263:12690–12697. [PubMed] [Google Scholar]
- Perkins SJ, Sim RB. Molecular modelling of human complement component C3 and its fragments by solution scattering. Eur. J. Biochem. 1986;157:155–168. doi: 10.1111/j.1432-1033.1986.tb09652.x. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat. Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- Sahu A, Soulika AM, Morikis D, Spruce L, Moore WT, Lambris JD. Binding kinetics, structure-activity relationship, and biotransformation of the complement inhibitor compstatin. J. Immunol. 2000;165:2491–2499. doi: 10.4049/jimmunol.165.5.2491. [DOI] [PubMed] [Google Scholar]
- Schuster MC, Chen H, Lambris JD. Hydrogen/deuterium exchange mass spectrometry: potential for investigating innate immunity proteins. Adv. Exp. Med. Biol. 2007;598:407–417. doi: 10.1007/978-0-387-71767-8_28. [DOI] [PubMed] [Google Scholar]
- Smith CA, Vogel CW, Muller-Eberhard HJ. MHC Class III products: an electron microscopic study of the C3 convertases of human complement. J. Exp. Med. 1984;159:324–329. doi: 10.1084/jem.159.1.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman JM, Holers VM. The Central Role of the Alternative Complement Pathway in Human Disease. J. Immunol. 2006;176:1305–1310. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- Volanakis JE, Frank M, editors. The Human Complement System in Health and Diseases. New York, NY: Marcel Dekker, Inc.; 1998. [Google Scholar]
- Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- Wiesmann C, Katschke KJ, Yin J, Helmy KY, Steffek M, Fairbrother WJ, McCallum SA, Embuscado L, DeForge L, Hass PE, van Lookeren Campagne M. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature. 2006;444:217–220. doi: 10.1038/nature05263. [DOI] [PubMed] [Google Scholar]
- Winters MS, Spellman DS, Lambris JD. Solvent accessibility of native and hydrolyzed human complement protein 3 analyzed by hydrogen/deuterium exchange and mass spectrometry. J. Immunol. 2005;174:3469–3474. doi: 10.4049/jimmunol.174.6.3469. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Marshall AG. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 1998;9:225–233. doi: 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]