

Abstract
Pancreatic adenocarcinoma is characterized by desmoplasia, local invasion, and metastasis. These features are regulated in part by MMP9 and SPARC. To explore the interaction of SPARC and MMP9 in cancer, we first established orthotopic pancreatic tumors in SPARC-null and wild-type mice with the murine pancreatic adenocarcinoma cell line, PAN02. MMP9 expression was higher in tumors from wild-type compared to SPARC-null mice. Coincident with lower MMP9 expression, tumors grown in SPARC-null mice were significantly larger, had decreased ECM deposition and reduced microvessel density compared to wild-type controls. In addition, metastasis was enhanced in the absence of host SPARC. Therefore, we next analyzed the orthotopic tumor growth of PAN02 cells transduced with MMP9 or a control empty vector. Forced expression of MMP9 by the PAN02 cells resulted in larger tumors in both wild-type and SPARC-null animals compared to empty vector controls and further diminished ECM deposition. Importantly, forced expression of MMP9 within the tumor reversed the decrease in angiogenesis and abrogated the metastatic potential displayed by control tumors grown in SPARC-null mice. Finally, contrary to the in vivo results, MMP9 increased cell migration in vitro, which was blocked by the addition of SPARC. These results suggest that SPARC and MMP9 interact to regulate many stages of tumor progression including ECM deposition, angiogenesis and metastasis.
Keywords: SPARC, MMP9, tumor microenvironment, ECM, pancreatic, metastasis
Introduction
SPARC is a highly conserved multifunctional glycoprotein that belongs to the matricellular class of proteins. Matricellular proteins function as adaptors that mediate cell-extracellular matrix (ECM) interactions (1) and are expressed in tissues undergoing repair or remodeling. For example, SPARC is expressed in healing wounds, areas of bone morphogenesis, developing embryonic tissue, as well as, sites of angiogenesis (2). Consistent with its function as a mediator of tissue remodeling, SPARC regulates the expression of proteins involved in ECM turnover and formation including collagens and matrix metalloproteinases (MMPs) (3). SPARC can also directly affect endothelial cell behavior by regulating proliferation, cell shape and response to different angiogenic growth factors including transforming growth factor (TGF-β), fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) (1, 4, 5).
MMPs are zinc-dependent endopeptidases that contribute to proteolytic events important for homeostasis, tissue remodeling and cancer progression (6). The targets of MMPs have traditionally been considered structural components of the ECM, although it is now clear that these enzymes have a broader array of substrates (7). For instance, MMP9 triggers the angiogenic switch by releasing ECM-associated VEGF, thus influencing subsequent tumor growth (8). MMPs may also influence tumor progression by facilitating events pivotal for neovascularization and establishment of distant metastasis including proliferation, survival and migration of endothelial, tumor and stromal cells, (9, 10).
SPARC expression is altered in many cancers. There is increased expression of SPARC in melanoma, glioma, colorectal, and breast carcinomas compared to their respective normal tissues (11). In these tumors, high levels of SPARC often correlate with enhanced invasion and metastasis (11). In contrast, increased expression of SPARC by ovarian carcinoma cells led to increased tumor cell apoptosis and correlated inversely with tumor progression in vivo (12, 13). In pancreatic tumors, expression of SPARC by cancer cells is limited due to promoter hypermethylation, while infiltrating stromal cells express increased levels of SPARC (14). Thus, the context of SPARC expression in the microenvironment is critical for understanding its influence on tumor growth and progression. Whether produced by tumor or stromal cells, SPARC protein is often found at tumor-stromal interfaces, tumor capsules, areas of desmoplasia and areas of angiogenesis and vascular remodeling (15). Given these findings, SPARC is well-placed to participate in the host response to tumor growth.
We showed previously that ectopic tumor growth is enhanced significantly in SPARC-null (SPARC-/-) mice compared to wild-type (WT) animals. These studies demonstrate that ECM deposition within and around the tumor is altered in the absence of host-derived SPARC and further suggest that SPARC is important in the host response to an implanted tumor (16, 17). In this context, it has been proposed that SPARC influences ECM production, deposition and function, in part, by modulating proteases or their inhibitors. In fact, SPARC has been shown to induce the expression of MMP9 in macrophages/monocytes, a predominant source of MMP9 in the tumor microenvironment (18). SPARC is also a substrate for MMP activity. Cleavage of SPARC between L196-L197 or L197-L198 by MMPs increases the affinity of SPARC for collagen I and IV (19, 20). Sage et al. (21) showed that SPARC can be cleaved into at least three bioactive polypeptides by MMP3. One of the polypeptides (designated Z2) has L198 at its COOH-terminus and promoted the migration of endothelial cells, but inhibited cell proliferation in vitro.
To explore the function of SPARC and MMP9 in the tumor microenvironment, we established orthotopic pancreatic tumors in WT and SPARC-/- mice with a mouse pancreatic adenocarcinoma cell line, PAN02, which was engineered to express MMP9. This study demonstrates that forced expression of MMP9 by PAN02 cells not only enhances primary tumor growth in WT and SPARC-/- mice, but also rescues angiogenesis and abrogates metastasis in tumors grown in SPARC-/- mice. These results demonstrate that SPARC and MMP9 interact to influence tumorigenesis by affecting angiogenesis, ECM deposition and degradation, and metastatic progression.
Materials and Methods
Tissue Culture
The murine pancreatic adenocarcinoma cell line (PAN02) was purchased from the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Frederick, MD), and grown in Dulbecco's Modified Eagles Medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with L-glutamine (2 mM), penicillin G (100 units/ml), streptomycin sulfate (100 μg/ml), and 5% fetal bovine serum (Life Technologies, Grand Island, NY). The PAN02 cell line was tested (Impact III PCR profiles; MU Research Animal Diagnostic Laboratory, Columbia, MO) and was found to be pathogen-free.
Recombinant MMP9 Chimeras and Cell Transduction
MMP9 was subcloned into the bicistronic plasmid pRV-IRES-GFP to obtain a recombinant retrovirus that was used to transduce PAN02 cells (22). Green fluorescent protein (GFP)-expressing cells were selected by fluorescence-activated cell sorting.
Cell Fractionation
Transduced cells were disrupted with a 30-gauge needle in 50 mM Tris, pH 7.5 in the presence of protease inhibitors. After centrifugation at 800 × g (10 min, 4°C) to remove nuclei, membrane and cytosolic fractions were obtained by centrifugation at 166,000 × g (1 h, 4°C). To verify the purity of the cell fractions, normalized protein amounts from each fraction were analyzed with anti-transferrin receptor (TfR; Zymed, San Francisco, CA) and anti-tubulin (Sigma, St. Louis, MO) by western blot. An anti-MMP9 polyclonal antibody (Calbiochem, San Diego, CA) and anti-SPARC antibody (R&D Systems, Minneapolis, MN) were employed to detect exogenous MMP9 and endogenous SPARC expression by Western blot analysis.
Immunocytochemistry
Transduced PAN02 cells were plated on chamber slides (BD Biosciences, San Diego, CA) coated with collagen I or IV. Cells were fixed with 3.7% paraformaldehyde (15 min on ice) and afterwards incubated (1 h, 4°C) with a rabbit anti-MMP9 polyclonal antibody (Calbiochem) or mouse anti-SPARC monoclonal antibody (303) (23) followed by Cy3-labeled anti-rabbit or anti-mouse antibody respectively (Jackson ImmunoResearch, West Grove, PA; 1 h; 4°C). Slides were mounted with Vectashield (Vector Laboratories, Burlingame, CA) and analyzed by confocal laser microscopy (Leica).
MMP9 Activity Assay
MMP9 activity was analyzed in cell extracts, serum samples and tumor extracts by gelatin zymography as described (24). For in situ zymography, cells plated on collagen I were incubated with 40 μg/ml of FITC-gelatin (DQ-Gelatin, Molecular Probes, Eugene, OR) at 37°C. After 1 h of incubation, cells were washed with PBS, fixed with 1% paraformaldehyde, incubated for 10 min at room temperature with To-Pro3 (Invitrogen) and analyzed by confocal laser microscopy. MMP9 activity of PAN02 cells over time was quantified by measuring the fluorescence released by cleavage of DQ-gelatin in multi-well format. Cells were incubated with 100 μg/ml of FITC-gelatin in 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM CaCl2 and 0.2 mM NaN3 for the indicated time (3, 5, 7, 22, or 27 hrs) at 37°C. Values represent the fluorescence increment after background subtraction.
Orthotopic Tumor Model
SPARC-/- mice were generated as described previously (25). The mice were housed in a pathogen-free facility and experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee of UT Southwestern Medical Center (Dallas, TX).
For injections, PAN02 cells either expressing MMP9 or the empty vector were harvested from subconfluent cultures (>90% viable), washed once in serum-free medium and resuspended in Hank's balanced salt solution (HBSS). Orthotopic tumor cell injection was carried out as described previously (26). Briefly, the mice were anesthetized with isoflurane and an incision was made in the left abdomen to expose the spleen and attached pancreas. Tumor cells (5×105 in 50 μl) were injected into the tail of the pancreas. Post-injection, mice were monitored for weight, signs of discomfort or morbidity and tumor size. Mice were euthanized thirty-five days after tumor cell injection and visually screened for extent of macroscopic tumor burden. The liver, entire gastrointestinal tract, lungs, mesentery, and other internal organs were visually inspected for the presence of surface metastases. Enlarged lymph nodes were counted and a subset was surveyed by H&E histology to confirm tumor burden. The entire pancreas, containing the tumor, was harvested and weighed. Half of the tissue was snap-frozen in liquid nitrogen while the other half was fixed in methyl Carnoys, paraffin-embedded, sectioned and stained with H&E or Masson's trichrome (Molecular Histopathology Laboratory at UT Southwestern Medical Center, Dallas, TX). Two independent experiments with 6 mice/group were performed.
Immunohistochemistry
Tissue sections were deparaffinized in a series of xylene and ethanol washes, then rehydrated in PBSt (0.2% Tween-20). Primary antibodies used were rabbit anti-collagen I (LF67, provided by Dr. Larry Fisher, NIH/NIDCR Matrix Biology Unit) (27, 28), rabbit anti-collagen type IV (BD Biosciences), rabbit anti-phosphorylated histone H3 (phospho-H3, Upstate Biotechnology, Inc., Lake Placid, NY), rat anti-mouse Gr-1 (clone RB6-8C5, Biolegend, San Diego, CA), rabbit anti full length MMP9 (Millipore, Billerica, MA), and rat anti-mouse endothelial cell, MECA-32 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) (29). For sections developed with diaminobenzidine (DAB) (Research Genetics, Huntsville, AL), endogenous peroxidases were blocked by incubating the samples in a 3% H2O2/methanol solution. Sections were then washed in PBSt and blocked in 20% AquaBlock (East Coast Biologics, Inc., North Berwick, ME). Incubation with primary antibodies was performed overnight at 4°C. Sections were washed in PBSt and incubated with peroxidase-conjugated secondary antibody for 1 h at room temperature. Lastly, sections were washed in PBSt, incubated with stable DAB for 5-20 minutes, counterstained with Meyer's Hematoxylin solution for 3 min and mounted in Permount (Fisher Scientific, Pittsburgh, PA). Alternatively, for fluorescence detection, following primary antibody incubation, sections were incubated for 1 h with fluorophore-conjugated (FITC or Cy3) secondary antibodies (Jackson Immunoresearch), washed in PBSt, and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen).
Tissue sections were analyzed with a Nikon Eclipse E600 microscope (Nikon, Lewisville, TX). DAB images were captured with a Nikon Digital Dx1200me camera using Act1 software (Universal Imaging Corporation, Downington, PA) while fluorescence images were captured with a Photometric Coolsnap HQ camera and MetaMorph software (Universal Imaging Corporation). Phospho-H3 and TUNEL staining were quantified by manually counting the number of positive cells in five random 400X fields per tumor section. MECA-32 and collagen IV staining were processed, analyzed and quantified using MetaMorph software. Fluorescent images were captured under identical conditions (exposure time, high and low limits, and scaling). Images were thresholded to exclude background signal from secondary antibody alone. Mean blood vessel counts, blood vessel area and blood vessel perimeter were measured using MetaMorph's “Integrated Morphometry Analysis” (IMA).
In Vitro Proliferation Assay
For the in vitro proliferation assay, PAN02 cells were plated at 27,000 cells per well in triplicate in a 24-well plate in DMEM containing 5% FBS. Cells were incubated at 37°C and counted at 24, 48 and 72 hrs after seeding. The total number of viable cells was determined by trypan blue exclusion and hand-counting cell density on a hemacytometer.
Migration Assay
The in vitro migration assays were performed using 8 μm transwell inserts formatted for 24 well tissue culture plates (Falcon). Wells were filled with 500 μl of serum-free DMEM +/- 10 μg rhSPARC. PAN02 cells (20,000 cells/well) were seeded onto the upper chamber of each transwell in serum-free DMEM. Cells were incubated at 37°C. After 24 h, cells were removed from the upper chamber and those cells that had migrated to the lower side of the membrane were fixed in 100% cold methanol and stained with hematoxylin (Sigma). The membranes were then cut from the transwell and mounted in crystal mount (Biomeda Corp. Foster City, CA). Cells were counted in five 100× fields on each membrane.
Intravasation Assay
For intravasation assays, PAN02 cells (1 × 106) were inoculated into chorioallantoic membranes (CAM) of 9-day-old chick embryos (30, 31). After 48 hrs incubation, CAMs were removed and total DNA isolated. The estimated percentage of intravasated cells was calculated by amplifying mouse B1 repeats in a quantitative real-time PCR (30). Data represents the estimated percentage of intravasated cells in CAM samples compared with a standard curve made with known concentrations of mouse DNA.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA) with assistance from The Center for Biostatistics and Clinical Sciences (UT-Southwestern Medical Center, Dallas, TX). Analyses include Mann-Whitney test, Kruskal-Wallis with Dunn multiple comparison results, Student's t test, or one way ANOVA with Tukey's multiple comparison test (32) where appropriate. Significance (p<0.05) was determined with 95% confidence.
Results
Orthotopic tumor growth is enhanced in the absence of host SPARC
We showed previously that PAN02 tumors grew larger after subcutaneous injection into SPARC-/- mice compared to WT littermates (16). To determine if this phenotype extended to tumors grown in the pancreas, we injected PAN02 cells underneath the capsule of the pancreas of SPARC-/- and WT animals. Figure 1A shows that orthotopic tumors were approximately 3-fold larger in SPARC-/- compared to WT mice (p<0.01). Tumors grown in SPARC-/- animals displayed a less robust capsule and were more invasive. Furthermore, the number of metastatic events increased in the absence of host SPARC (data not shown). Given that MMP9 expression is linked to ECM remodeling and tumor cell metastasis, we evaluated the level of expression of MMP9 and its cell distribution by immunohistochemistry in subcutaneous and orthotopic PAN02 tumors grown in WT and SPARC-/- animals. The number of cells positive for MMP9 was reduced in the absence of host-derived SPARC, such that there were 53.0 +/- 14.7 and 33.8 +/- 5.0 MMP9 positive cells in orthotopic tumors grown in WT and SPARC-/- mice, respectively (Fig. 1B, p<0.05). Immunofluorescence analysis of MMP9 does reveal some stromal-associated MMP9 but the majority of the staining is cell-associated. Neutrophils are known to infiltrate tumors and to express MMP9 (33). Therefore, we evaluated the tumors for neutrophils and MMP9 (Fig. 2). The level of Gr-1 positive cells was reduced in tumors grown in SPARC-/- animals. Image analysis of co-localization of Gr-1 and MMP9 demonstrates that the majority of MMP9 produced in PAN02 tumor microenvironment was associated with Gr-1 positive cells. We anticipate that the MMP9 detected represents newly produced protein or MMP9 in the secretory pathway. However, an alternative explanation is that the immuno-positive cells have tethered MMP9 to the cell surface via CD44 (34) or other alternative cell surface receptors.
Figure 1.
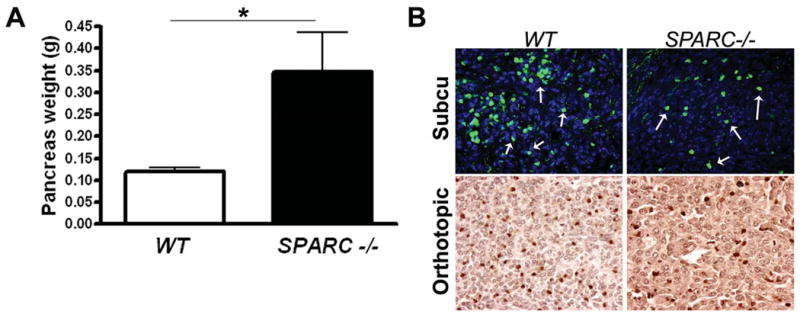
Pancreatic tumor growth is increased in the absence of host SPARC. A) 5 ×105 murine pancreatic adenocarcinoma (PAN02) cells were injected orthotopically into wild-type (WT, n=9) and SPARC-/- (SPARC-/-, n=5) mice. The mice were sacrificed 28 days after tumor cell injection and the weight (g) of the entire pancreas including tumor was determined. The mean pancreas weight +/- SEM for WT and SPARC-/- mice are displayed. *, p<0.01. B) Orthotopic and subcutaneous PAN02 tumors from WT and SPARC-/- mice were harvested, fixed in methyl Carnoys or formalin, respectively, sectioned and evaluated for MMP9 expression by immunohistochemistry. MMP9 levels (green) in subcutaneous tumors were developed with FITC-conjugated secondary antibody and sections counterstained with DAPI to identify nuclei (blue). Arrows indicate cells positive for MMP9. MMP9 levels in orthotopic tumors were developed with a peroxidase-conjugated secondary antibody with subsequent reaction with the chromagen DAB. MMP9 positive cells were hand counted in a minimum of 10 fields/group to be 53.0 +/- 14.7 and 33.8 +/- 5.0 in WT and SPARC-/- mice, respectively (p<0.05). Total magnification is 400×.
Figure 2.
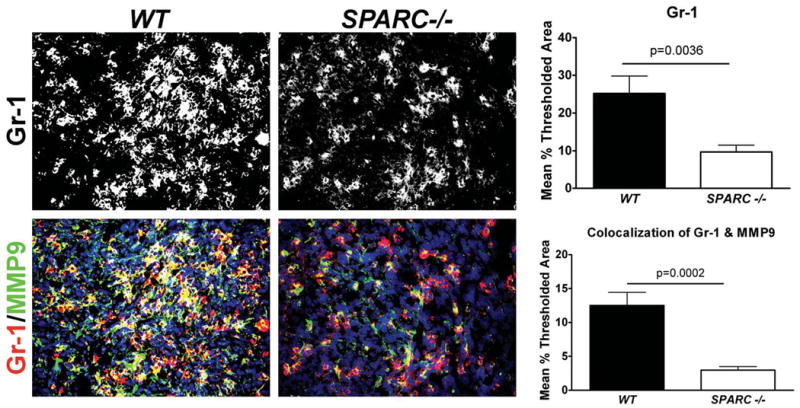
Neutrophils express MMP9 and their infiltration into PAN02 tumors is reduced in the absence of host SPARC. Paraffin-embedded sections of orthotopic PAN02 tumors grown in WT or SPARC-/- mice were evaluated immunohistochemically for Gr-1 (red) and MMP9 (green) and sections were counterstained with DAPI to identify nuclei. The level of Gr-1 staining and the co-localization of Gr-1 with MMP9 was quantified using Metamorph software. Total magnification is 400×.
Forced MMP9 expression confers PAN02 cells with increased proteolytic activity
To determine the effect of forced expression of MMP9 on tumor growth and if host SPARC had any effect on tumor-derived MMP9, PAN02 cells were transduced with recombinant retroviruses containing either empty vector (PRV) or native human MMP9 (MMP9). Zymographic analysis of total cell extracts from PAN02 cells transduced with MMP9 (PAN02-MMP9) detected a double gelatinolytic band corresponding to the pro and activated form of MMP9 (Fig. 3A). In empty vector transduced cells (PAN02-PRV), no gelatinolytic activity was observed. As expected for a secreted protein, MMP9 was expressed abundantly in conditioned medium of PAN02-MMP9 cells (Fig. 3B). In addition, cell fractionation of PAN02-MMP9 cells confirmed that MMP9 associates with both the cytosol and the cell membrane (Fig. 3C). The membrane protein transferrin receptor (TfR) was used as a marker of the membrane fraction and tubulin (Tub) was used as a marker of the cytosol.
Figure 3.
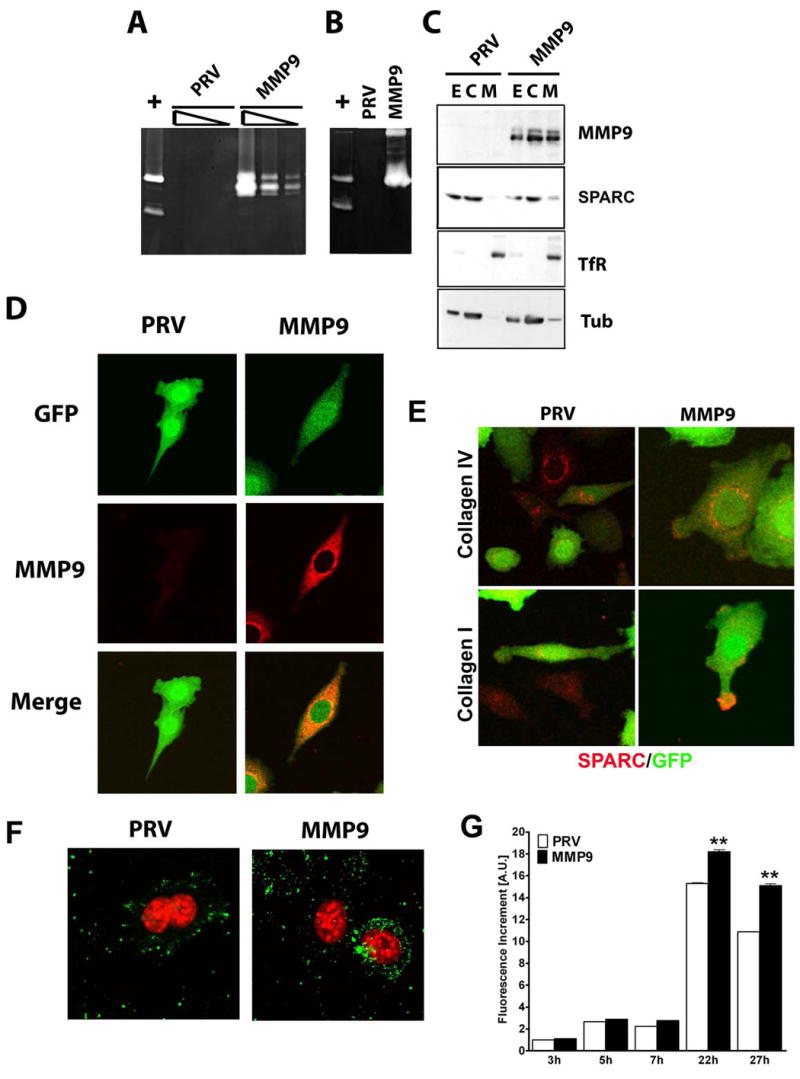
Characterization of MMP9 and SPARC expression in PAN02-PRV and PAN02-MMP9 cells. A) Gelatinolytic activity in total cell extracts of PAN02-PRV (PRV) and PAN02-MMP9 (MMP). Three serial dilutions of RIPA extracts corresponding to 5, 1, and 0.2 μg of protein were loaded on an 8% gelatin-containing SDS gels. B) Gelatinolytic activity in conditioned medium from the same cells as in (A) after 24 h of serum depletion. In panel A and B HT1080 conditioned media (+) was loaded as a positive control of gelatinolytic activity and shows MMP9 and MMP2 bands. C) Membrane and cytosolic fractions were obtained from total cell lysates. 25 μg of total extracts (E) and cytosolic (C) fraction and 1/6 of the membrane fraction (M) were analyzed on a 10% SDS gel. Western blot analysis of MMP9 and SPARC was performed; anti-TfR (membrane marker) and anti-tubulin (cytosolic marker) were used as controls for fractionation. D) Confocal microscopy of MMP9 (red) in PAN02 cells, cells transduced with the indicated construct are positive for GFP (green). E) PAN02-PRV (PRV) and PAN02-MMP9 (MMP) cells plated on either collagen I or collagen IV slides were analyzed for SPARC (red) expression by immunocytochemistry. Cells transduced with the indicated construct are positive for GFP (green). F) PAN02-PRV (PRV) and PAN02-MMP9 (MMP) cells were analyzed for gelatinase activity (green) by incubating with DQ-gelatin and subsequent confocal microscopy. To-Pro3 cell tracker (red) was used to localize nuclei. G) Quantification of gelatinase activity in PAN02-PRV (PRV) and PAN02-MMP9 (MMP9). Cells were incubated with DQ-gelatin for the indicated time and the fluorescence increment was represented. Values are normalized to PAN02-PRV cells after 3h of incubation (** p<0.01, Students t test).
The cellular localization of MMP9 in PAN02-MMP9 cells plated on tissue culture plastic or collagen IV was determined by immunocytochemistry (Fig. 3D). Consistent with the zymographic (Fig. 3A, B) and biochemical (Fig. 3C) analyses, PAN02-PRV cells showed no MMP9 staining, while PAN02-MMP9 cells exhibited staining distributed between internal membranes and the cytosol (Fig. 3D).
We found no significant difference in the expression level of SPARC in parental and modified PAN02 cells by immunocytochemistry. However the subcellular distribution of SPARC was altered in PAN02-MMP9 cells. We found SPARC in the membrane fraction in PAN02-MMP9 but not PAN02-PRV cells by Western blot analysis (Fig. 3C). Furthermore, we observed SPARC at the tip of cell extensions more frequently in PAN02-MMP9 compared to PAN02-PRV cells (Fig. 3E). To analyze in vitro proteolytic activity of cells expressing MMP9, we performed in situ zymography with DQ-gelatin. Incubation of PAN02-PRV or PAN02-MMP9 cells with DQ-gelatin for 1hr resulted in a similar punctate staining (Fig. 3F). However, longer incubation with the substrate, especially at 22hrs, revealed that PAN02-MMP9 cells display significantly greater gelatinolytic activity compared to PAN02-PRV cells, possibly as a result of increased MMP9 expression (Fig 3G).
Host SPARC contributes to MMP9-mediated effects on tumor growth
To recapitulate the events of pancreatic cancer progression, we used an orthotopic pancreatic cancer model where PAN02-PRV and PAN02-MMP9 cells were injected directly into the tail of the pancreas of age-matched WT and SPARC-/- mice (n=6/group) in two independent experiments. One hundred percent of the mice in each genotype developed primary pancreatic tumors with a reproducible pattern of growth. Figure 4A shows that PAN02-PRV tumors were larger in SPARC-/- mice compared to WT counterparts, which is consistent with previous results (16, 17). Tumors initiated with PAN02-MMP9 cells were significantly larger in both genotypes compared to PAN02-PRV tumors (WT mice, p<0.01; SPARC-/- mice, p<0.001). Tumors grown in the absence of host SPARC were more invasive such that the tumor margin was more difficult to identify grossly and there was a higher frequency of involvement of local organs (e.g., spleen, stomach, intestine, peritoneal wall). The invasive character of tumors grown in SPARC-/- animals was confirmed by analysis of H&E stained tumor tissue. Overall, these results demonstrate that tumor cell MMP9 and host SPARC both contribute to promotion or control of tumor growth, respectively. It is also evident that the absence of host SPARC and increased MMP9 expression is additive in terms of promoting tumor size.
Figure 4.
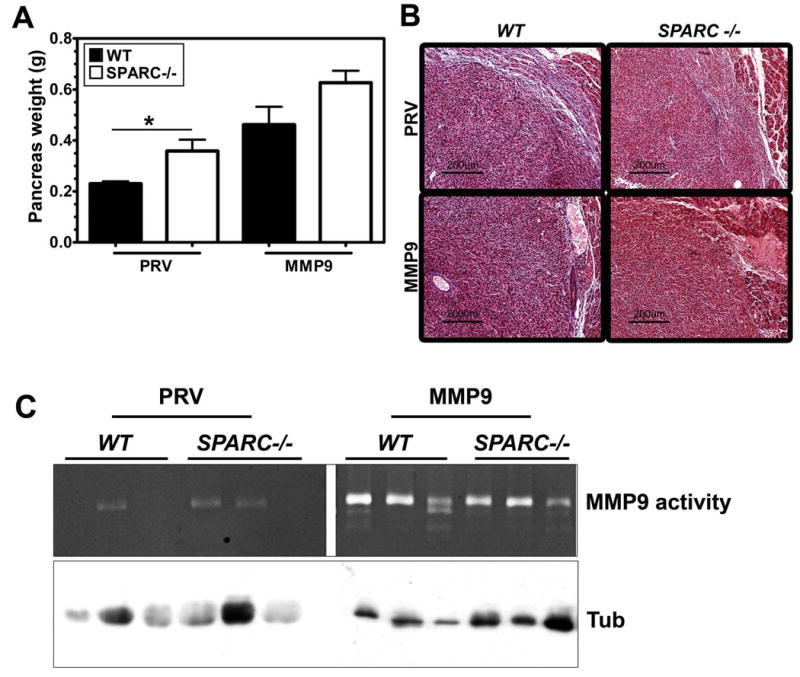
Forced expression of MMP9 or the absence of host SPARC increases PAN02 tumor growth. A) PAN02-PRV (PRV) and PAN02-MMP9 (MMP9) cells (5 × 105) were implanted orthotopically in the pancreas of age- and sex-matched SPARC-/- and WT mice (n= 6/group). After 6 weeks, the entire pancreas including tumor was weighed (g). A comparison of mean +/- SD pancreas weight is shown (* p<0.05, Student's t test). One way ANOVA with Tukey's multiple comparison test revealed significant differences across tumor groups as well. PAN02-PRV tumors in WT mice were significantly smaller than PAN02-MMP9 tumors grown in WT mice (p<0.01) or SPARC-/- mice (p<0.001) while PAN02-PRV tumors in SPARC-/- mice were significantly different from PAN02-MMP9 in SPARC-/- mice (p<0.001). B) Masson's trichrome staining of paraffin-embedded tumors revealed decreased collagen fibers (blue) in tumors grown in SPARC-/- mice compared with those in WT mice. Forced expression of MMP9 resulted in an even further reduction in collagen deposition in response to tumor growth. C) Tumor extracts from PAN02-PRV (PRV) and PAN02-MMP9 (MMP9) grown in WT and SPARC -/- mice were analyzed by gelatin zymography. The bands corresponding to MMP9 are indicated. Western blot analysis of tubulin in equivalent amount of extracts was performed as a loading control.
Consistent with previous studies, the deposition of collagen as shown by Masson's trichrome (Fig. 4B) and immunohistochemistry (data not shown) was reduced in and around pancreatic tumors grown in SPARC-/- mice. There were, however, notable differences between PAN02-PRV and PAN02-MMP9 tumors. PAN02-MMP9 tumors displayed less collagen staining than PAN02-PRV tumors in WT and SPARC-/- animals. The Masson's trichrome staining was consistent with the local invasiveness and gross appearance of the tumors upon necropsy.
We evaluated the level of MMP activity in tumors from a subset of each group of animals (Fig. 4C) and found that there was a significant increase in the level of enzyme activity in PAN02-MMP9 compared to PAN02-PRV tumors. However, the presence or absence of host SPARC did not alter the level of enzyme activity detected by zymography of tumor lysates.
We also evaluated the growth of the cells in vitro (Fig. 5A) and in vivo (Fig. 5B). Under normal in vitro growth conditions PAN02-MMP9 cells proliferated more rapidly than control transfected PAN02-PRV cells (Fig. 5A). To evaluate in vivo growth characteristics we quantified the number of proliferating and apoptotic cells in tumors grown in WT and SPARC-/- mice by detecting nuclei positive for phosphorylated histone H3 (phospho-H3) or terminal deoxynucleotidyl transferase mediated dUTP nick-end-labeling (TUNEL), respectively. We found that the overall balance of proliferating and apoptotic cells was very similar regardless of MMP9 status or genotype (Fig. 5B). These data support the idea that the proliferation and death of PAN02 cells is not the principal reason for an increase in tumor size in the absence of SPARC or in the presence of elevated levels of MMP9.
Figure 5.
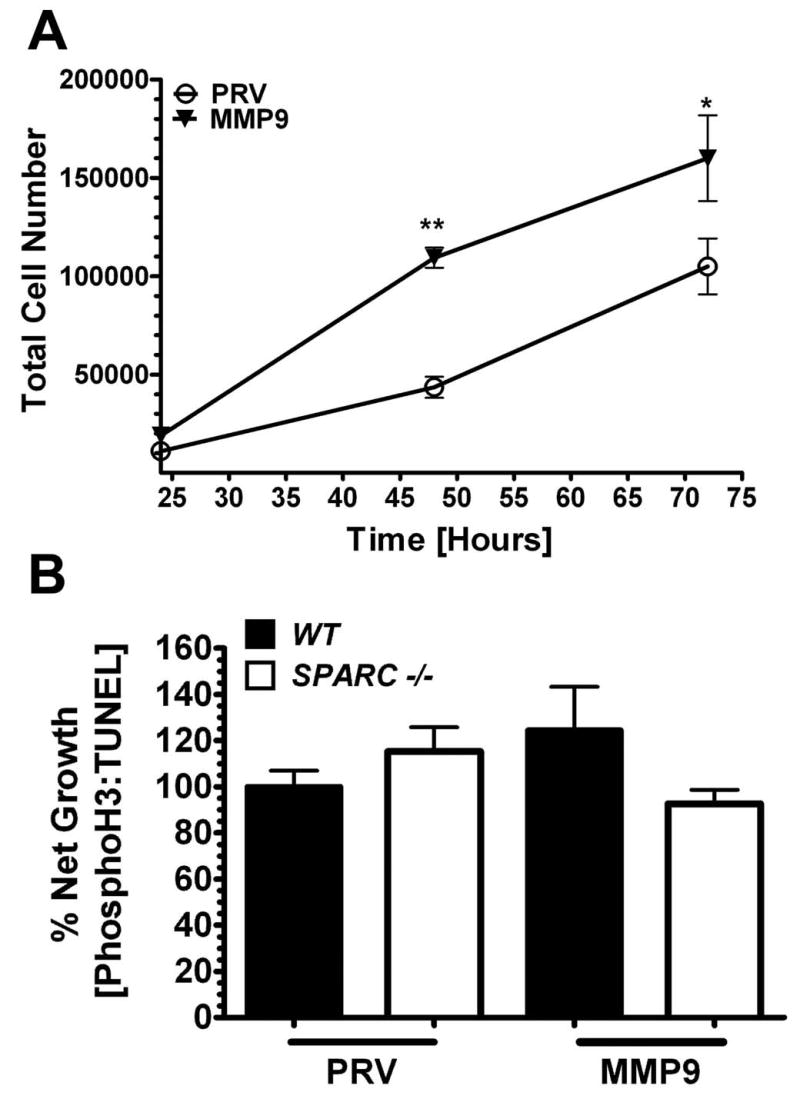
PAN02 proliferation and apoptosis in vitro and in vivo. A) In vitro proliferation of PAN02-PRV (PRV) and PAN02-MMP9 (MMP9) cells at 24, 48 and 72 hrs after seeding in DMEM containing 5% FBS. (*, p<0.05; **, p<0.01; two-way ANOVA with Bonferroni post-test) B) Net growth of tumors from PAN02-PRV (PRV) and PAN02-MMP9 (MMP9) cells grown in WT and SPARC -/- mice. Proliferating and apoptotic cells in tumor sections were counted by detecting nuclei positive for phosphorylated histone H3 (PhosphoH3) or terminal deoxynucleotidyl transferase mediated dUTP nick-end-labeling (TUNEL), respectively. The ratio of PhosphoH3 to TUNEL positive cells (% Net Growth) was calculated and normalized to PAN02-PRV (PRV) cells from WT mice. There was no significant difference between any groups (one-way ANOVA with Tukey's multiple comparison test)
In summary, tumor growth was controlled least in tumors that were either grown in the absence of SPARC or produced and secreted MMP9. Furthermore, MMP9 expression and the absence of SPARC were additive in terms of tumor size. Overall, these in vivo tumor growth assays demonstrate that MMP9 and SPARC have the capacity to impact primary tumor size, local invasion and tumor survival.
Angiogenesis profile
We examined the number and morphology of blood vessels in tumors by immunohistochemical identification of endothelial cells (MECA-32) and vascular basement membrane (collagen IV). For all of the analyses, we used Metamorph software to perform integrated morphometric analysis (IMA) of immunofluorescence images. Figure 6A shows an example of an image that has been rendered for morphometric analysis. The highlighted structures (green) are blood vessels that fulfill blood vessel specific parameters based on shape and size. In addition to improving accuracy, this method of analysis allows the measurement of pixel-based number, perimeter and area of antibody-bound blood vessels.
Figure 6.
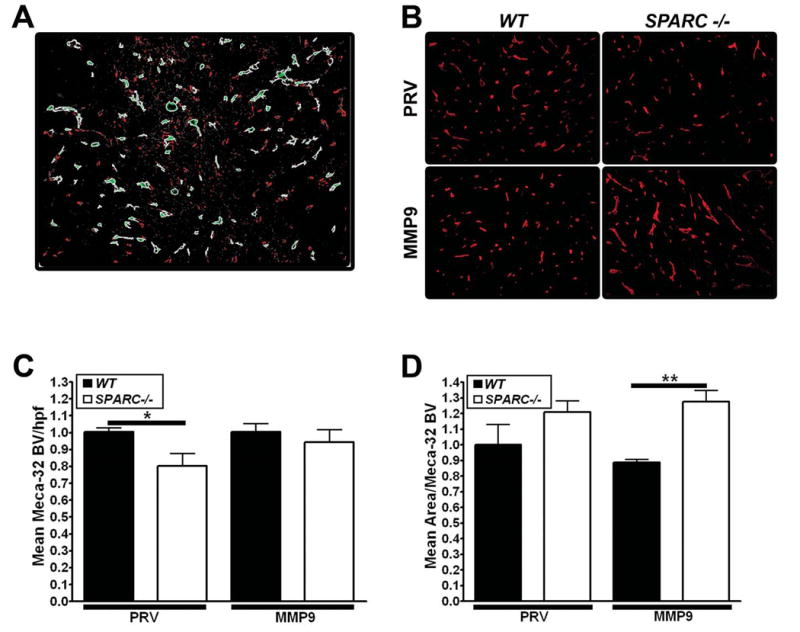
Analysis of angiogenesis in PAN02 tumors grown in SPARC-/- and WT mice. The number and size of blood vessels in tumors from PAN02-PRV (PRV) and PAN02-MMP9 (MMP9) cells were analyzed by immunohistochemistry of paraffin-embedded sections of tumors grown in WT or SPARC-/- mice. A) An example of integrated morphometric analysis (IMA) of immunofluorescence reactivity using MetaMorph software on Meca32 staining. An intensity and size threshold was set and maintained for every tumor section analyzed. Examples of objects meeting the size and intensity restrictions of blood vessels are shown in light green staining. B) MECA-32 immunofluorescence C) Mean blood vessel number/hpf and D) mean area/blood vessel were determined by IMA. C, D) Vascular parameters were normalized to PAN02-PRV tumors grown in WT mice to ease comparison between groups (*, p<0.05; **, p<0.01; one-way ANOVA with Tukey's multiple comparison test).
IMA analysis shows that PAN02-PRV tumors grown in SPARC-/- mice had significantly fewer MECA-32 positive vessels per high power field (hpf) compared to PAN02-PRV tumors grown in WT mice (Fig. 6B) but there was no change in blood vessel size (Fig. 6C). Unlike the PAN02-PRV tumors, the number of MECA-32 positive vessels/hpf was similar in PAN02-MMP9 tumors grown in WT and SPARC-/- mice (Fig. 6B). Furthermore, the number of MECA-32 positive blood vessels was almost identical in PAN02-PRV and PAN02-MMP9 tumors grown in WT mice. Interestingly, MECA-32 positive blood vessels were significantly larger in PAN02-MMP9 tumors grown in SPARC-/- vs WT mice (Fig. 6C). This is due to a decrease in the mean area/blood vessel in PAN02-MMP9 tumors grown in WT mice and is also reflected in the mean perimeter of the vessels (data not shown).
Collagen IV is a primary constituent of vascular basement membranes and a prominent substrate for MMP9. Figure 7 displays chromagen (DAB) immunohistochemistry for collagen IV in each tumor type. Immunohistochemical detection of collagen IV and IMA of fluorescent images in PAN02-PRV tumors showed a decrease in the number and size of collagen IV-positive blood vessels in tumors grown in SPARC-/- animals (Fig. 7B, C).
Figure 7.
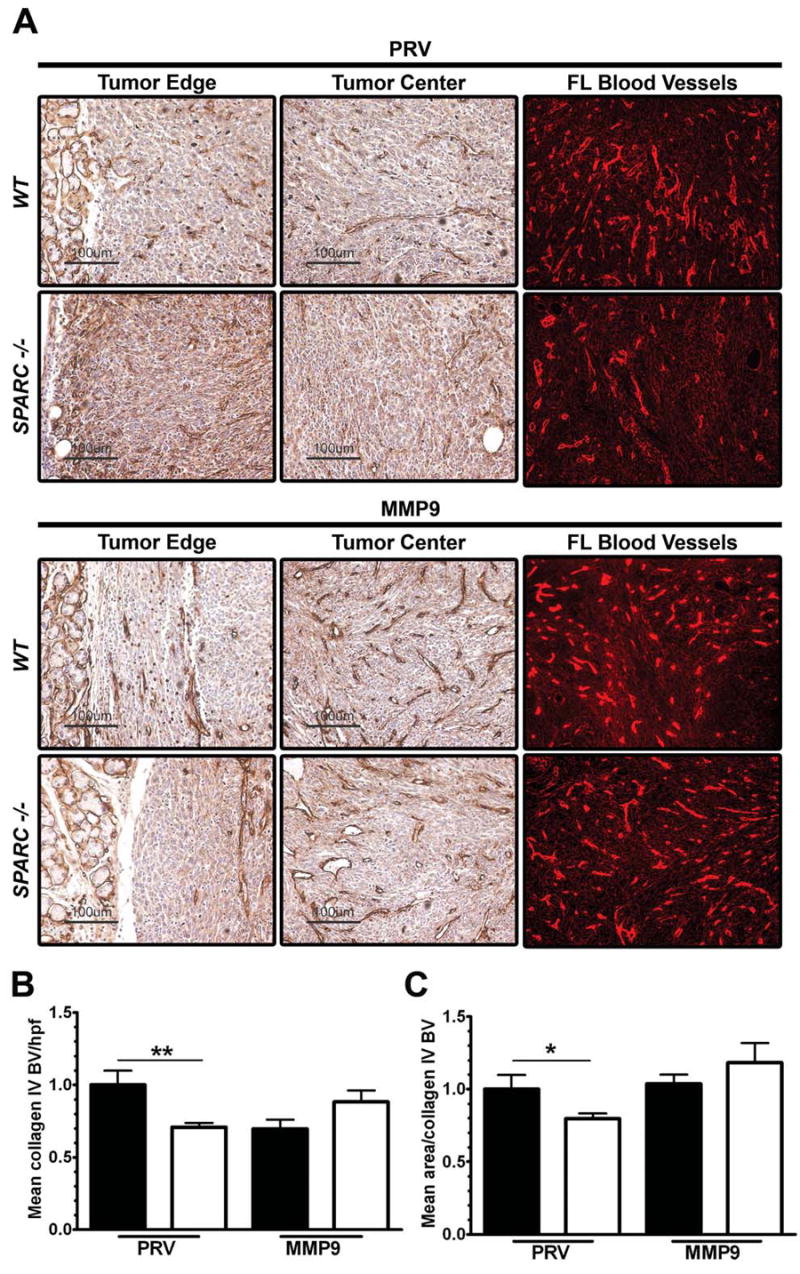
Collagen IV deposition in vascular basement membranes is altered by tumor cell expression of MMP9. A) The level and localization of collagen IV was determined immunohistochemically in PAN02-PRV (PRV) and PAN02-MMP9 (MMP9) tumors grown in WT and SPARC-/- mice. Collagen IV Immunofluorescent staining was evaluated by integrated morphometric analysis (IMA) for B) Mean blood vessel number/hpf and C) mean area/blood vessel. Total magnification of the fluorescent images is 200×. Vascular parameters were normalized to PAN02-PRV tumors grown in WT mice to ease comparison between groups (*, p<0.05; **, p<0.01; one-way ANOVA with Tukey's multiple comparison test).
The number of collagen IV-positive blood vessels decreased significantly in WT animals when PAN02 cells expressed MMP9 (Fig. 7B). Interestingly, there was an increase, although not statistically significant, in the number of collagen IV-positive blood vessels in PAN02-MMP9 tumors grown in SPARC-/- animals (Fig. 7B). Furthermore, the mean area/blood vessel as determined by collagen IV reactivity increased in PAN02-MMP9 compared to PAN02-PRV tumors grown in SPARC-/- mice (Fig. 7C). Thus, forced expression of MMP9 by PAN02 tumor cells resulted in a decrease in the number of collagen IV-positive blood vessels in WT animals but an increase in the number (MECA-32 and collagen IV) and size (collagen IV) of blood vessels grown in the absence of SPARC.
Furthermore, collagen IV was almost exclusively found associated with the vascular basement membrane in PAN02-MMP9 tumors (Fig. 7A and data not shown). In summary, analysis of the effect of host SPARC and MMP9 on vasculature in orthotopic pancreatic tumors demonstrates that MMP9 expression can rescue the lack of host-derived SPARC with respect to the number and size of blood vessels.
Host SPARC is a dominant factor in tumor cell migration and metastasis
Both SPARC and MMP9 have been shown to have effects on cell migration in vitro and in vivo. To better understand how each protein might effect cell migration of PAN02 cells, we first evaluated the migration of PAN02-PRV and PAN02-MMP9 cells in the presence and absence of SPARC in an in vitro cell migration assay. Forced expression of MMP9 conferred significantly increased cell migration in a transwell assay (Fig. 8A). However, the addition of SPARC decreased cell migration by greater than 80% in each cell type (Fig. 8A).
Figure 8.
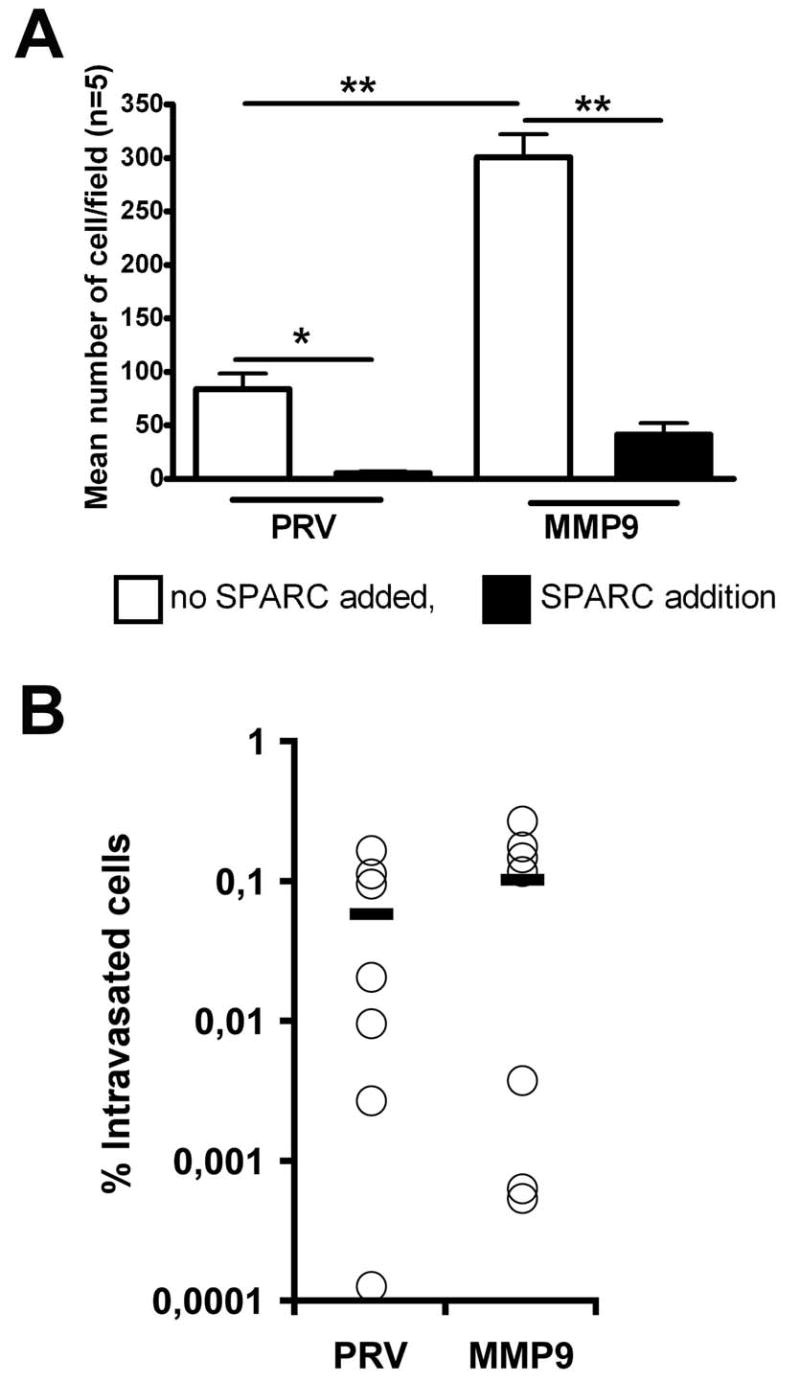
MMP9 expression confers increased PAN02 cell migration in vitro but not in vivo. A) PAN02-PRV (PRV) and PAN02-MMP9 (MMP9) cells in serum-free media were added to the top of an uncoated transwell insert. Serum-free media was added to the chamber below the insert. After 18 hours the inserts were removed, fixed, stained with hematoxylin, and the number of cells that migrated across the insert were counted. The mean +/- SEM number of cells/field (n=5/condition) is displayed. Addition of rSPARC (10 μg/ml) to the wells significantly decreased the number of cells that migrated (*, p<0.05; **, p<0.01). B) Cell invasion was evaluated by quantitative PCR of mouse DNA purified from CAM inoculated with PAN02 cells. Data represent the percentage of intravasated mouse cells in each sample. Mean value for PAN02-PRV and PAN02-MMP9 cells were 0.059 and 0.104 respectively. No significant differences in mean values were found (Mann-Whitney test).
To better understand the function of MMP9 in PAN02 cell invasion we tested whether forced expression of MMP9 provides PAN02 cells with a selective advantage for metastasis. PAN02 cells were incubated on the top of a chick embryo chorioallantoic membrane (CAM) and intravasation was assessed by collection of the distal CAM followed by mouse specific PCR (30, 35). Surprisingly, PAN02-MMP9 cells showed no increased intravasation compared to parental PAN02-PRV cells (Fig. 8B). These results are in contrast to the in vitro migration assay (Fig. 8A) and suggest that constitutive MMP9 activity alone does not confer greater metastatic capacity for these cells.
To validate these results, the incidence and total metastatic events was determined after orthotopic implantation of the tumor cells into WT and SPARC-/- mice (Table 1). The absence of host-derived SPARC increased the incidence of lymphatic metastasis but not the overall incidence of metastasis of PAN02-PRV tumors (Table 1). However, the total metastatic events were higher in SPARC-/- mice compared to their WT counterparts (Table 1). Furthermore, there was an increase in tumor-related complications including bloody ascites and bowel obstructions in SPARC-/- mice bearing PAN02-PRV tumors. We anticipated that forced expression of MMP9 by PAN02 cells would result in increased tumor cell invasion and metastasis in WT and SPARC-/- mice. However, PAN02-MMP9 tumors showed a decrease in total number of metastatic events in WT and SPARC-/- animals compared to PAN02-PRV tumors. Although the PAN02-MMP9 tumors were large and locally invasive, distant metastases were limited.
Table 1.
Host SPARC regulates metastasis
| Metastatic Incidence (%) | Metastatic Events | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Cell Type | n= | Lymph | Peritoneal | Liver | Other | Total | Lymph | Peritoneal | Liver | Other | Total |
| WT | PRV | 6 | 16.7 | 66.7 | 16.7 | 0 | 66.7 | 1 | 10 | 1 | 0 | 12 |
| SPARC -/- | PRV | 6 | 66.7 | 83.3 | 0 | 33.3 | 83.3 | 14 | 34 at, bt | 0 | 1 | 49at, bt |
| WT | MMP9 | 5 | 20.0 | 60.0 | 0 | 20.0 | 80.0 | 1 | 4 | 0 | 1 | 6 |
| SPARC -/- | MMP9 | 6 | 33.3 | 16.7 | 0 | 0 | 50.0 | 5 | 4 | 0 | 0 | 9 |
The influence of host SPARC and tumor-derived MMP9 on metastasis rate was evaluated after orthotopic implantation of pancreatic PAN02 tumors in WT and SPARC-/- mice. The total metastatic events and metastatic incidence in specific target organs was determined in one experiment at the time of sacrifice by visual inspection of each organ with a dissection scope under illumination with blue light to excite GFP in the tumor cells (n=number of animals in each group). Metastatic incidence is displayed as the percentage (%) of animals/group with a metastatic lesion in the target organ. The incidence of metastasis was compared between treatment groups were using a Tukey-type multiple comparison test for proportions (42). Statistical significance was set at 5%. Metastatic events are displayed as the total number of metastatic lesion in the target organ for the group. The number of metastatic events per mouse per organ were compared to median events per organ for the sample population using a Tukey-type multiple comparison test for medians (42). Trends (p<0.10) detected are indicated by at, vs. WT MMP9; bt, vs. SPARC MMP9.
Discussion
Pancreatic adenocarcinoma is the fourth leading cause of cancer-related death in the Western industrialized world, owing to rapid primary tumor growth and ensuing metastasis (36) (http://seer.cancer.gov). The metastatic cascade is dependent upon angiogenesis and cell migration, two processes that are regulated acutely by the local microenvironment (37-39). Therefore, understanding the interaction of pancreatic tumor cells with stromal components is critical to developing improved therapeutic options for patients. We sought to determine if two proteins linked to the desmoplastic response of pancreatic cancer interact at a functional level in the progression of this disease. SPARC is a matricellular protein implicated in tumor growth (11) with an in vivo function in the desmoplastic response, characteristic of pancreatic cancer. MMP9 on the other hand is associated with ECM turnover and cell migration through the ECM.
Our results show for the first time that orthotopic pancreatic tumors grow larger and more aggressively in the absence of host SPARC. Furthermore, forced expression of MMP9 by tumor cells increases tumor size but does not increase metastasis in WT and SPARC-/- animals. We also identified that SPARC regulates MMP9 expression in vivo and that SPARC and MMP9 both impact ECM deposition and angiogenesis in the tumor microenvironment. We found that, in general, tumor size correlated inversely with collagen deposition and that the lack of SPARC or the increased expression of MMP9 resulted in reduced collagen.
The expression of SPARC in tumor tissue from patients with pancreatic cancer has been shown to correlate with a worse prognosis (40). In particular, patients whose tumor had fibroblasts that expressed SPARC by immunohistochemistry had a median survival of 15 months whereas patients whose tumor stroma did not express SPARC had a median survival of 30 months. The authors conclude that, after controlling for other prognostic factors, the relative hazard for patients with stromal expression of SPARC was 1.89 (40). These studies support the idea that SPARC contributes to the progression of pancreatic cancer in humans, which is contrary to our results in SPARC-deficient animals. The reason(s) underlying this difference is unclear but might be due to the fact that the ECM of tumors from SPARC-/- animals is likely quite different from the ECM of human tumors from patients that have low or hard to detect SPARC protein. It is possible that defects inherent to the ECM of tumors grown in SPARC-/- mice are not replicated effectively by low or absent SPARC expression by stromal cells present in human tumors.
There are conflicting reports on whether SPARC is a substrate for MMP9. Sasaki et al. (41) found that MMP9 could cleave SPARC while Sage et al. (21) found that MMP3 but not MMP9 was the relevant MMP that mediated cleavage of SPARC. We did not determine if there was an increase in SPARC cleavage in PAN02-MMP9 tumors. It is possible however, to evaluate if SPARC is cleaved at L197 or L196 using neo-epitope specific antibodies (20), these studies are now in progress. Our results are however, consistent with previous studies (42-44) that have demonstrated that SPARC can increase the expression of MMPs.
Metastasis is the leading cause of death in pancreatic cancer patients. We provide evidence here that metastatic events occur more frequently in SPARC-/- versus WT animals. Since the initial steps in the metastatic cascade occur in the context of the ECM, we hypothesize that the compromised ECM formed in the absence of host SPARC favors invasion and metastasis. SPARC expression by pancreatic tumors cells has been implicated in tumor cell migration in vitro and as a possible factor contributing to metastasis in vivo (45). Our results suggest that an ECM formed in the absence of host SPARC can also facilitate metastasis. It is important to note that PAN02 cells produce SPARC. Therefore, we cannot exclude the possibility that tumor cell-derived SPARC contributes to metastatic spread in WT or SPARC-/- animals.
MMP9 expression is frequently associated with metastasis and is thought to facilitate tumor cell invasion (39). However, results from this study supported by previous compelling reports suggest that the underlying mechanisms and the function of MMP9 in metastasis are more complex (31, 46, 47). Our study demonstrates that orthotopic implantation of tumor cells forced to express MMP9 results in a decrease in metastatic burden compared to parental PAN02 tumor cells. Altogether, these results are in striking contrast to our own in vitro cell migration data and the generally perceived function of MMP9 as a promoter of metastasis but consistent with, Deryugina et al. (31) who showed that down-regulation of MMP9 in HT1080 cells resulted in increased intravasation and metastasis in the CAM.
These findings highlight the complex nature of MMP9 activity in the tumor microenvironment and might be partly explained by the paradigm that excess MMP9 expression inhibits tumor progression by generating endothelial cell inhibitors from both ECM and non-ECM sources, such as angiostatin and tumstatin (46, 48). Thus, the outcome of MMP activity in the tumor microenvironment may be dependent on a variety of factors including ECM deposition, the presence of other proteases and cytokines, the time, level, and site of MMP production, and the level and activity of adaptor proteins such as SPARC.
Acknowledgments
We thank present and former members of the Brekken Laboratory of advice and suggestions. The hybridoma MECA-32, developed by Dr. Eugene C. Butcher, was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of NICHD and maintained by The University of Iowa, Department of Biological Sciences (Iowa City, IA 52242). This study was supported in part by grants from The National Pancreas Foundation, The American Cancer Society, and the NIH (to RAB) as well as the Spanish Ministry of Science and Education (to S Mañes). RAB is the Effie Marie Cain Scholar in Angiogenesis Research.
Supported in part by The Effie Marie Cain Scholarship in Angiogenesis Research (to RAB.), The National Pancreas Foundation (grant 47431 to RAB.), The American Cancer Society (grant 47411/56540 to RAB.), and the NIH (R01 CA118240 to RAB). SA was supported in part by a training grant from the NIH (GM007062). The Department of Immunology and Oncology was founded and is supported by the Spanish Council for Scientific Research (CSIC) and by Pfizer. This work was in part supported by the grant SAF2005-00241 from the Spanish Ministry of Science and Education (to S Mañes).
References
- 1.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19:816–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- 2.Bornstein P. Cell-matrix interactions: the view from the outside. Methods Cell Biol. 2002;69:7–11. doi: 10.1016/s0091-679x(02)69003-x. [DOI] [PubMed] [Google Scholar]
- 3.Lane TF, Sage EH. The biology of SPARC, a protein that modulates cell-matrix interactions. Faseb J. 1994;8:163–173. [PubMed] [Google Scholar]
- 4.Liaw L, Crawford HC. Functions of the extracellular matrix and matrix degrading proteases during tumor progression. Braz J Med Biol Res. 1999;32:805–812. doi: 10.1590/S0100-879X1999000700002. [DOI] [PubMed] [Google Scholar]
- 5.Motamed K, Sage EH. Regulation of vascular morphogenesis by the matricellular protein SPARC. Kidney Int. 1997;51:1383–1387. doi: 10.1038/ki.1997.189. [DOI] [PubMed] [Google Scholar]
- 6.Forget MA, Desrosiers RR, Beliveau R. Physiological roles of matrix metalloproteinases: implications for tumor growth and metastasis. Can J Physiol Pharmacol. 1999;77:465–480. [PubMed] [Google Scholar]
- 7.Himelstein BP, Canete-Soler R, Bernhard EJ, Dilks DW, Muschel RJ. Metalloproteinases in tumor progression: the contribution of MMP-9. Invasion Metastasis. 1994;14:246–258. [PubMed] [Google Scholar]
- 8.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70:561–573. doi: 10.1046/j.1432-0436.2002.700909.x. [DOI] [PubMed] [Google Scholar]
- 10.Chantrain CF, Shimada H, Jodele S, Groshen S, Ye W, Shalinsky DR, Werb Z, Coussens LM, DeClerck YA. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res. 2004;64:1675–1686. doi: 10.1158/0008-5472.can-03-0160. [DOI] [PubMed] [Google Scholar]
- 11.Framson PE, Sage EH. SPARC and tumor growth: where the seed meets the soil? J Cell Biochem. 2004;92:679–690. doi: 10.1002/jcb.20091. [DOI] [PubMed] [Google Scholar]
- 12.Mok SC, Chan WY, Wong KK, Muto MG, Berkowitz RS. SPARC, an extracellular matrix protein with tumor-suppressing activity in human ovarian epithelial cells. Oncogene. 1996;12:1895–1901. [PubMed] [Google Scholar]
- 13.Brown TJ, Shaw PA, Karp X, Huynh MH, Begley H, Ringuette MJ. Activation of SPARC expression in reactive stroma associated with human epithelial ovarian cancer. Gynecol Oncol. 1999;75:25–33. doi: 10.1006/gyno.1999.5552. [DOI] [PubMed] [Google Scholar]
- 14.Sato N, Fukushima N, Maehara N, Matsubayashi H, Koopmann J, Su GH, Hruban RH, Goggins M. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene. 2003;22:5021–5030. doi: 10.1038/sj.onc.1206807. [DOI] [PubMed] [Google Scholar]
- 15.Koukourakis MI, Giatromanolaki A, Brekken RA, Sivridis E, Gatter KC, Harris AL, Sage EH. Enhanced expression of SPARC/osteonectin in the tumor-associated stroma of non-small cell lung cancer is correlated with markers of hypoxia/acidity and with poor prognosis of patients. Cancer Res. 2003;63:5376–5380. [PubMed] [Google Scholar]
- 16.Puolakkainen PA, Brekken RA, Muneer S, Sage EH. Enhanced growth of pancreatic tumors in SPARC-null mice is associated with decreased deposition of extracellular matrix and reduced tumor cell apoptosis. Mol Cancer Res. 2004;2:215–224. [PubMed] [Google Scholar]
- 17.Brekken RA, Puolakkainen P, Graves DC, Workman G, Lubkin SR, Sage EH. Enhanced growth of tumors in SPARC null mice is associated with changes in the ECM. J Clin Invest. 2003;111:487–495. doi: 10.1172/JCI16804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. 2005;23:953–964. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 19.Sasaki T, Hohenester E, Gohring W, Timpl R. Crystal structure and mapping by site-directed mutagenesis of the collagen-binding epitope of an activated form of BM- 40/SPARC/osteonectin. Embo J. 1998;17:1625–1634. doi: 10.1093/emboj/17.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasaki T, Miosge N, Timpl R. Immunochemical and tissue analysis of protease generated neoepitopes of BM-40 (osteonectin, SPARC) which are correlated to a higher affinity binding to collagens. Matrix Biol. 1999;18:499–508. doi: 10.1016/s0945-053x(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 21.Sage EH, Reed M, Funk SE, Truong T, Steadele M, Puolakkainen P, Maurice DH, Bassuk JA. Cleavage of the matricellular protein SPARC by matrix metalloproteinase 3 produces polypeptides that influence angiogenesis. J Biol Chem. 2003;278:37849–37857. doi: 10.1074/jbc.M302946200. [DOI] [PubMed] [Google Scholar]
- 22.Mira E, Lacalle RA, Gonzalez MA, Gomez-Mouton C, Abad JL, Bernad A, Martinez AC, Manes S. A role for chemokine receptor transactivation in growth factor signaling. EMBO Rep. 2001;2:151–156. doi: 10.1093/embo-reports/kve027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sweetwyne MT, Brekken RA, Workman G, Bradshaw AD, Carbon J, Siadak AW, Murri C, Sage EH. Functional analysis of the matricellular protein SPARC with novel monoclonal antibodies. J Histochem Cytochem. 2004;52:723–733. doi: 10.1369/jhc.3A6153.2004. [DOI] [PubMed] [Google Scholar]
- 24.Quesada AR, Barbacid MM, Mira E, Fernandez-Resa P, Marquez G, Aracil M. Evaluation of fluorometric and zymographic methods as activity assays for stromelysins and gelatinases. Clin Exp Metastasis. 1997;15:26–32. doi: 10.1023/a:1018480222301. [DOI] [PubMed] [Google Scholar]
- 25.Norose K, Clark JI, Syed NA, Basu A, Heber-Katz E, Sage EH, Howe CC. SPARC deficiency leads to early-onset cataractogenesis. Invest Ophthalmol Vis Sci. 1998;39:2674–2680. [PubMed] [Google Scholar]
- 26.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernstein EF, Chen YQ, Kopp JB, Fisher L, Brown DB, Hahn PJ, Robey FA, Lakkakorpi J, Uitto J. Long-term sun exposure alters the collagen of the papillary dermis. Comparison of sun-protected and photoaged skin by northern analysis, immunohistochemical staining, and confocal laser scanning microscopy. J Am Acad Dermatol. 1996;34:209–218. doi: 10.1016/s0190-9622(96)80114-9. [DOI] [PubMed] [Google Scholar]
- 28.Bernstein EF, Fisher LW, Li K, LeBaron RG, Tan EM, Uitto J. Differential expression of the versican and decorin genes in photoaged and sun-protected skin. Comparison by immunohistochemical and northern analyses. Lab Invest. 1995;72:662–669. [PubMed] [Google Scholar]
- 29.Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 1995;202:325–332. doi: 10.1002/aja.1002020402. [DOI] [PubMed] [Google Scholar]
- 30.Mira E, Lacalle RA, Gomez-Mouton C, Leonardo E, Manes S. Quantitative determination of tumor cell intravasation in a real-time polymerase chain reaction-based assay. Clin Exp Metastasis. 2002;19:313–318. doi: 10.1023/a:1015563031769. [DOI] [PubMed] [Google Scholar]
- 31.Deryugina EI, Zijlstra A, Partridge JJ, Kupriyanova TA, Madsen MA, Papagiannakopoulos T, Quigley JP. Unexpected effect of matrix metalloproteinase down-regulation on vascular intravasation and metastasis of human fibrosarcoma cells selected in vivo for high rates of dissemination. Cancer Res. 2005;65:10959–10969. doi: 10.1158/0008-5472.CAN-05-2228. [DOI] [PubMed] [Google Scholar]
- 32.Zar JH. Biostatistical Analysis. Fourth. 1999. [Google Scholar]
- 33.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–12498. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Hinsbergh VW, Engelse MA, Quax PH. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol. 2006;26:716–728. doi: 10.1161/01.ATV.0000209518.58252.17. [DOI] [PubMed] [Google Scholar]
- 35.Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- 36.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 37.Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524–6536. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- 38.Raghunand N, Gatenby RA, Gillies RJ. Microenvironmental and cellular consequences of altered blood flow in tumours. Br J Radiol. 2003;76(Spec No 1):S11–22. doi: 10.1259/bjr/12913493. [DOI] [PubMed] [Google Scholar]
- 39.Ludwig T. Local proteolytic activity in tumor cell invasion and metastasis. Bioessays. 2005;27:1181–1191. doi: 10.1002/bies.20306. [DOI] [PubMed] [Google Scholar]
- 40.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C, Goggins M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–325. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki T, Gohring W, Mann K, Maurer P, Hohenester E, Knauper V, Murphy G, Timpl R. Limited cleavage of extracellular matrix protein BM-40 by matrix metalloproteinases increases its affinity for collagens. J Biol Chem. 1997;272:9237–9243. doi: 10.1074/jbc.272.14.9237. [DOI] [PubMed] [Google Scholar]
- 42.McClung HM, Thomas SL, Osenkowski P, Toth M, Menon P, Raz A, Fridman R, Rempel SA. SPARC upregulates MT1-MMP expression, MMP-2 activation, and the secretion and cleavage of galectin-3 in U87MG glioma cells. Neurosci Lett. 2007;419:172–177. doi: 10.1016/j.neulet.2007.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilles C, Bassuk JA, Pulyaeva H, Sage EH, Foidart JM, Thompson EW. SPARC/osteonectin induces matrix metalloproteinase 2 activation in human breast cancer cell lines. Cancer Res. 1998;58:5529–5536. [PubMed] [Google Scholar]
- 44.Fujita T, Shiba H, Sakata M, Uchida Y, Nakamura S, Kurihara H. SPARC stimulates the synthesis of OPG/OCIF, MMP-2 and DNA in human periodontal ligament cells. J Oral Pathol Med. 2002;31:345–352. doi: 10.1034/j.1600-0714.2002.00188.x. [DOI] [PubMed] [Google Scholar]
- 45.Guweidhi A, Kleeff J, Adwan H, Giese NA, Wente MN, Giese T, Buchler MW, Berger MR, Friess H. Osteonectin influences growth and invasion of pancreatic cancer cells. Ann Surg. 2005;242:224–234. doi: 10.1097/01.sla.0000171866.45848.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pozzi A, LeVine WF, Gardner HA. Low plasma levels of matrix metalloproteinase 9 permit increased tumor angiogenesis. Oncogene. 2002;21:272–281. doi: 10.1038/sj.onc.1205045. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Su Y, Fingleton B, Acuff H, Matrisian LM, Zent R, Pozzi A. Increased plasma MMP9 in integrin alpha1-null mice enhances lung metastasis of colon carcinoma cells. Int J Cancer. 2005;116:52–61. doi: 10.1002/ijc.20997. [DOI] [PubMed] [Google Scholar]
- 48.Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. Physiological levels of tumstatin, a fragment of collagen IV [alpha]3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via [alpha]V[beta]3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]