

Abstract
Fibroblast-mediated collagen gel contraction has been used as an in vitro model of tissue remodeling. Thrombin is one of the mediators present in the milieu of airway inflammation and may be involved in airway tissue remodeling. We have previously reported that thrombin stimulates fibroblast-mediated collagen gel contraction partially through the PAR1/PKCε signaling pathway (Fang et al, ERJ, 2004; 24: 918-924). Here we further report that the delta-isoform of PKC (PKCδ) is also activated by thrombin and involved in the thrombin-mediated augmentation of collagen gel contraction. Thrombin (10nM) significantly increased PKCδ activity (over 5-fold increase after 15-30 min stimulation) and stimulated phosphorylation of PKCδ. Rottlerin, a PKCδ inhibitor, completely inhibited activation of PKCδ and partially blocked collagen gel contraction stimulated by thrombin. Similarly, PKCδ -specific siRNA significantly inhibited PKCδ activation without affecting PKCε expression and activation. Furthermore, suppression of PKCδ by siRNA resulted in partial blockade of thrombin-augmented collagen gel contraction. These results suggest that thrombin contributes to the tissue remodeling in inflammatory airways and lung diseases at least partially through both PKCδ and PKCε signaling.
Introduction
The architectural changes in lung and bronchi are thought to contribute to the loss of lung function in many chronic airway and lung diseases, such as asthma, chronic obstructive pulmonary disease (COPD) and interstitial lung disease. Fibroblast proliferation and migration and extracellular matrix accumulation within the airway wall are characteristic pathologic changes in airway remodeling [1-4]. Fibroblast-mediated collagen gel contraction is recognized as an in vitro model of tissue remodeling, specifically of the contraction that characterizes both fibrotic scar tissue and normal wound healing. Many mediators including thrombin, platelet-derived growth factor (PDGF), transforming growth factor (TGF) and prostaglandin D2 (PGD2) that are believed to be involved in the tissue repair processes simulate collagen gel contraction [5-8]. However, the mechanisms that mediate collagen gel contraction are not fully understood.
Thrombin is a serine protease activated from pro-thrombin as part of the clotting cascade. In addition to its proteolytic effect, thrombin can initiate many cellular effects through activating protease-activated receptors [9-11]. We have previously reported that thrombin, through binding to its receptor PAR1, stimulated collagen gel contraction partially through the novel ε-isoform of protein kinase C (PKCε) [5].
The family of PKC enzymes regulates diverse cellular functions in a variety of tissues including lungs. Twelve different isozymes in 3 groups have been described so far [12]. These PKC isozymes have been reported to play roles not only in maintaining normal lung structure and function, but also in the pathophysiology of many lung diseases including pulmonary edema, adult respiratory distress syndrome, interstitial lung disease, asthma and lung cancer [12-16]. PKCδ is one isozyme of the novel PKC subfamily that includes PKCδ, ε, η and θ. Recent studies suggest that PKCδ plays an important role in many cellular functions including cell survival and apoptosis, cell proliferation and migration, T cell functions, wound repair and fibrotic tissue formation [17-19]. The current study, therefore, was designed to determine whether PKCδ also regulates thrombin-augmented collagen gel contraction by lung fibroblasts.
Materials and methods
Materials
Type I collagen was extracted from rat tail tendons (RTTC) as previously described [20].
Thrombin from human plasma was purchased from Sigma (St. Louis, MO). Rottlerin was obtained from Calbiochem (San Diego, CA, USA). Tissue culture supplements, fetal calf serum (FCS) and media were purchased from GIBCO (Life Technologies, Grand Island, NY). SMARTpool siRNA targeting PKCδ was purchased from Dharmacon.
Cell culture
Human fetal lung fibroblasts (HFL-1) were obtained from the American Type Culture Collection (Rockville, MD) and were cultured as described [5]. Fibroblasts used in these experiments were between cell passage 14 and 19.
Collagen gel preparation and contraction assay
Collagen gels were prepared and contraction assays performed as described previously [20]. The areas of floating gels were measured using an image analyzer (Optomax , Burlington, MA). Each experiment included triplicate gels and each experiment was performed on no less than three separate experiments for each unique parameter.
Transfection of siRNA targeting PKCδ
Small interfering RNA (siRNA) targeting PKCδ was purchased from Dharmacon (Lafayette, CO). Transfection with TransIT-TKO (Mirus Corporation, Madison WI, USA) was performed as described previously [5]. Twenty-four hours after transfection, cells were used for either assessing the efficacy of silence by Western blot, or PKC activity and collagen gel contraction assays.
Cell viability and toxicity assay
Cell viability was evaluated by calcein AM and ethidium homodimev-1, a two-color fluorescence-based method, using the LIVE/DEAD Kit, following the manufacturer’s instructions (Invitrogen, Eugene, Oregon).
Immunoblot
Immunoblotting was performed as previously described [5] and probed with anti-PKCδ, anti-PKCε or anti-p-PKCδ mAb (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or anti-ß-actin antibody (Sigma, St. Louis, MO). Target proteins were subsequently detected using horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG in conjunction with enhanced chemiluminescence detection system (ECL; GE Life Sciences Products, Piscataway, NJ).
PKCδ and PKCε activity assay
Enzymatic activity of PKCδ and PKCε was determined in crude whole-cell fractions. The assay employed was a modification [21] of procedures previously described [22] using 900 μM PKCδ or PKCε specific substrate peptide (Calbiochem), 8 μM phosphatidyl-L-serine, 24 μg/ml phorbol myristate acetate, 30 mM dithiothreitol, 150 μM ATP, 45 mM magnesium acetate, and 10 μCi/ml [γ-32P] ATP in a Tris-HCl buffer (pH7.5). Kinase activity was expressed in relationship to total cellular protein assayed and calculated in picomoles phosphate transferred to the peptide substrate per minute per mg of total cellular protein assayed (pmol/min/mg). All samples were assayed in triplicate, and each experiment was repeated on no less than three separate occasions.
Statistical analysis
Individual experiments included triplicate gels within an experiment for all experimental conditions. Results were always confirmed by repeating each experiment on separate occasions at least three times. Statistical comparisons were made from all experiments, including both the within and between group variance. PKCδ and PKCε activity were expressed as fold change in activation compared to control, untreated cells for all experiments. Group data were analyzed by one-way ANOVA. Differences between series of data that appeared statistically different were corrected by Tukey’s test. P<0.05 was considered significant.
Results
Thrombin stimulates activation and phosphorylation of PKCδ
Two separate approaches were used to evaluate the effect of thrombin on PKCδ activation in human lung fibroblasts. First, PKCδ catalytic activity was determined in response to thrombin stimulation of lung fibroblasts cultured in monolayer. Thrombin (10nM) stimulated PKCδ activity in a time-dependent manner (Fig 1A). After 15min and 30min of thrombin stimulation, PKCδ activity was greatly increased (6.2±1.0 and 6.4±1.0 fold of control, p<0.01, Fig 1A). After 60 minutes thrombin stimulation, however, the PKCδ activity had declined to baseline levels (Fig 1A). Second, PKCδ activation was also evaluated by immunoblotting phosphorylated PKCδ. Consistent with the observed increase in kinase activity, phosphorylation of PKCδ was significantly stimulated by thrombin (10nM) and the maximum phosphorylation was observed at 15min and 30 min after stimulation with thrombin (Fig 1B). After 60min of stimulation, PKCδ phosphorylation was reduced although it was still higher than that of baseline (Fig 1B).
Figure 1. Effect of thrombin on PKCδ activity and phosphorylation.
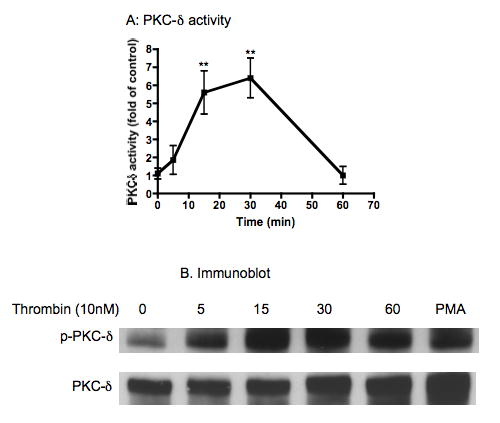
HFL-1 fibroblasts were stimulated with thrombin (10nM) for up to 60min. PKCδ activity was measured as described in methods (Panel A) and PKCδ phosphorylation was evaluated by immunoblot (Panel B). Panel A: Vertical axis: PKCδ activity (fold of control). Horizontal axis: Time (min). ** p<0.01 compared to time 0. Data presented are the mean ± standard error of the mean for three separate experiments (n=3). Panel B: p-PKCδ: phosphorylated PKCδ. PMA: phorbol 12-myristate 13-acetate (100nM, 30min) was used as positive control.
Rottlerin and PKCδ siRNA inhibit thrombin-induced PKCδ activation, but not PKCε activation
Both a pharmacologic inhibitor of PKCδ and RNA interference targeting of PKCδ were used to suppress PKCδ activation in response to thrombin stimulation in human lung fibroblasts. Rottlerin (1 μM) significantly inhibited 10nM and 20nM thrombin-induced PKCδ activity (p<0.01, Fig 2A), but had no effect on PKCε activity (Fig 2B). To confirm the specificity of rottlerin as a pharmacologic kinase inhibitor in our assay, molecular interference with siRNA was also used to suppress PKCδ. PKCδ -specific siRNA significantly decreased both baseline PKCδ activity as well as thrombin-stimulated PKCδ activity (Fig 2C, p<0.001), but tended to slightly increase PKCε activity both in the presence and absence of thrombin, although this effect was not statistically significant (Fig 2D, p>0.05). Neither rottlerin nor PKCδ siRNA altered cell viability assessed by LIVE/DEAD assay (data not shown).
Figure 2. Effect of inhibition of PKCδ on thrombin-induced PKCδ and PKCε activity.
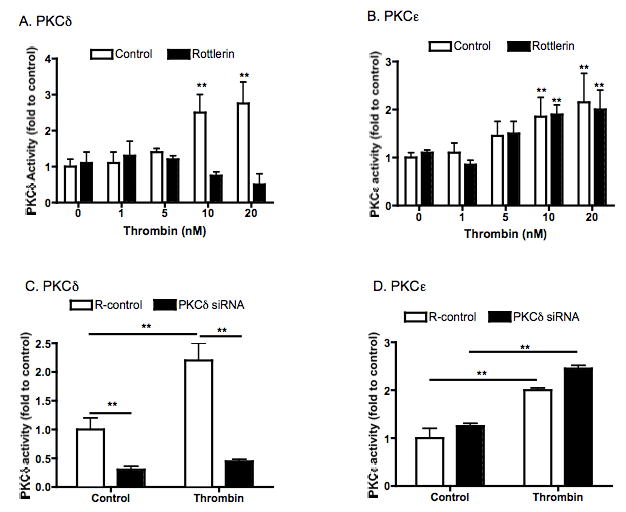
Panel A and B: Pharmacologic inhibition with rottlerin. HFL-1 fibroblasts were stimulated with varying concentrations of thrombin with or without rottlerin (1μM) for 30min. PKCδ (Panel A) and PKCε (Panel B) activity was measured as described in methods. Vertical axes: PKCδ or PKCε activity (fold of control). Horizontal axes: thrombin concentration (nM). ** p<0.01 compared to 0 nM thrombin. Panel C and D: RNAi suppression. Sub-confluent HFL-1 fibroblasts were transfected with or without specific siRNA that targeting PKCδ. After 24 hours, cells were then treated with thrombin for 30min. PKCδ (Panel C) or PKCε (Panel D) activity was then measured as described in the methods. Vertical axes: PKCδ or PKCε activity (fold of control). Horizontal axes: thrombin treatment (10nM) ** p<0.01. Data presented are the mean ± standard error of the mean for three separate experiments (n=3).
Inhibition of PKCδ by rottlerin or by siRNA results in partial blockade of thrombin-augmented collagen gel contraction
Fibroblast-populated collagen gels were allowed to contract in the presence or absence of thrombin (10nM), one of the factors known to stimulate collagen gel contraction by fibroblasts [5]. Two approaches were used to evaluate the role of PKCδ in mediating thrombin-augmented collagen gel contraction.
First, the effect of rottlerin, an inhibitor of the PKCδ isozyme, on thrombin-augmented collagen gel contraction was investigated. Thrombin significantly stimulated collagen gel contraction mediated by HFL-1 fibroblasts in a time-dependent manner (Fig 3A). Rottlerin (1μM) alone had no effect on gel contraction on day 1, 2 and 3, but significantly inhibited on day 4 (85.1±0.2% vs 71.2±0.1% of control, p<0.05) and day 5 (86.1±1.0% vs 69.1±0.2% of control, p<0.01, Fig 3A). In the presence of thrombin, rottlerin significantly inhibited thrombin-augmented collagen gel contraction from day 2 (78.5±0.8% vs 62.5±1.3% of thrombin, p<0.01, Fig 3A). The inhibitory effect of rottlerin on thrombin-augmented collagen gel contraction was also concentration-dependent (Fig 3B). Rottlerin at concentrations of 1, 2.5 and 5μM significantly inhibited thrombin-augmented collagen gel contraction; the gel size was 73.5±1.5%, 79.3±0.4% and 87.7±0.7% of original gel size, respectively, (p<0.05 or 0.01, compared to the corresponding gel size stimulated by thrombin, Fig 4B). Rottlerin at its maximum concentration (10μM) completely inhibited collagen gel contraction stimulated by thrombin (99.0±0.8% vs 62.5±0.5% of thrombin alone, p<0.01, Fig 3B).
Figure 3. Effect of PKCδ inhibitor (rottlerin) on thrombin-induced collagen gel contraction.
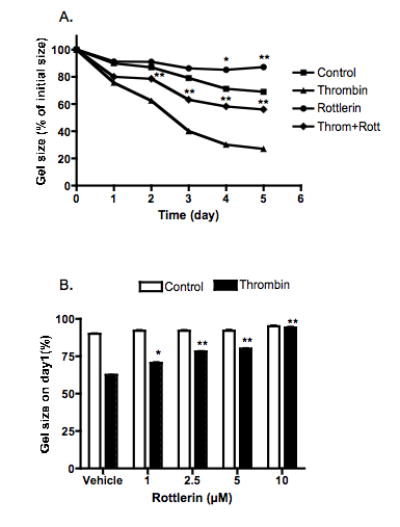
Panel A: Time-dependence. Fibroblasts were cast into collagen gels which were released and cultured in serum-free DMEM with or without thrombin (10nM) and /or rottlerin (1μM) for 5 days. Gel size was measured daily with an image analyzer. Vertical axis: gel size expressed as percentage of initial gel size (%). Horizontal axis: Time (days). Panel B: Concentration-dependence. Gels were released into serum-free DMEM with varying concentrations of rottlerin as indicated in the presence or absence of thrombin (10nM). Gel size was measured on day 1 with an image analyzer. Vertical axis: gel size expressed as percentage of initial gel size (%). Horizontal axis: Rottlerin concentration (μM). Data presented are the mean ± standard error of the mean for three separate experiments, each of which included triplicate gels for each condition. * p<0.05, ** p<0.01 compared to Thrombin or control alone.
Figure 4. PKCδ suppression by siRNA.
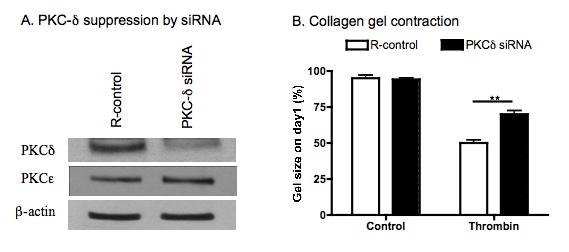
Sub-confluent HFL-1 cells were treated with transfection reagent only (R-control) or transfected with PKCδ specific siRNA. Panel A: Suppression of PKCδ expression was assessed by immunoblot using anti-PKCδ or anti-PKCε antibodies. Blots were probed using anti-β-actin as a loading control. Panel B: Effect on collagen gel contraction. After transfection of PKCδ siRNA, the cells were cast into collagen gels and allowed to contract in the presence or absence of thrombin (10nM). Gel size was measured on day 1. Vertical axis: gel size expressed as percent of initial size (%). Horizontal axis: treatment.
Second, expression of PKCδ was suppressed by PKCδ targeting siRNA in human lung fibroblasts following which the fibroblasts were cast into collagen gels and allowed to contract in the presence or absence of thrombin (10nM). PKCδ -specific siRNA significantly suppressed PKCδ expression as determined by immunoblot (Fig 4A), but had no effect on PKCε expression (Fig 4A). In addition, consistent the effect of rottlerin on collagen gel contraction, suppression of PKCδ by siRNA resulted in partial blockade of collagen gel contraction in response to thrombin stimulation (p<0.01, Fig 4B).
Discussion
The current study investigated the role of PKCδ in thrombin-induced collagen gel contraction as mediated by human lung fibroblasts. Thrombin significantly stimulated both PKCδ and PKCε activity, and rottlerin specifically inhibited PKCδ activity, but not PKCε activity. Thrombin-augmented collagen gel contraction mediated by fibroblasts was partially, but significantly, inhibited by rottlerin. Furthermore, suppression of PKCδ expression by siRNA resulted in partial blockade of thrombin-augmented collagen gel contraction. These results indicate that PKCδ mediates thrombin-augmented collagen gel contraction by human lung fibroblasts, and by this mechanism, PKCδ may be involved in tissue repair and remodeling following airways inflammatory injury.
Thrombin is formed in the blood coagulation cascade, which is commonly activated following tissue injury, and during inflammation and repair. Through activating the protease-activated receptor1 (PAR1), thrombin has been shown to stimulate cell proliferation and affect extracellular matrix deposition and degradation. Our previous study has shown that activation of PKCε, downstream of PAR1, is one mechanism that contributes to thrombin-induced collagen gel contraction mediated by HFL-1 fibroblasts. In the current study, we further investigate another novel class isozyme of PKC, the delta isoform (PKCδ), in mediating collagen gel contraction in response to thrombin stimulation. Similar to the effect of PKCε, PKCδ also partially modulates fibroblast-mediated collagen gel contraction in response to thrombin.
Whether PKCδ plays a role in “constitutive” contraction of collagen gels mediated by fibroblasts was not assessed in the current study. A modest, but significant effect was observed with rottlerin under baseline conditions. While this could be due to “constitutive” PKCδ, it is not possible to rule out a non-specific effect. No effect was observed with siRNA under baseline conditions. However, like most siRNAs, suppression was not complete. Thus, the lack of effect of the siRNA does not rigorously exclude a role for PKCδ under baseline conditions. While the role of PKCδ under baseline conditions remains undefined, a role in the thrombin stimulated contraction is supported by the consistent inhibition by both rottlerin and siRNA for PKC.
PKC is comprised of a family of serine/threonine kinases that play important roles in variety of cell functions including proliferation, apoptosis and differentiation. There are 9 PKC genes, coding 11 PKC isoforms, which are classified into 3 subfamilies: classical PKCs (cPKCs: PKCα, ßI, ßII and γ); novel PKCs (nPKCs: PKCε, δ, η, θ); and atypical PKC (aPKCs: PKCζ and τ) [23]. The mechanisms of PKC activation in a variety of cell types have been extensively studied [24, 25]. PKC activity is regulated by several mechanisms including phosphorylation [26]. In the present study, activation of PKCδ was associated with enzyme phosphorylation. In general PKC activity is transient. There are several mechanisms for inactivation, including dephosphorylation, translocation, proteolysis and formation of covalent linkage on the active site of the enzyme [27-30]. While the loss of PKCδ catalytic activity at 60 minutes with preservation of phosphorylation, the mechanisms of this phenomenon were not directly evaluated in the current study.
Both PKCδ and PKCε belong to the novel PKC (nPKC) subfamily and they are expressed in human lung fibroblasts [14, 31]. We have reported that thrombin stimulates human lung fibroblast-mediated collagen gel contraction partially through PKCε signaling [5]. Here we found that both a PKCδ inhibitor (rottlerin) and PKCδ specific siRNA partially blocked fibroblast-mediated collagen gel contraction in response to thrombin, indicating that PKCδ has an effect similar to that previously shown for PKCε in mediating collagen gel contraction, an in vitro model of tissue repair. In contrast to the parallel effects observed in the current study, the biological functions of these two isozymes in cancer or cardiac injury are opposite. PKCδ is a pro-apoptotic or growth inhibitory PKC, and many types of apoptotic stimuli can induce PKCδ translocation to mitochondria, leading to cytochrome c release and caspase-3 activation [23, 32]. In contrast, PKCε promotes cell survival in many cell types through activation of Akt pathways and up-regulation of anti-apoptotic proteins [23, 33]. In a model of myocardial ischemia/reperfusion injury, PKCδ and PKCε play opposing roles. Activation of PKCδ during reperfusion induces cell death through the mechanism of mitochondria-mediated apoptosis, whereas activation of PKCε before ischemia protects mitochondrial function and inhibits cell death [34].
Fibroblast-mediated collagen gel contraction is considered to be an in vitro model of wound healing and tissue remodeling [20, 35]. The current study demonstrated that PKCδ mediates thrombin signaling in fibroblasts cultured in the three-dimensional collagen gels. This finding suggests that PKCδ signaling plays a role in wound repair and tissue remodeling. Consistent with our findings, recent studies reported that asbestos-induced peribronchiolar cell proliferation, cytokine release (IL-4, IL-6 and IL-13) and inflammatory cells were reduced in PKCδ knock out mice compared to wild type mice [15]. Similar to the seemingly redundant PKCδ and PKCε effects on gel contraction found in our studies, PKCε signaling also plays an important role in lung cytokine release [21]. These results indicate that both PKCδ and PKCε can have multiple, and sometimes overlapping, effects on pro-inflammatory and pro-fibrotic reactions in the lungs.
In summary, thrombin is capable of activating PKCδ in human lung fibroblasts cultured in three dimensional collagen gels. Rottlerin and PKCδ specific siRNA partially blocked thrombin-augmented collagen gel contraction. These results support the concept that thrombin could contribute to the pathogenesis of fibrotic tissue formation in a variety of lung and airway diseases through activating PKCδ signaling. Thus, targeted interruption of PKCδ may provide a potential therapeutic option for the blockade of peri-bronchiolar fibrosis in lung diseases such as asthma and COPD.
Acknowledgments
Funding agency: NIH grant 1R01 HL-6408804 (SIR); the Larson Endowment, University of Nebraska (SIR); VA Merit grant (TAW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tschumperlin DJ, Drazen JM. Mechanical stimuli to airway remodeling. Am J Respir Crit Care Med. 2001;164:S90–94. doi: 10.1164/ajrccm.164.supplement_2.2106060. [DOI] [PubMed] [Google Scholar]
- 2.Choe MM, Sporn PH, Swartz MA. Extracellular matrix remodeling by dynamic strain in a three-dimensional tissue-engineered human airway wall model. Am J Respir Cell Mol Biol. 2006;35:306–313. doi: 10.1165/rcmb.2005-0443OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James A. Airway remodeling in asthma. Curr Opin Pulm Med. 2005;11:1–6. doi: 10.1097/01.mcp.0000146779.26339.d8. [DOI] [PubMed] [Google Scholar]
- 4.Rennard SI. Epithelial cells and fibroblasts. Novartis Found Symp. 2001;234:104–112. discussion 112-109. [PubMed] [Google Scholar]
- 5.Fang Q, Liu X, Abe S, Kobayashi T, Wang XQ, Kohyama T, Hashimoto M, Wyatt T, Rennard SI. Thrombin induces collagen gel contraction partially through PAR1 activation and PKC-epsilon. Eur Respir J. 2004;24:918–924. doi: 10.1183/09031936.04.00005704. [DOI] [PubMed] [Google Scholar]
- 6.Gullberg D, Tingstrom A, Thuresson AC, Olsson L, Terracio L, Borg TK, Rubin K. Beta 1 integrin-mediated collagen gel contraction is stimulated by PDGF. Exp Cell Res. 1990;186:264–272. doi: 10.1016/0014-4827(90)90305-t. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Wen FQ, Kobayashi T, Abe S, Fang Q, Piek E, Bottinger EP, Roberts AB, Rennard SI. Smad3 mediates the TGF-beta-induced contraction of type I collagen gels by mouse embryo fibroblasts. Cell Motil Cytoskeleton. 2003;54:248–253. doi: 10.1002/cm.10098. [DOI] [PubMed] [Google Scholar]
- 8.Kohyama T, Wyatt TA, Liu X, Wen FQ, Kobayashi T, Fang Q, Kim HJ, Rennard SI. PGD(2) modulates fibroblast-mediated native collagen gel contraction. Am J Respir Cell Mol Biol. 2002;27:375–381. doi: 10.1165/rcmb.4830. [DOI] [PubMed] [Google Scholar]
- 9.Coughlin SR. Protease-activated receptors in the cardiovascular system. Cold Spring Harb Symp Quant Biol. 2002;67:197–208. doi: 10.1101/sqb.2002.67.197. [DOI] [PubMed] [Google Scholar]
- 10.Reed CE, Kita H. The role of protease activation of inflammation in allergic respiratory diseases. J Allergy Clin Immunol. 2004;114:997–1008. doi: 10.1016/j.jaci.2004.07.060. quiz 1009. [DOI] [PubMed] [Google Scholar]
- 11.Hollenberg MD, Houle S. Proteinases as hormone-like signal messengers. Swiss Med Wkly. 2005;135:425–432. doi: 10.4414/smw.2005.11037. [DOI] [PubMed] [Google Scholar]
- 12.Mackay HJ, Twelves CJ. Targeting the protein kinase C family: are we there yet? Nat Rev Cancer. 2007;7:554–562. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 13.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 14.Luzina IG, Highsmith K, Pochetuhen K, Nacu N, Rao JN, Atamas SP. PKCalpha mediates CCL18-stimulated collagen production in pulmonary fibroblasts. Am J Respir Cell Mol Biol. 2006;35:298–305. doi: 10.1165/rcmb.2006-0033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shukla A, Lounsbury KM, Barrett TF, Gell J, Rincon M, Butnor KJ, Taatjes DJ, Davis GS, Vacek P, Nakayama KI, Nakayama K, Steele C, Mossman BT. Asbestos-induced peribronchiolar cell proliferation and cytokine production are attenuated in lungs of protein kinase C-delta knockout mice. Am J Pathol. 2007;170:140–151. doi: 10.2353/ajpath.2007.060381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park JA, He F, Martin LD, Li Y, Chorley BN, Adler KB. Human neutrophil elastase induces hypersecretion of mucin from well-differentiated human bronchial epithelial cells in vitro via a protein kinase C{delta}-mediated mechanism. Am J Pathol. 2005;167:651–661. doi: 10.1016/s0002-9440(10)62040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altman A, Villalba M. Protein kinase C-theta (PKC theta): a key enzyme in T cell life and death. J Biochem. 2002;132:841–846. doi: 10.1093/oxfordjournals.jbchem.a003295. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi K, Altman A. Protein kinase C theta (PKCtheta): a key player in T cell life and death. Pharmacol Res. 2007;55:537–544. doi: 10.1016/j.phrs.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farhadi A, Keshavarzian A, Ranjbaran Z, Fields JZ, Banan A. The role of protein kinase C isoforms in modulating injury and repair of the intestinal barrier. J Pharmacol Exp Ther. 2006;316:1–7. doi: 10.1124/jpet.105.085449. [DOI] [PubMed] [Google Scholar]
- 20.Liu XD, Umino T, Ertl R, Veys T, Skold CM, Takigawa K, Romberger DJ, Spurzem JR, Zhu YK, Kohyama T, Wang H, Rennard SI. Persistence of TGF-beta1 induction of increased fibroblast contractility. In Vitro Cell Dev Biol Anim. 2001;37:193–201. doi: 10.1290/1071-2690(2001)037<0193:POTIOI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 21.Wyatt TA, Slager RE, Devasure J, Auvermann BW, Mulhern ML, Von Essen S, Mathisen T, Floreani AA, Romberger DJ. Feedlot dust stimulation of interleukin-6 and -8 requires protein kinase Cepsilon in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1163–1170. doi: 10.1152/ajplung.00103.2007. [DOI] [PubMed] [Google Scholar]
- 22.Hannun YA, Loomis CR, Bell RM. Activation of protein kinase C by Triton X-100 mixed micelles containing diacylglycerol and phosphatidylserine. J Biol Chem. 1985;260:10039–10043. [PubMed] [Google Scholar]
- 23.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 24.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. Embo J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C delta (PKC delta): activation mechanisms and functions. J Biochem. 2002;132:831–839. doi: 10.1093/oxfordjournals.jbchem.a003294. [DOI] [PubMed] [Google Scholar]
- 27.Lee JY, Hannun YA, Obeid LM. Functional dichotomy of protein kinase C (PKC) in tumor necrosis factor-alpha (TNF-alpha) signal transduction in L929 cells. Translocation and inactivation of PKC by TNF-alpha. J Biol Chem. 2000;275:29290–29298. doi: 10.1074/jbc.M000170200. [DOI] [PubMed] [Google Scholar]
- 28.Lee JY, Hannun YA, Obeid LM. Ceramide inactivates cellular protein kinase Calpha. J Biol Chem. 1996;271:13169–13174. doi: 10.1074/jbc.271.22.13169. [DOI] [PubMed] [Google Scholar]
- 29.Ward NE, Gravitt KR, O’Brian CA. Irreversible inactivation of protein kinase C by a peptide-substrate analog. J Biol Chem. 1995;270:8056–8060. doi: 10.1074/jbc.270.14.8056. [DOI] [PubMed] [Google Scholar]
- 30.Domenicotti C, Paola D, Vitali A, Nitti M, Cottalasso D, Melloni E, Poli G, Marinari UM, Pronzato MA. Mechanisms of inactivation of hepatocyte protein kinase C isoforms following acute ethanol treatment. Free Radic Biol Med. 1998;25:529–535. doi: 10.1016/s0891-5849(98)00079-3. [DOI] [PubMed] [Google Scholar]
- 31.Kucich U, Rosenbloom JC, Abrams WR, Rosenbloom J. Transforming growth factor-beta stabilizes elastin mRNA by a pathway requiring active Smads, protein kinase C-delta, and p38. Am J Respir Cell Mol Biol. 2002;26:183–188. doi: 10.1165/ajrcmb.26.2.4666. [DOI] [PubMed] [Google Scholar]
- 32.Assender JW, Gee JM, Lewis I, Ellis IO, Robertson JF, Nicholson RI. Protein kinase C isoform expression as a predictor of disease outcome on endocrine therapy in breast cancer. J Clin Pathol. 2007;60:1216–1221. doi: 10.1136/jcp.2006.041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aziz MH, Manoharan HT, Sand JM, Verma AK. Protein kinase Cepsilon interacts with Stat3 and regulates its activation that is essential for the development of skin cancer. Mol Carcinog. 2007;46:646–653. doi: 10.1002/mc.20356. [DOI] [PubMed] [Google Scholar]
- 34.Churchill EN, Mochly-Rosen D. The roles of PKCdelta and epsilon isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem Soc Trans. 2007;35:1040–1042. doi: 10.1042/BST0351040. [DOI] [PubMed] [Google Scholar]
- 35.Kamamoto F, Paggiaro AO, Rodas A, Herson MR, Mathor MB, Ferreira MC. A wound contraction experimental model for studying keloids and wound-healing modulators. Artif Organs. 2003;27:701–705. doi: 10.1046/j.1525-1594.2003.07277.x. [DOI] [PubMed] [Google Scholar]