

Abstract
Recent studies are reviewed indicating that the transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFβ1, PTEN, p53 and fibronectin. The downstream pathways of these factors display multiple nodes of interaction with each other suggesting the existence of a functional network of suppressor factors that serves to maintain normal growth regulation and resist the emergence of transformed variants. Paradoxically, Egr-1 is oncogenic in prostate cancer. In the majority of these cancers PTEN and/or p53 is inactive. It is suggested that these defects in the tumor suppressor network allow for the unopposed induction of TGFβ1 and fibronectin, which favor transformation and survival of prostate tumor epithelial cells, explain the role of Egr1 in prostate cancer. Egr1 is a novel and logical target for intervention by gene therapy methods and targeting methods are discussed.
Keywords: cancer, suppressor network, anoikis, prostate cancer, gene therapy, tumor progression, anchorage-independent
Introduction
Egr1 or the early growth response–1 gene product is a zinc-finger transcription factor of 59,000 Daltons that, unusually, appears to activate transcription by binding to DNA as a monomer. The role of Egr1 in cancer was last reviewed in these pages in 1998 1. The thrust of the review was the emerging evidence indicating that Egr1 has significant tumor suppressor properties. Two mechanisms – the direct induction of the epithelial cell suppressor TGFβ1 and direct induction of p53 in melanoma cells to promote apoptosis – were emphasized. Numerous other genes implicated in the regulation of cancer such as members of the PDGF and IGF families have promoter elements consistent with regulation by Egr1 and the evidence that Egr1 functionally regulates these genes was summarized. Since then, a wealth of new information has extended our mechanistic knowledge of the suppressor theory especially for the role of the TGFβ1, IGF-II, PTEN, fibronectin, p53, and p73 genes and has added the complication that Egr1 plays an important role in the progression of prostate cancer. These topics are briefly reviewed and we offer the hypothesis that Egr1 participates in the regulation of a network of suppressor gene products. In the case of prostate cancer however, specific defects in this network may lead to the unopposed action of Egr1 on TGFβ1 that favors tumor progression.
TGFβ1
Strong evidence indicates that under physiological conditions TGFβ1 is growth inhibitory for cells of epithelial origins that bear an intact TGFβ1 RII receptor signal transduction pathway 2, 3. The promoter of the human TGFβ1 gene contains at least two Egr1 binding sites both of which can bind Egr1 leading to the activation of transcription 4, 5. The significance of this interaction in human tumor cells was shown by re-expression of Egr1 in human HT1080 fibrosarcoma cells, which do not normally express Egr1 but do exhibit functional TGFβ1 RII receptors 5. HT1080 cells that have been made to express Egr-1 acquire the ability to synthesize and secrete precursor TGFβ1 that becomes activated and acts in an autocrine fashion to suppress growth. Chromatin immunoprecipitation of Egr1-DNA complexes has confirmed the regulation of TGFβ1 by Egr1 in living cells 6. Thus, Egr1-induced TGFβ1 is a growth suppressor in HT1080 cells in spite of the apparent mesenchymal origin of these cells. A number of additional studies have documented functional TGFβ1 expression in correlation with or following expression of Egr1 in other cells types as well as in vivo (Table 1). In addition to HT1080 cells, this suppression mechanism has been observed in human glioblastoma cells 7 and in the mouse 8. In connective tissue cells TGFβ1-induced fibrosis in a variety of settings also may be attributed to Egr1 9, 10, 11. Thus, TGFβ1 appears to be a general effector of Egr1-dependent growth regulation.
Table 1.
Evidence for the Regulation of Key Suppressor and Growth Modulating Factors by Egr-1
| Suppressor Factor/Growth Regulator | System | Mechanism1 | References |
|---|---|---|---|
| TGFβ1 | Human fibrosarcoma cells, H T1080 | Direct | Liu 1996; de Belle 2000 5, 6 |
| Transgenic mouse lung tissue | Indirect | Lee 2004 11 | |
| Vascular smooth muscle cells | Indirect | Fu 2003 83 | |
| In vivo mouse arterial stress model | Indirect | Sho 2002 84 | |
| Human nonsmall cell lung carcinoma cells | Indirect | Kane 2002 85 | |
| Airway smooth muscle cell preparation | Indirect | McKay 1998 86 | |
| Monkey kidney, CV-1 cells | Direct | Dey 1994 4 | |
|
| |||
| PTEN | Human 293T fibroblasts | Direct | Virolle 2001 19 |
| Mouse Embryo Fibroblasts | Direct | Virolle 2001 19 | |
| Human squamous carcinoma cells (SDCCTF) | Indirect | Okamura 2005 24 | |
| normal FRTL-5 rat thyroid cells, vs. thyroid tumor cell lines, ARO & BCPAP | Indirect | Tell 2004 23 | |
| Fetal kidney epithelial cells | Indirect | Tsugawa 2003 20 | |
| Mouse mammary gland in vivo | Direct | Moorehead 2003 21 | |
| Human prostate, DU145 & lung H226 carcinoma cells | Indirect | Han 2003 22 | |
|
| |||
| BCL-2 | v-sis-transformed mouse NIH-3T3 cells | Indirect | Huang 1998 87 |
|
| |||
| p53 | Wild type and Egr-1 (−/−) mouse embryo fibroblasts | Direct | Krones-Herzig, Mittal 2005 35 |
| Wild type and Egr-1 (−/−) mouse embryo fibroblasts | Indirect | Krones-Herzig 2003 34 | |
| Fresh surgical human glioblastoma series | Indirect | Calogero 2001 14 | |
| Wild type and Egr-1 (−/−) mouse embryo fibroblasts | Indirect | Krones-Herzig 2001 88 | |
| B-cell lymphoma cells BKS-2 from CBA mice | Indirect | Han 1999 89 | |
| Human A375-C6 melanoma cells | Direct | Ahmed 1996 90 | |
| Mouse NIH-3T3 cells | Indirect | Huang 1995 91 | |
|
| |||
| p73 | Human genomic DNA | Indirect | Ding 1999 37 |
| Murine cells, Neuro-2A cells | Indirect | Pignatalli 2003 38 | |
| Human cells, HI299 | Indirect | Kartecheva 2003 39 | |
|
| |||
| Fibronectin | Human fibrosarcoma cells, HT1080 | Direct | Liu 1999 7 |
| Human Glioblastoma, U251 cells | Direct | Liu 2000 13 | |
| Human surgical glioblastoma and primary cell lines | Indirect | Calogero 2004 15 | |
| Human fibrosarcoma cells, HT1080 | Indirect | de Belle 1999 43 | |
“Direct” that there is experimental evidence that Egr1 binds the promoter in question at regulatory sequences to affect transcription whereas as indirect refers to published data consistent with a regulator role of Egr1 over the level of the factor without explicit demonstration of the mechanism of regulation.
Fibronectin and plasminogen activator inhibitor-1 (PAI-I)
Potential targets of the TGFβ1/Smad signal transduction pathway that may explain the transformation-suppressor activity of TGFβ1 include fibronectin and PAI-1 12, 3. This possibility was examined in several human glioblastoma cells. Established human cell lines 13 and surgical specimens of glioblastoma 14 or primary cultures of the tumors 15 commonly exhibit low or undetectable levels of Egr1 consistent with a role of Egr1 in the cause or development of glioblastoma 13, 14. Re-expression of Egr1 in several human glioblastoma cell lines such as U251 led to increased expression of TGFβ1, fibronectin and PAI-1 13. Surprisingly, direct addition of activated TGFβ1 to the cells induced expression of PAI-1 but not fibronectin. This led to the discovery that fibronectin is a direct target gene of Egr1 via two closely spaced Egr1-binding sequences in the proximal promoter 13. Both fibronectin and PAI-1 induced by Egr1/TGFβ1 are secreted and become part of the extracellular matrix where they greatly increase attachment, an effect that is blocked by addition of RGD-containing peptides or neutralizing antibodies. The results are associated with slower growth and a flattened more epithelioid phenotype. Consistent with these results, expression of fibronectin alone in HT1080 cells reverses key aspects of transformation 16 similar to the effects of Egr1 expression 17. Indeed, Egr1 stimulates coordinated expression of TGFβ1, fibronectin, and PAI-1. All of three have been found to participate in the restoration of a normal phenotype in HT1080 cells 7. Comparable examples have been observed by others (Table 1) indicating that this mechanism may be general.
Fibronectin and PAI-1 are known to contribute to anchorage-dependent growth control 18. One mechanism involves the interaction of fibronectin with specific integrins leading to activation of the Protein Kinase-B/Akt (PKB/AkT) pathway (Figure 1). This pathway is a major site of action of the tumor suppressor PTEN thereby suggesting a potential crosstalk downstream of fibronectin as described below.
Figure 1.
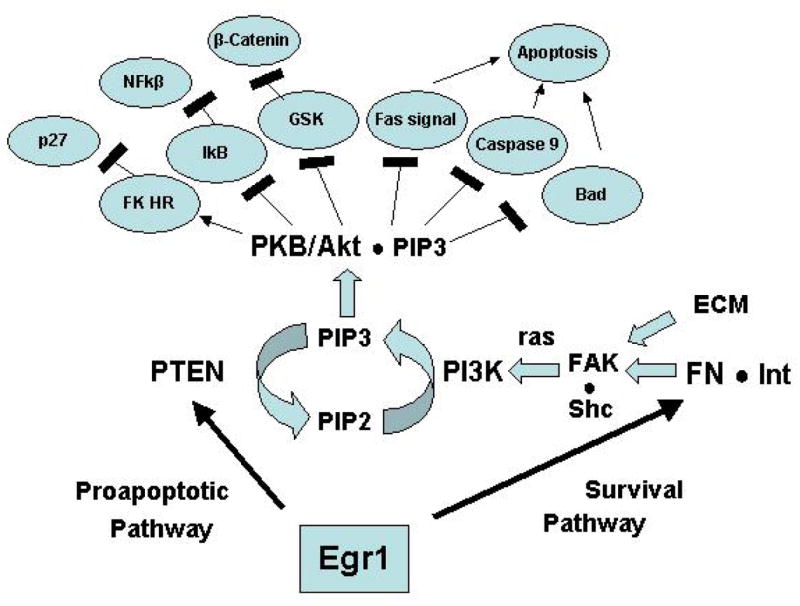
Potential interaction between two known Egr1 target genes, PTEN and fibronectin achieves a delicate balance between attachment and apoptosis of detached cells (anoikis). It is known that Egr1 directly stimulates the expression and secretion of functional fibronectin leading to RGD-dependent attachment and FAK activation. This pathway activates Akt in other systems thereby inhibiting apoptosis and promoting survival. Cells with decreased attachment properties – a manifestation of transformation - experience relatively increased PTEN-mediated inhibition of Akt and increased apoptosis (anoikis). Thus Egr1 favors attachment and survival of normal cells but apoptosis of abnormal forms with deficient attachment properties.
PTEN
The tumor suppressor gene, PTEN/MMAC1/TEP1, is a proapoptotic factor that is frequently suppressed or mutated in a variety of human cancers (reviewed in ref. 19). Egr1 is one of the first transcription factors shown to directly regulate the expression of PTEN and the first shown to regulate PTEN in living cells 19. The proximal promoter of PTEN is GC-rich and contains one functional Egr1 binding site. We have examined the regulation of PTEN in a variety of human cell types and the Egr1-null mouse system (Table 1). In 293T human fetal kidney cells, Egr1 strongly activates a PTEN-promoter-driven luciferase reporter system, which is inhibited by low concentrations of Egr1 antisense oligonucleotides. EMSA, mutation analysis of DNA-binding sequence and chromatin immunoprecipitation of Egr1-PTEN promoter complexes from living cells have been used to test the functional role of this site. These results indicate that Egr1 is a direct and functional regulator of the PTEN gene.
Irradiation of wild type mice strongly induces Egr1 and PTEN mRNA, however PTEN expression is not induced in irradiated Egr1-null mice indicating that Egr1 regulates the expression of PTEN in the whole animal 19. The functional significance of PTEN regulation by Egr1 is suggested further by the observation that MEFs from Egr1-null mice are resistant to apoptosis following UV irradiation whereas MEFs from wild type littermates rapidly undergo apoptosis following UV-irradiation. The apoptosis response could be rescued by exogenous expression of Egr1 in MEFs from the Egr1-null mice. These observations provide an additional mechanism for the tumor suppressor activity of Egr1.
In addition to the mouse system and human cells (MEFs, HT1080, 293T 19), consistent significant effects have been observed in other cells and tissues 20, 21, 22, 23, 24 (Table 1). Moorehead et al. 21 observed that Egr1 is induced by and required for IGF-II-dependent induction of PTEN in mouse mammary gland, an effect that is absent in Egr1-null mice. Human squamous carcinoma cells SCCTF, can be stimulated to apoptosis by treatment with calyculin A, a potent inducer of apoptosis in several cell lines 24. Treatment of these cells with Calyculin A causes elevation of Egr1 expression as well as its phosphorylation, which is known to increase its transactivation potential 25, 26. This coincides with an elevation of PTEN expression 24. RNA interference specific for the EGR1 gene inhibited not only EGR1 expression but also PTEN expression in these cells. These observations also support the view that EGR1 regulates PTEN expression during the initial steps of the apoptotic pathway. Silencing of Egr1 gene expression has also been revealing in the case of rat follicular thyroid cell transformation 23. RT-PCR experiments performed on human thyroid tumors showed that the absence of EGR1 mRNA is always paralleled by the absence of PTEN mRNA, suggesting that EGR1-dependent mechanisms may play a role in the absence of PTEN gene expression that occurs during thyroid cell transformation 23.
EGR1 protein levels are suppressed or absent in a variety of human tumor cell lines 27, 13 and tumors including breastcarcinoma 27, glioblastoma 14, 15, lung cancer 28, 29, and lymphoma 30. Moreover, in the case of human non-small-cell lung cancer, Ferraro et al. (2005) examined an extensive series of samples from over 125 patients and observed that Egr1 expression predicts both PTEN level and survival to a high degree of significance. Low levels of Egr1 expression are associated with poor outcome and may have tumors resistant to therapy secondary to loss of pathways such as PTEN. It would be interesting to determine if low EGR1 expression is correlated with decreased PTEN expression in these cancer cells.
p53Family: p53
Egr1 directly induces the transcription of p53 and may also bind with p53 in a cytoplasmic complex, although the significance of this interaction is not clear 31. The stability of p53 is under extensive and complex regulation largely owing to intricate cytoplasmic and nuclear interactions especially with Mdm2 and p14ARF family members and related proteins (Figure 2) (reviewed in references 33–34). Thus, significant regulation of p53 expression by direct induction of transcription may be unusual. Direct binding of the p53 promoter by Egr1 was first shown in human melanoma A375-C6 cells during thapsigargin-induced apoptosis 32. Studies of the Egr1-null mouse system have revealed that Egr1 normally controls p53 expression leading to cell cycle arrest, senescence, and - for cells that survive “crisis”-transformation. Freshly prepared primary fibroblasts (MEFs) from Egr1-null embryos exhibit a surprising phenotype characterized by rapid and apparent immortal growth with no hint of replication-dependent growth arrest characteristic of MEFs from wild type mice33. These cells express little or no p53 although the gene is intact. Thus, the Egr1-null cells were found to be insensitive to genotoxic stress and do not arrest following DNA damage as wild type MEFs do. The properties of replicative senescence and DNA damage-induced growth arrest can be rescued by infection of the Egr-1-null MEFs with a retrovirus expressing either p53 or Egr1. However the Egr1-expressing retrovirus is ineffective at restoring replicative senescence or the DNA damage response in p53-null MEFs, indicating that Egr1 acts upstream of p53. Expression of Egr1 in Egr1-null MEFs is indeed accompanied by increased p53 protein. These studies indicate that Egr1 is a key regulator of p53. Lack of Egr1 in MEFs leads to suppression of key p53-dependent growth regulation mechanisms including replicative senescence and DNA damage-induced growth arrest. Thus, regulation of p53 expression by Egr1 plays a central functional role in p53-dependent growth control in the mouse 34.
Figure 2.
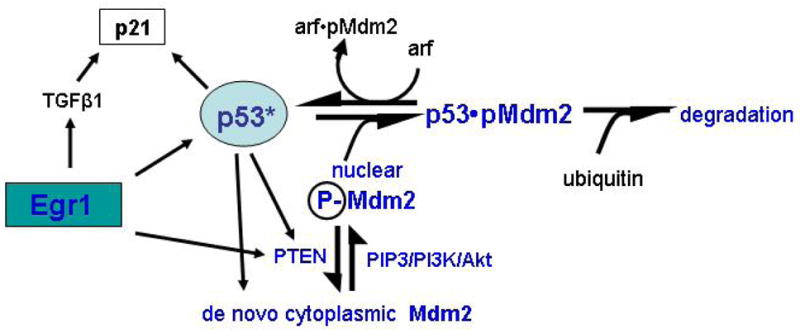
Interactions between the two known Egr1 target genes, PTEN and p53. p53 controls the expression of PTEN and MdM2 which in turns binds to and limits the amount of active p53. p53 controls the expression of the cell cycle inhibitor p21 which also regulated by Egr1 through direct induction of TGFβ1. Thus, Egr1, TGFβ1, and p21 work to suppress cell cycling while the Egr1/PTEN path limits the role of p53.
Consistent results have been observed in a variety of human cell and tissue systems (Table 1). For example, p53 is mutated in the majority of fresh human glioblastoma and these tumors exhibit near normal levels of Egr1 14. Conversely, in the minority of cases with wild type p53, Egr1 mRNA and protein is significantly suppressed or absent 14 suggesting that progression of human glioblastoma is associated with selection for either silencing of Egr1 expression or inactivating mutations of p53 but not both.
The mechanistic basis for these finding has recently be suggested Krones-Herzig, Mittal et al. 2005 35. The mouse and human promoters of p53 both contain at least two sequences consistent with Egr-1 binding sites. In the case of mouse, two sites have been shown to bind nuclear extracts of wild type Egr1 cells but not from extracts of Egr1 null MEFs. Antibody and oligonucleotide studies confirm the specificity of binding of these sites by Egr1. Moreover, chromatin immunoprecipitation of Egr1-DNA complexes from living wild type but not null MEFs and human tumor cells treated with Egr1 inducers but not from untreated cells have been isolated indicating that Egr1 is an in vivo regulator of p53 in mouse and human. Expression analysis of the wild type MEFS reveals significantly increased transcript levels of p53 and nine known target genes of p53 compared to Egr1 null cells. Many additional transcript increases and decreases also were observed of which 143 were known genes with the same significance level as for the p53-regulated target genes. Using a bioinformatics approach, it was found that 66 of these occur in the reported literature in correlation with changes in Egr1. Program assisted pathway construction showed that all of these informative observations could be accommodated on a single regulatory pathway consisting of only four branch points. In addition to p53, TGFβ1, IGF-1 and IL-6 form branch points leading to up or down regulation of all informative significant transcript changes of the expression analysis. The experimental observations of the direct regulation of p53 in MEFs by Egr1 together with the consistency of the expression analysis with the available literature supports the concept that Egr1 is a functional regulator of p53 and p53-regulated genes in mouse MEFs.
p53 Family: p73 and p63
Several of the Egr1-dependent properties of p53 are reflected in the functions of p73, a member of the p53 family which undergoes multiple splicing effects producing multiple isoforms. The regulation of p73 at the transcriptional level is controlled by two promoters that produce two main forms; one is named TAp73 because it contains the transactivating domain (TA) that gives it activities similar to those of p53. The other promoter regulates the expression of a set of isoforms known as ΔNp73, which lack the same transactivating domain and therefore generates effects distinct from those of p53. In fact, many of the roles of ΔNp73 are oncogenic, which is possibly related to a distinct set of target genes that can be transcriptionally regulated 36.
Ding et al. 37 noticed that there were putative Egr1 binding sites in the p73 promoter and later it was shown that Egr1 can upregulate the p73 gene to induce the apoptosis of neuroblastoma cells 38. Kartasheva et al. 39 noted that ΔNp73 can also regulate the expression of a variety of genes independently of p53 and that Egr1 is an important player in this effect. Our recent results in this regard (unpublished data of Jianxiu Yu and E.D. Adamson) indicate that a complicated set of feedback loops come into play between the p53 and p73 promoters and Egr1, further underlying the importance of a network of stress response genes that induce growth arrest, stress response and apoptosis. The p63 family of gene products that are related to p53 may also play a role but they appear not to be regulated by Egr1. The p63 and p73 genes are infrequently mutated in cancers and this is also true for Egr1. Therefore TAp73 and TAp63 could act in place of p53 in p53-null cancer cells when an apoptosis response is required to remove damaged cells.
Egr1 is a suppressor in vivo
Egr1 null mice are prone to induction of skin tumors Krones-Herzig, Mittal et al. 2005 35. In these studies wild type and Egr1 null mice were challenged with the two-step skin carcinogenesis model by treatment of skin with the mutagen DMBA (7,12-dimethylbenzanthracene) followed by treatment with the tumor promoter tetraphorbol acetic acid. Wild type mice developed tumors on average 11 weeks after induction whereas mice treated with either agent alone or vehicle whether wild type, null or heterozygous for the Egr1 gene did not develop tumors. In contrast, Egr1 null mice exposed to the complete protocol uniformly developed tumors with an average time of appearance of 6 weeks – a significant difference and a time when no tumors were apparent in the wild type control mice Krones-Herzig, Mittal et al. 2005 35.
The suppressor network concept
PTEN-Mdm2 interactions
A major regulator of cellular levels of active p53 is the formation of p53-protein complexes in the nucleus involving the mouse double minute2 gene product (Mdm2) or the human homolog, the p53-binding protein. Mdm2 is an oncoprotein that leads to sequestration of active p53 and also, owing to the ligase activity of Mdm2, ubiquitination and proteosomal degradation thereby further limiting the amount of active p53 (reviewed in refs. 40–41) (Figure 2). Recent observations show that phosphoinositide 3-kinase (PtdIns 3-kinase, PI3K)-Akt signaling promotes the phosphorylation and translocation of the Mdm2 oncoprotein into the nucleus thus facilitating the downregulation of p53 40, 41. Donner and coworkers observed that PTEN inhibits activation of Akt by dephosphorylation of the PtdIns 3-kinase activator phospho-inositol triphosphate (PIP3) hence restricting Mdm2 to the cytoplasm in human U87 glioblastoma cells 40, 41. Restriction of Mdm2 to the cytoplasm preserves p53 function and promotes growth arrest and apoptosis of the cancer cells following DNA-damaging chemotherapy. It was suggested that there is a direct connection between these two major tumor suppressors that act together to respond to stresses and transformation. Moreover, as shown here, both p53 and PTEN are direct targets of the Egr1 transcription factor in a variety of cell types.
p53-PTEN interactions
Another example of network effects is illustrated by the observation that p53 itself is a strong transactivator of the PTEN promoter 42. This induction is augmented by Egr1 (unpublished data of I. de Belle and E. D. Adamson). Thus the crosstalk between PTEN and p53 is expected to be favored by an increase in Egr1 and disrupted by suppression of Egr1 – a common circumstance in many human cancers. The possibility that Egr1-deficiency has effects in vivo has been shown by the analysis of Egr1-null mice. In Egr1-null mice, the level of PTEN expression is reduced in all tissues except thymus and is uninducible even in the thymus 19. This observation provides strong support for the thesis that deficiencies of Egr1 expression in vivo may have consequences on the tumor suppressor function of PTEN in vivo.
PTEN and TGFβ1/fibronectin compete for regulation of apoptosis
At least two additional Egr1 target genes, fibronectin and TGFβ1, display downstream signaling activities that potentially interact with the p53-PTEN network. Fibronectin, by virtue of its binding and activation of specific integrins such as β1 is a major mediator of cell attachment, activation of focal adhesion kinase (FAK) and cell survival 18. Integrin binding leads to activation of the antiapoptotic pathway Akt via activation of FAK 18. As described earlier, expression of Egr1 in HT1080 human fibrosarcoma cells results in increased secretion of functional fibronectin 7, 13 and activation of FAK 43. Activated Akt is a major modulator of apoptosis at several levels (reviewed in refs. 18 and 46) (Figure 1). It phosphorylates and inactivates the proapoptotic factors BAD and Caspase 8. Akt prevents the release of cytochrome c from mitochondria and inactivates the forkhead transcription factors (FKLR) which are known for their proapoptotic effects. Moreover, Akt mediates integrin-induced activation of the antiapoptotic factor Bcl-2. Thus, activation of Akt constitutes an important cell survival mechanism. Concomitantly the activity of Akt is opposed by the action of PTEN. These influences establish a critical balance (Figure 1). In the absence of PTEN, any alteration that give rise to autonomous activation of the integrin/FAK/Akt pathway may favor the survival of unattached cells. In the presence of PTEN, the Akt survival pathway is opposed by the PTEN-catalyzed hydrolysis of PIP3 thereby increasing apoptosis of unattached cells, a phenomenon termed “anoikis” (Gr., oikos, home; anoikos, homelessness) by Frisch and Ruoslahti 18,44. This mechanism protects normal cells from the emergence of anchorage-independent populations. Cells that gain autonomous Akt activity such as occurs upon LOH, or complete deletion or inactivating mutations of PTEN may escape this mechanism. Indeed genetically altered mice that are heterozygous for inactivating mutations of PTEN exhibit constitutively elevated Akt activity in a variety of tumors including thyroid, endometrium, breast, and prostate carcinomas as well as T-cell lymphomas 45. We propose that Egr1, by inducing the expression of functional PTEN as well as functional fibronectin and other attachment factors such as PAI-1, contributes to the maintenance of the critical balance of survival vs. apoptosis determined by Akt activity (Figure 1).
TGFβ1 interacts at several levels
The TGFβ1 signaling pathway also interacts with the network. First, as noted, TGFβ1-induced PAI-1 is secreted and functions to facilitate cell attachment 13. Second, several growth inhibitors such as p27Kip1Cip1 46, 47, 48 and p21Cip1/Waf1 49, 50, 51 are regulated by the TGFβ1 signal transduction pathway. p21Waf1 is also under the direct regulation of p53 indicating a potential cooperation between Egr1-induced p53 and Egr1-induced TGFβ1 in controlling cell growth. In certain cell types such as HT1080 52 fibronectin is known to be a direct target gene of the TGFβ1 signal transduction pathway thereby cooperating with Egr1-induced fibronectin. Thus, Egr1-induced TGFβ1 may lead to several cooperative interactions with tumor suppressor networks (Figure 1).
In summary, recent studies indicate that Egr1 is a direct regulator of at least four major suppressors: TGFβ1, PTEN, p53 family members, and fibronectin. These factors have several overlapping functions suggesting that cooperative interactions occur that favor the maintenance of the normal cell phenotype and work to eliminate the emergence of transformed cells. The combined direct regulatory effects of Egr1 and the potential downstream effects are summarized in Figure 3.
Figure 3.
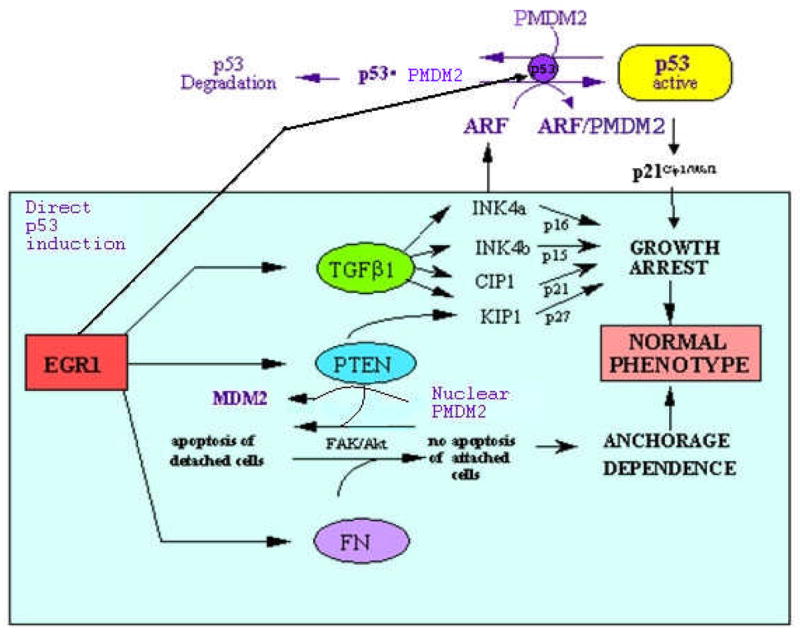
Summary of the overall network of suppression factors that are under the direct control of Egr-1: TGFβ1, PTEN, p53 and fibronectin. The solid arrows from Egr1 indicate regulation of target genes by direct promoter binding known in one or more human cell types to be under the direct regulation by Egr1. The subsequent downstream interactions have been observed in one or more of these systems except the potential roles of the Inks, Akt, and Kip1 which are taken from known interactions in other systems.
Egr1-induced oncogenesis of prostate cancer
Consistent with a role as a tumor suppressor, deletion of the Egr1-containing 5q31 region has been associated with other malignant conditions such lymphoma of Bloom’s Syndrome 30 and small cell lung carcinoma (SCLC) 28. Decreased or absent Egr1 protein expression occurs in NSCLC 29 and glioblastoma 14. The observations indicate that Egr1 expression is commonly suppressed in tumors as has been observed in cells lines 27, 13.
Paradoxically, increased expression of Egr1 is consistently observed in prostate cancer where growing evidence indicates that Egr1 is oncogenic. Russell and coworkers 53 observed that Egr1 mRNA levels are elevated in 12 of 12 organ confined prostate cancer but not in breast or ovarian cancers, or in rapidly dividing rat ventral prostate cells. Egr1 mRNA was detected in epithelial and stromal cells at tumor margins but not in lymph node metastases. The correlation was extended to protein expression. In addition, Egr1 expression was significantly increased in tumors with Gleason scores of 8–10 54. The protein NAB2 (NGF-1A binding protein), which represses the transcriptional activity of Egr1, is down-regulated in primary prostate carcinomas 55. Thus, both up-regulation of Egr1 and loss of its repressor NAB2 may contribute to increased levels of Egr1 activity in human prostate cancer. The functional effects were demonstrated by Milbrandt and coworkers who examined the TRAMP mouse model of prostate cancer following crossing these mice with Egr1-null mice 56. TRAMP mice spontaneously develop prostate cancer owing to the expression of the SV40 T-antigen under the control of a prostate specific probasin promoter. Due to SV40 T-antigen expression, it is likely that p53 is inactivated. Tumor initiation and tumor growth rate were not affected by the lack of Egr1; however, Egr1 deficiency significantly delayed the progression from prostatic intra-epithelial neoplasia to invasive carcinoma. Further evidence for a role of Egr-1 in oncogenesis was provided by experiments using specific high-affinity Egr1 antisense oligonucleotides 57, 58, 59. It was shown that in a series of mouse and human prostate cancer cells Egr1 antisense inhibited cell proliferation, colony formation and growth in soft agar, which are hallmarks of transformed cells in vitro59, 60. Thus, Egr1 antisense reversed the transformed phenotype of the cancer cells. Moreover, TRAMP mice have been treated systemically with antisense Egr1 or control oligonucleotide by I.P. injection every other day from 10 weeks of age – a time when tumors are established – to 20 weeks of age. The antisense oligonucleotides reduced Egr1 protein expression in vivo and significantly reduced the frequency and delayed the occurrence of prostate tumors 59, 60. The observations strongly indicate a unique role for Egr1 in regulating the transition from localized, carcinoma in situ to invasive carcinoma.
The basis of the oncogenic role of Egr1 in prostate cancer is not known in detail. Two studies have suggested possible Egr1 target genes in prostate cancer cells. Expression analysis performed on prostate cancer cells LAPC4 where Egr1 overexpression was driven by adenovirus-mediated transfection identified a number of growth factors, such as IGF-II, PDGF-A and TGFβ1 61. Interestingly, several genes identified in this screen are associated with neuroendocrine differentiation, which is known to occur frequently in murine prostate cancer. Another Affymetrix microarray analysis compared mouse prostate cancer cells TRAMP-C2 in the presence or absence of Egr1 antisense 62. A total of 960 genes were regulated by endogenous Egr1 and several new targets were identified. Some of these play important roles in cell proliferation or apoptosis, such as cyclin D2, p19INK, Fas and TGFβ1. Direct regulation of the expression of these genes was confirmed by quantitative PCR, western analysis and chromatin immunoprecipitation.
The network concept of tumor suppressors provides additional suggestions for the basis of the oncogenic properties of Egr1 in prostate cancer. For example, in the case of prostate cancer, PTEN is the most commonly altered gene known. The region on chromosome 10q that harbors the PTEN gene is hemizygously deleted in many human cancers, with a frequency reaching 60%–80% in the case of prostate cancer 63, 64, 65. Interestingly, even in prostate cancer cells that display PTEN normally, Egr1 was unable to regulate PTEN expression 59 (and our unpublished results). This may be because of hypermethylation of PTEN promoter, which prevents Egr-1 from binding (our unpublished results). PTEN hypermethylation occurs in about 50% of prostate cancer cases 66. Thus, in prostate tumors where PTEN is not mutated or deleted, hypermethylation of its promoter may preclude regulation by Egr-1. In addition, p53 is inactivated in up to 25 – 50% of prostate cancer cases 67. Therefore PTEN and/or p53 are inactivated in the majority of prostate cancers. In these cases Egr1 may have an unopposed effect on Akt activation by continued induction of fibronectin. Fibronectin has long been associated with the stimulation of anchorage-independent growth 68. The expression of fibronectin correlates with features of aggression such a metastases in prostate cancer 69, 70 and is a known activator of the Akt pathway in prostate cancer cells 71, 72, 73.
An important feature of the suppressor network in prostate tumors is Egr1-induced TGFβ1 59, 61, 62. TGFβ1 has diverse roles and may have the net effect of promoting the progression of prostate cancer (reviewed in refs. 71–72). TGFβ1 may also facilitate detachment and metastasis 74. An additional important mechanism of tumor growth promotion by TGFβ1 may be via autocrine stimulation of endothelin-1, implicated in prostate tumor progression 75. Thus, it may be speculated that in the absence of PTEN and/or p53, Egr1 becomes an oncogenic agent in prostate cancer via the effects of its target genes fibronectin and TGFβ1. Furthermore, the oncogenic effect of TGFβ is not restricted to prostate cancer. Recently the progression and metastases of breast cancer in the transgenic mouse was shown to be driven by autocrine TGFβ1 secretion especially during the stage when epithelial breast cells are transforming into mesenchymal cell types 76.
While it is of interest that the tumor suppressor network can both accommodate growth suppression and oncogenic role of Egr1, this hypothetical basis for an oncogenic role of Egr1 remains untested. The preponderance of experimental evidence, however, strongly indicates that in formed prostate carcinomas, Egr1 is oncogenic. We suggest that Egr1 is a valid target for suppression in prostate cancer by gene therapy methods.
Targeting Egr1 for gene therapy of prostate cancer
How can one exploit the indications that Egr1 promotes prostate cancer for therapy in view of the arguments that Egr1 is a suppressor factor in normal tissue via regulation of a net work of well known suppressor factors? Two approaches are apparent. First, in the whole animal, temporary suppression of Egr1 in nonprostatic tissue may not be disastrous. For example, Egr1 null mice – unlike PTEN and p53 null mice – do not experience an increased incidence of tumors. This may be related to the fact that other Egr1 family members compensate for the absence of Egr1 77, 78, 79. For example, Egr-4 can compensate for the deficiency of Egr1 which causes failure of LH receptor expression and sterility owing to failure of luteinizing hormone regulation and Leydig cell steroidogenesis. If this compensatory mechanism functions “acutely”, systemic agents that inhibit the expression of Egr1 may be appropriate at least for the relatively short term that therapy would be applied. Effective high affinity antisense agents that block Egr1 expression in the mouse are known 59, 60.
Second localized irradiation therapy may be combined with radiosensitive vectors that express proapoptic agents or Egr1 antisense or iRNA. Intriguingly, this approach may best utilize the Egr1 promoter itself. In a series of studies, Weischselbaum and coworkers have exploited the multiple CG-rich so-called CarG-boxes of the Egr1 promoter to prepare a variety of expression vectors that are induced by irradiation 80 81, 82. For example, the radio-inducible DNA sequences from the CarG elements of the Egr-1 promoter have been cloned upstream of a cDNA encoding TNFα and expressed as an adenoviral construct. Thus, expression of either an anti-Egr1 factor or general proapoptotic factor may be localized by combination with localized (or target directed?) radiotherapy. Moreover, expression by the Ad.Egr-TNF construct may be induced by a variety of clinically important chemotherapeutic agents such as cisplatin and doxorubicin. Resistance of PC-3 human prostate carcinoma to doxorubicin in vivo was reversed by combining doxorubicin with Ad.Egr-TNF and resulted in significant antitumor effects 81. These approaches suggest that Egr1 is a theoretical and practical target for gene therapy of prostate cancer.
Acknowledgments
We acknowledge the support of the USPHS for funding as NIH RO1 67888 (EDA), The U.S. Army Medical Research and Materiel Command DOD, DAMD-17-01-1-0005 and 0165, (EDA), NIH/NCI for UO1 CA084998 (DAM), RO1 CA084107 (DAM) and 1RO1 CA102688 (VB). We thank Connie White for the graphics and formatting.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Liu C, Rangnekar VM, Adamson E, Mercola D. Suppression of growth and transformation and induction of apoptosis by EGR-1. Cancer Gene Ther. 1998;5:3–28. [PubMed] [Google Scholar]
- 2.Akhurst RJ, Balmain A. Genetic events and the role of TGF beta in epithelial tumour progression. J Pathol. 1999;187:82–90. doi: 10.1002/(SICI)1096-9896(199901)187:1<82::AID-PATH248>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 3.Shi H, et al. Oligonucleotide-based microarray for DNA methylation analysis: Principles and applications. J Cell Biochem. 2003;88:138–143. doi: 10.1002/jcb.10313. [DOI] [PubMed] [Google Scholar]
- 4.Dey BR, et al. Repression of the transforming growth factor-beta 1 gene by the Wilms’ tumor suppressor WT1 gene product. Mol Endocrinol. 1994;8:595–602. doi: 10.1210/mend.8.5.8058069. [DOI] [PubMed] [Google Scholar]
- 5.Liu C, Adamson E, Mercola D. Transcription factor EGR-1 suppresses the growth and transformation of human HT-1080 fibrosarcoma cells by induction of transforming growth factor beta 1. Proc Natl Acad Sci U S A. 1996a;93:11831–11836. doi: 10.1073/pnas.93.21.11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Belle I, Mercola D, Adamson ED. Method for cloning in vivo targets of the Egr-1 transcription factor. Biotechniques. 2000;29:162–169. doi: 10.2144/00291rr03. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, et al. The transcription factor EGR-1 suppresses transformation of human fibrosarcoma HT1080 cells by coordinated induction of transforming growth factor-beta1, fibronectin, and plasminogen activator inhibitor-1. J Biol Chem. 1999;274:4400–4411. doi: 10.1074/jbc.274.7.4400. [DOI] [PubMed] [Google Scholar]
- 8.McCaffrey TA, et al. High-level expression of Egr-1 and Egr-1-inducible genes in mouse and human atherosclerosis. J Clin Invest. 2000;105:653–662. doi: 10.1172/JCI8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han DC, Isono M, Hoffman BB, Ziyadeh FN. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: mediation by autocrine activation of TGF- beta. J Am Soc Nephrol. 1999c;10:1891–1899. doi: 10.1681/ASN.V1091891. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura H, et al. Introduction of DNA enzyme for Egr-1 into tubulointerstitial fibroblasts by electroporation reduced interstitial alpha-smooth muscle actin expression and fibrosis in unilateral ureteral obstruction (UUO) rats. Gene Ther. 2002;9:495–502. doi: 10.1038/sj.gt.3301681. [DOI] [PubMed] [Google Scholar]
- 11.Lee CG, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hocevar BA, Howe PH. Analysis of TGF-beta-mediated synthesis of extracellular matrix components. Methods Mol Biol. 2000;142:55–65. doi: 10.1385/1-59259-053-5:55. [DOI] [PubMed] [Google Scholar]
- 13.Liu C, Yao J, Mercola D, Adamson E. The transcription factor EGR-1 directly transactivates the fibronectin gene and enhances attachment of human glioblastoma cell line U251. J Biol Chem. 2000a;275:20315–20323. doi: 10.1074/jbc.M909046199. [DOI] [PubMed] [Google Scholar]
- 14.Calogero A, et al. The early growth response gene EGR-1 behaves as a suppressor gene that is down-regulated independent of ARF/Mdm2 but not p53 alterations in fresh human gliomas. Clin Cancer Res. 2001;7:2788–2796. [PubMed] [Google Scholar]
- 15.Calogero A, et al. Inhibition of cell growth by EGR-1 in human primary cultures from malignant glioma. Cancer Cell Int. 2004;4:1. doi: 10.1186/1475-2867-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akamatsu H, et al. Suppression of transformed phenotypes of human fibrosarcoma cells by overexpression of recombinant fibronectin. Cancer Res. 1996;56:4541–4546. [PubMed] [Google Scholar]
- 17.Liu C, et al. EGR-1, the reluctant suppression factor: EGR-1 is known to function in the regulation of growth, differentiation, and also has significant tumor suppressor activity and a mechanism involving the induction of TGF-beta1 is postulated to account for this suppressor activity. Crit Rev Oncog. 1996b;7:101–125. [PubMed] [Google Scholar]
- 18.Ruoslahti E. Fibronectin and its integrin receptors in cancer. Adv Cancer Res. 1999;76:1–20. doi: 10.1016/s0065-230x(08)60772-1. [DOI] [PubMed] [Google Scholar]
- 19.Virolle T, et al. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol. 2001;3:1124–1128. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 20.Tsugawa K, et al. Abnormal PTEN expression in portal hypertensive gastric mucosa: a key to impaired PI 3-kinase/Akt activation and delayed injury healing? Faseb J. 2003;17:2316–2318. doi: 10.1096/fj.02-1107fje. [DOI] [PubMed] [Google Scholar]
- 21.Moorehead RA, et al. Insulin-like growth factor-II regulates PTEN expression in the mammary gland. J Biol Chem. 2003;278:50422–50427. doi: 10.1074/jbc.M306894200. [DOI] [PubMed] [Google Scholar]
- 22.Han B, et al. Regulation of constitutive expression of mouse PTEN by the 5′-untranslated region. Oncogene. 2003;22:5325–5337. doi: 10.1038/sj.onc.1206783. [DOI] [PubMed] [Google Scholar]
- 23.Tell G, et al. Control of phosphatase and tensin homolog (PTEN) gene expression in normal and neoplastic thyroid cells. Endocrinology. 2004;145:4660–4666. doi: 10.1210/en.2004-0282. [DOI] [PubMed] [Google Scholar]
- 24.Okamura H, Yoshida K, Morimoto H, Haneji T. PTEN expression elicited by EGR-1 transcription factor in calyculin A-induced apoptotic cells. J Cell Biochem. 2005;94:117–125. doi: 10.1002/jcb.20283. [DOI] [PubMed] [Google Scholar]
- 25.Grover-Bardwick A, Adamson E, Mercola D. Transformation-specific pattern of phosphorylation of c-Jun, Jun-B, Jun- D and Egr-1 in v-sis transformed cells. Carcinogenesis. 1994;15:1667–1674. doi: 10.1093/carcin/15.8.1667. [DOI] [PubMed] [Google Scholar]
- 26.Huang RP, Adamson ED. The phosphorylated forms of the transcription factor, Egr-1, bind to DNA more efficiently than non-phosphorylated. Biochem Biophys Res Commun. 1994a;200:1271–1276. doi: 10.1006/bbrc.1994.1588. [DOI] [PubMed] [Google Scholar]
- 27.Huang RP, et al. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int J Cancer. 1997a;72:102–109. doi: 10.1002/(sici)1097-0215(19970703)72:1<102::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 28.Goguel AF, et al. Evolution of chromosomal alterations and biologic features in two small cell lung carcinoma cell lines established from one patient during the course of the disease. Cancer Genet Cytogenet. 1995;80:47–54. doi: 10.1016/0165-4608(94)00154-4. [DOI] [PubMed] [Google Scholar]
- 29.Levin WJ, et al. Expression patterns of immediate early transcription factors in human non-small cell lung cancer. The Lung Cancer Study Group. Oncogene. 1995;11:1261–1269. [PubMed] [Google Scholar]
- 30.Fundia A, Gorla N, Larripa I. Non-random distribution of spontaneous chromosome aberrations in two Bloom Syndrome patients. Hereditas. 1995;122:239–243. doi: 10.1111/j.1601-5223.1995.00239.x. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, et al. Physical interaction between p53 and primary response gene Egr-1. Int J Oncol. 2001b;18:863–870. doi: 10.3892/ijo.18.4.863. [DOI] [PubMed] [Google Scholar]
- 32.Nair P, et al. Early growth response-1-dependent apoptosis is mediated by p53. J Biol Chem. 1997;272:20131–20138. doi: 10.1074/jbc.272.32.20131. [DOI] [PubMed] [Google Scholar]
- 33.Krones-Herzig A, Adamson Eileen, Mercola Dan. “Gatekeeper” of the p53 Tumor Suppressor in Replicative Senescence. Cancer Gene Therapy. 2002;9(12 Suppl 1):S7. (abstract no.”18) 2002: 9(Suppl 1):s7. [Google Scholar]
- 34.Krones-Herzig A, Adamson Eileen, Mercola Dan. EGR1, a novel upstream gatekeeper of the p53 tumor suppressor, controls replicative senescence. Proc Natl Acad Sci U S A. 2003;100:3233–3238. doi: 10.1073/pnas.2628034100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krones-Herzig A, Mittal Shalu, Yule Kelly, Liang Hongyan, English Chris, Urcis Rafael, Soni Tarun, Adamson Eileen D, Mercola Dan. Egr1 acts as a tumor suppressor in vivo and in vitro via regulation of p53. Cancer Research. 2005 doi: 10.1158/0008-5472.CAN-04-3742. in press. [DOI] [PubMed] [Google Scholar]
- 36.Moll UMSN. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–386. [PubMed] [Google Scholar]
- 37.Ding Y, et al. Molecular cloning and functional characterization of the upstream promoter region of the human p73 gene. DNA Res. 1999;6:347–351. doi: 10.1093/dnares/6.5.347. [DOI] [PubMed] [Google Scholar]
- 38.Pignatelli M, et al. The transcription factor early growth response factor-1 (EGR-1) promotes apoptosis of neuroblastoma cells. Biochem J. 2003;373:739–746. doi: 10.1042/BJ20021918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kartasheva NN, et al. DeltaNp73 can modulate the expression of various genes in a p53-independent fashion. Oncogene. 2003;22:8246–8254. doi: 10.1038/sj.onc.1207138. [DOI] [PubMed] [Google Scholar]
- 40.Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002b;27:462–467. doi: 10.1016/s0968-0004(02)02166-7. [DOI] [PubMed] [Google Scholar]
- 41.Mayo LD, et al. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem. 2002a;277:5484–5489. doi: 10.1074/jbc.M108302200. [DOI] [PubMed] [Google Scholar]
- 42.Stambolic V, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 43.de Belle I, et al. p53 and Egr-1 additively suppress transformed growth in HT1080 cells but Egr-1 counteracts p53-dependent apoptosis. Oncogene. 1999;18:3633–3642. doi: 10.1038/sj.onc.1202696. [DOI] [PubMed] [Google Scholar]
- 44.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 45.Kishimoto H, et al. Physiological functions of pten in mouse tissues. Cell Struct Funct. 2003;28:11–21. doi: 10.1247/csf.28.11. [DOI] [PubMed] [Google Scholar]
- 46.Koff A, et al. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- 47.Polyak K, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994a;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 48.Polyak K, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994b;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 49.Datto MB, Yu Y, Wang XF. Functional analysis of the transforming growth factor beta responsive elements in the WAF1/Cip1/p21 promoter. J Biol Chem. 1995b;270:28623–28628. doi: 10.1074/jbc.270.48.28623. [DOI] [PubMed] [Google Scholar]
- 50.Datto MB, et al. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci U S A. 1995a;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rorke EA, et al. TGF-beta-mediated cell cycle arrest of HPV16-immortalized human ectocervical cells correlates with decreased E6/E7 mRNA and increased p53 and p21(WAF-1) expression. Exp Cell Res. 2000;259:149–157. doi: 10.1006/excr.2000.4953. [DOI] [PubMed] [Google Scholar]
- 52.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. Embo J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thigpen AE, et al. Increased expression of early growth response-1 messenger ribonucleic acid in prostatic adenocarcinoma. J Urol. 1996;155:975–981. [PubMed] [Google Scholar]
- 54.Eid MA, et al. Expression of early growth response genes in human prostate cancer. Cancer Res. 1998;58:2461–2468. [PubMed] [Google Scholar]
- 55.Abdulkadir SA, et al. Frequent and early loss of the EGR1 corepressor NAB2 in human prostate carcinoma. Hum Pathol. 2001a;32:935–939. doi: 10.1053/hupa.2001.27102. [DOI] [PubMed] [Google Scholar]
- 56.Abdulkadir SA, et al. Impaired prostate tumorigenesis in Egr1-deficient mice. Nat Med. 2001b;7:101–107. doi: 10.1038/83231. [DOI] [PubMed] [Google Scholar]
- 57.Baron V, De Gregoria Giorgia, Mercola Dan. Toward the Development of High Affinity and Specific Antisense Egr-1. Cancer Gene Therapy. 2001;8:S5. abstract PD-15. [Google Scholar]
- 58.Baron VB, De Gregorio Giorgia, Krones-Herzig Anja, Virolle Thierry, Calogero Antonella, Urcis Rafael, Mercola Dan. INHIBITION OF EGR-1 EXPRESSION RESTRAINS TRANSFORMATION OF PROSTATE CANCER CELLS AND DELAYS CANCER PROGRESSION. Cancer Gene Therapy. 2002;9(Suppl 1):S57. doi: 10.1038/sj.onc.1206560. [DOI] [PubMed] [Google Scholar]
- 59.Baron V, Giorgia De Gregorio, Anja Krones-Herzig, Thierry Virolle, Antonella Calogero, Rafael Urcis, Mercola Dan. Inhibition of Egr-1 Expression Reverses Transformation of Prostate Cancer Cells in vitro AND in vivo. Oncogene. 2003a doi: 10.1038/sj.onc.1206560. in press: in press. [DOI] [PubMed] [Google Scholar]
- 60.Baron V, Duss S, Rhim J, Mercola D. Antisense to the early growth response-1 gene (Egr-1) inhibits prostate tumor development in TRAMP mice. Ann N Y Acad Sci. 2003b;1002:197–216. doi: 10.1196/annals.1281.024. [DOI] [PubMed] [Google Scholar]
- 61.Svaren J, et al. EGR1 target genes in prostate carcinoma cells identified by microarray analysis. J Biol Chem. 2000;275:38524–38531. doi: 10.1074/jbc.M005220200. [DOI] [PubMed] [Google Scholar]
- 62.Virolle T, et al. Egr1 promotes growth and survival of prostate cancer cells. Identification of novel Egr1 target genes. J Biol Chem. 2003;278:11802–11810. doi: 10.1074/jbc.M210279200. [DOI] [PubMed] [Google Scholar]
- 63.Bruckheimer EM, Gjertsen BT, McDonnell TJ. Implications of cell death regulation in the pathogenesis and treatment of prostate cancer. Semin Oncol. 1999;26:382–398. [PubMed] [Google Scholar]
- 64.Dong JT. Chromosomal deletions and tumor suppressor genes in prostate cancer. Cancer Metastasis Rev. 2001;20:173–193. doi: 10.1023/a:1015575125780. [DOI] [PubMed] [Google Scholar]
- 65.Stiles B, et al. PTENless means more. Dev Biol. 2004;273:175–184. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 66.Yea Whang. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 1998;95:5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Downing SR, Russell PJ, Jackson P. Alterations of p53 are common in early stage prostate cancer. Can J Urol. 2003;10:1924–1933. [PubMed] [Google Scholar]
- 68.Chung LW, et al. Human prostate cancer model: roles of growth factors and extracellular matrices. J Cell Biochem Suppl. 1992;16H:99–105. doi: 10.1002/jcb.240501222. [DOI] [PubMed] [Google Scholar]
- 69.Sonmez H, et al. Tissue fibronectin levels of human prostatic cancer, as a tumor marker. Cancer Biochem Biophys. 1995;15:107–110. [PubMed] [Google Scholar]
- 70.Albrecht M, et al. Fibronectin in human prostatic cells in vivo and in vitro: expression, distribution, and pathological significance. Histochem Cell Biol. 1999;112:51–61. doi: 10.1007/s004180050391. [DOI] [PubMed] [Google Scholar]
- 71.Zheng DQ, Woodard AS, Tallini G, Languino LR. Substrate specificity of alpha(v)beta(3) integrin-mediated cell migration and phosphatidylinositol 3-kinase/AKT pathway activation. J Biol Chem. 2000;275:24565–24574. doi: 10.1074/jbc.M002646200. [DOI] [PubMed] [Google Scholar]
- 72.Liu J, et al. Molecular heterogeneity and function of EWS-WT1 fusion transcripts in desmoplastic small round cell tumors. Clin Cancer Res. 2000c;6:3522–3529. [PubMed] [Google Scholar]
- 73.Morgan M, Saba S, Gower W. Fibronectin influences cellular proliferation and apoptosis similarly in LNCaP and PC-3 prostate cancer cell lines. Urol Oncol. 2000;5:155–159. doi: 10.1016/s1078-1439(99)00058-7. [DOI] [PubMed] [Google Scholar]
- 74.Matuo Y, et al. Potential role of HBGF (FGF) and TGF-beta on prostate growth. Adv Exp Med Biol. 1992;324:107–114. doi: 10.1007/978-1-4615-3398-6_11. [DOI] [PubMed] [Google Scholar]
- 75.Le Brun G, et al. Upregulation of endothelin 1 and its precursor by IL-1beta, TNF-alpha, and TGF-beta in the PC3 human prostate cancer cell line. Cytokine. 1999;11:157–162. doi: 10.1006/cyto.1998.0407. [DOI] [PubMed] [Google Scholar]
- 76.Muraoka-Cook RS, et al. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004;64:9002–9011. doi: 10.1158/0008-5472.CAN-04-2111. [DOI] [PubMed] [Google Scholar]
- 77.Lee SL, et al. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1) Science. 1996;273:1219–1221. doi: 10.1126/science.273.5279.1219. [DOI] [PubMed] [Google Scholar]
- 78.Tourtellotte WG, et al. Infertility associated with incomplete spermatogenic arrest and oligozoospermia in Egr4-deficient mice. Development. 1999;126:5061–5071. doi: 10.1242/dev.126.22.5061. [DOI] [PubMed] [Google Scholar]
- 79.Tourtellotte WG, Nagarajan R, Bartke A, Milbrandt J. Functional compensation by Egr4 in Egr1-dependent luteinizing hormone regulation and Leydig cell steroidogenesis. Mol Cell Biol. 2000;20:5261–5268. doi: 10.1128/mcb.20.14.5261-5268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weichselbaum RR, Kufe DW, Advani SJ, Roizman B. Molecular targeting of gene therapy and radiotherapy. Acta Oncol. 2001;40:735–738. doi: 10.1080/02841860152619151. [DOI] [PubMed] [Google Scholar]
- 81.Lopez CA, et al. Chemoinducible gene therapy: a strategy to enhance doxorubicin antitumor activity. Mol Cancer Ther. 2004;3:1167–1175. [PubMed] [Google Scholar]
- 82.Greco O, et al. Gene therapy vectors containing CArG elements from the Egr1 gene are activated by neutron irradiation, cisplatin and doxorubicin. Cancer Gene Ther. 2005 doi: 10.1038/sj.cgt.7700834. [DOI] [PubMed] [Google Scholar]
- 83.Fu M, et al. Early stimulation and late inhibition of peroxisome proliferator-activated receptor gamma (PPAR gamma) gene expression by transforming growth factor beta in human aortic smooth muscle cells: role of early growth-response factor-1 (Egr-1), activator protein 1 (AP1) and Smads. Biochem J. 2003;370:1019–1025. doi: 10.1042/BJ20021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sho E, et al. Arterial enlargement in response to high flow requires early expression of matrix metalloproteinases to degrade extracellular matrix. Exp Mol Pathol. 2002;73:142–153. doi: 10.1006/exmp.2002.2457. [DOI] [PubMed] [Google Scholar]
- 85.Kane S, et al. Differential induction of early response genes by adrenomedullin and transforming growth factor-beta1 in human lung cancer cells. Anticancer Res. 2002;22:1433–1444. [PubMed] [Google Scholar]
- 86.McKay S, de Jongste JC, Saxena PR, Sharma HS. Angiotensin II induces hypertrophy of human airway smooth muscle cells: expression of transcription factors and transforming growth factor- beta1. Am J Respir Cell Mol Biol. 1998;18:823–833. doi: 10.1165/ajrcmb.18.6.2924. [DOI] [PubMed] [Google Scholar]
- 87.Huang RP, et al. Egr-1 inhibits apoptosis during the UV response: correlation of cell survival with Egr-1 phosphorylation. Cell Death Differ. 1998;5:96–106. doi: 10.1038/sj.cdd.4400322. [DOI] [PubMed] [Google Scholar]
- 88.Krones-Herzig A, Adamson E, Mercola Dan. Gene Chip Analyses with Egr-1 Knockout MEF Reveal Mediators of Growth Suppression by Egr-1. Cancer Gene Therapy. 2001;8:S7. (abstract PD-23)(unsolicited award: AACR-AFLAC Scholar in Cancer Research award to AKH, 12/2001) [Google Scholar]
- 89.Han SS, et al. CpG oligodeoxynucleotides rescue BKS-2 immature B cell lymphoma from anti-IgM-mediated growth inhibition by up-regulation of egr-1. Int Immunol. 1999a;11:871–879. doi: 10.1093/intimm/11.6.871. [DOI] [PubMed] [Google Scholar]
- 90.Ahmed MM, et al. EGR-1 induction is required for maximal radiosensitivity in A375-C6 melanoma cells. J Biol Chem. 1996;271:29231–29237. doi: 10.1074/jbc.271.46.29231. [DOI] [PubMed] [Google Scholar]
- 91.Huang RP, Adamson ED. A biological role for Egr-1 in cell survival following ultraviolet irradiation. Oncogene. 1995;10:467–475. [PubMed] [Google Scholar]