

Abstract
Activation of nicotinic acetylcholine receptors (nAChRs) on neurons engages calcium-dependent signaling pathways regulating numerous events. Receptors containing α7 subunits (α7-nAChRs) are prominent in this because of their abundance and high relative calcium permeability. We show here that EphB2 receptors are co-localized with postsynaptic α7-nAChRs on chick ciliary ganglion neurons and that treatment of the cells with an ephrinB1 construct to activate the EphB receptors exerts physical restraints on both classes of receptors, diminishing their dispersal after spine retraction or lipid raft disruption. Moreover, the ephrinB1/EphB receptor complex specifically enhances the ability of α7-nAChRs to activate the transcription factor CREB, acting through a pathway including a receptor tyrosine kinase, a Src family member, PI3 kinase, and protein kinase A most distally. The enhancement does not appear to result from a change in the α7-nAChR current amplitude, suggesting a downstream target. The results demonstrate a role for ephrin/EphB action in nicotinic signaling.
Keywords: nicotinic, EphB2, ephrin, receptor, ciliary
Introduction
EphB receptor tyrosine kinases are widely expressed in the brain (Yamaguchi and Pasquale, 2004). Activation of postsynaptic EphB2 receptors (EphB2Rs) by their presynaptic ligand ephrinB helps organize glutamatergic synapses. The extracellular portion of EphB2Rs interacts directly with NMDA receptors, promoting receptor clustering and synapse formation/maturation (Dalva et al., 2000). The intracellular portion of EphB2Rs is instrumental in augmenting calcium signaling by NMDA receptors and downstream effects, including altered gene expression (Takasu et al., 2002). Moreover, ephrinB/EphB2R interactions can recruit AMPA receptors to synaptic locations and support dendritic spine formation (Kayser et al., 2006).
A role for EphB2Rs in nicotinic signaling pathways has not been reported. The chick parasympathetic ciliary ganglion (CG) offers a useful preparation for examining the possibility. The CG contains ciliary neurons that innervate striated muscle in the iris and ciliary body, and choroid neurons that innervate smooth muscle in the choroid layer (Dryer, 1994). Both neuronal populations receive cholinergic input from the accessory oculomotor nucleus in the midbrain which activates homopentameric α7-containing nicotinic acetylcholine receptors (α7-nAChRs) and heteromeric α3*-nAChRs on the neurons. On ciliary neurons, the α7-nAChRs are concentrated on somatic spines arranged in clumps and embedded in lipid rafts (Shoop et al., 1999, 2002; Bruses et al., 2001). The α3*-nAChRs are concentrated in postsynaptic densities (Jacob et al., 1984; Wilson Horch and Sargent, 1995; Williams et al., 1998). Both can be activated by transmitter released from the large preganglionic caylx engulfing individual ciliary neurons (Zhang et al., 1996; Ullian et al., 1997; Coggan et al., 2005).
While both types of nicotinic receptors can participate in synaptic signaling in the ganglion (Chang and Berg, 1999), the α7-nAChRs due to their location and high relative calcium permeability (Bertrand et al., 1993; Seguela et al., 1993) also mediate local changes in calcium levels (Shoop et al., 2002). This calcium influx can have a number of consequences, including control of gene expression (Chang and Berg, 2001). Scaffold proteins, notably members of the PSD-95 family and APC, interact with either or both types of receptors (Conroy et al., 2003; Temburni et al., 2004; Farias et al., 2007), offering mechanisms for tethering signal-transducing components. And recently, the synaptogenic cell adhesion molecules neuroligin and L1 have both been shown to be expressed by CG neurons, to co-localize with nicotinic receptors on the neurons, and to influence synapse formation/stabilization (Triana-Baltzer et al., 2006; Conroy et al., 2007; Ross and Conroy, in press). These components suggest a molecular organization at nicotinic synapses that has unexpected features in common with glutamatergic synapses.
We show here that CG neurons express EphB2Rs, which co-localize with α7-nAChRs on the cell surface. Though EphB2Rs do not appear to interact directly with the α7-nAChRs, EphBR activation physically constrains α7-nAChRs on the neurons and enhances their downstream signaling. These effects can be elicited by treating with an ephrinB1 construct which serves as an agonist of EphBRs, and they are specific for the α7-nAChR. Further, we identify a multi-component pathway of kinases necessary for the ephrinB/EphBR-mediated enhancement of α7-nAChR function.
Results
EphB2Rs Co-Distribute with α7-nAChRs in Chick Ciliary Ganglia
EphB2Rs are expressed in the chick ciliary ganglion (CG) and reach their highest relative levels during the period of naturally-occurring neuronal cell death in the ganglion, i.e. embryonic day (E) 8-E14 (Landmesser and Pilar, 1974). This was seen by performing quantitative Western blot analysis on extracts prepared from CGs taken at various times and normalizing either for the amount of ganglionic protein or number of neurons present (Fig. 1). The EphB2R component was detected with an anti-EphB2R antibody (Ab) raised against the chick species and affinity purified for specificity (Holash and Pasquale, 1995; and see Methods). The slight shift in EphB2R distribution towards older times when normalized per neuron, as opposed to protein, reflects the fact that only half as many neurons are present at later times but the ganglion contains more protein (larger neurons, more nonneuronal cells).
Figure 1.
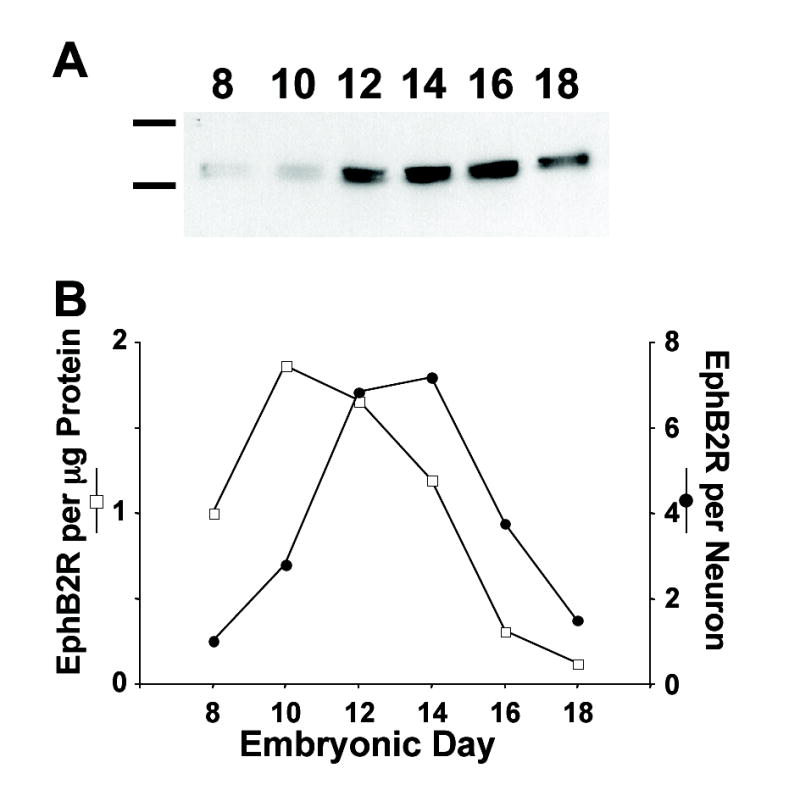
Developmental timecourse for EphB2R appearance in chick ciliary ganglia. (A) Western blots of embryonic ciliary ganglia dissected at the indicated times, solubilized, electrophoresed, and probed with anti-EphB2R Ab. Size standards (left): 116 (βgalactosidase) and 200 (myosin) kD. (B) Quantification of the Western blots, normalizing for the amount of ganglionic protein (open squares) or for the number of neurons present at each age (filled circles). Peak levels of EphB2R occur at E12–E14 and subsequently decline.
Immunostaining for EphB2Rs shows that the receptors are arranged in large clusters on the neurons. Co-staining for α7-nAChRs reveals that the two receptor types co-distribute on CGs. Highest levels of EphB2R staining are found at E14, consistent with the Western blot analysis, but even at earlier and later times, the receptors largely co-distribute with α7-nAChRs (Fig. 2). The staining for EphB2Rs using the anti-chick EphB2R Ab cited above is specific; no staining was seen when a nonimmune immunoglobulin was substituted for the primary anti-EphB2R Ab.
Figure 2.
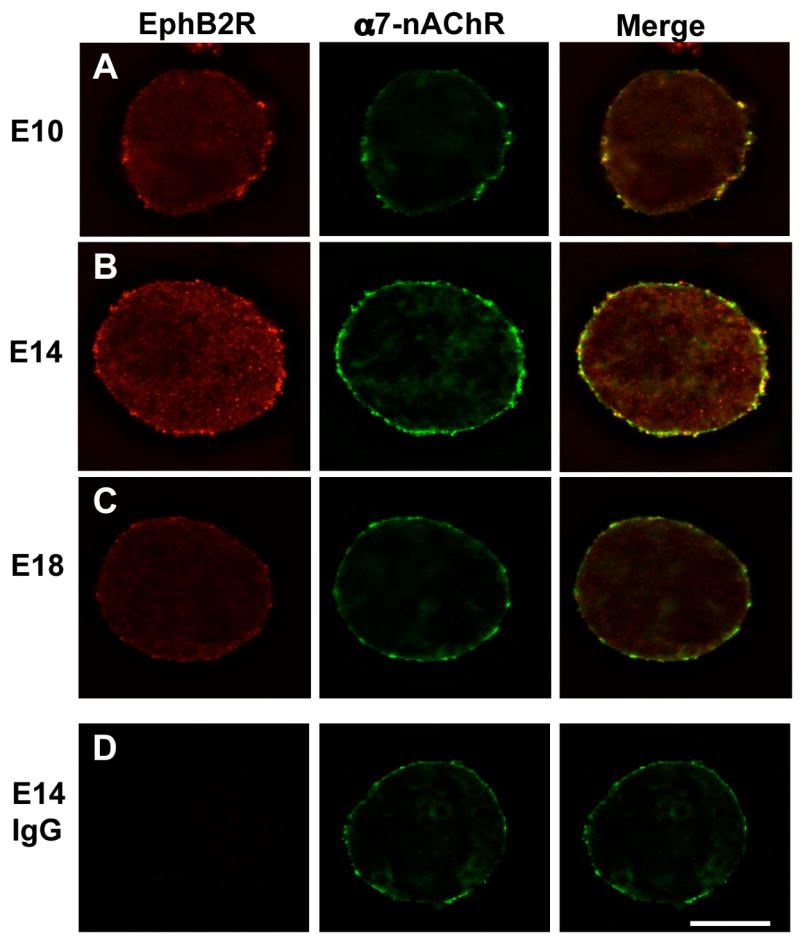
Surface distribution of EphB2R on CG neurons. Embryonic ganglia of the indicated ages were dissociated and co-stained for EphB2Rs (left; red) and α7-nAChRs (middle; green) , and the images overlaid (right; yellow). (A) E10. (B) E14. (C) E18. (D) E14 with non-immune IgG substituted for the anti-EphB2R Ab as a negative control. Scale bar: 10 μm. EphB2Rs co-distribute with α7-nAChRs on the neuron surface, and are most prominent at E14.
EphB2R/α7-nAChR Interactions
The fact that EphB2Rs have previously been shown to interact directly with NMDA receptors via extracellular domains (Dalva et al., 2000) led us to test whether the same might occur with α7-nAChRs. Numerous attempts to demonstrate co-immunoprecipitation of EphB2Rs and α7-nAChRs from detergent-solubilized E14 CGs, however, failed to detect association (data not shown). The same conditions had previously been successful in demonstrating linkage between α7-nAChRs and PDZ-containing proteins (Conroy et al, 2003) and were able to reveal a small amount of specific co-immunoprecipitation between α7-nAChRs and a Src family member that co-distributes with the receptors in lipid rafts on the neurons (data not shown).
Despite the lack of co-immunoprecipitation, a specific link between EphBRs and α7-nAChRs was revealed in another way. The large clusters of α7-nAChRs on E14 CG neurons represent groups of somatic spines embedded with the receptors and encompassed by lipid rafts (Shoop et al., 1999, 2002; Bruses et al., 2001). Treatment with latrunculin A to disassemble filamentous actin (F-actin) and collapse the spines causes the α7-nAChR clusters to disperse. Similarly, treatment with methyl-β-cyclodextrin (MβCD) to extract cholesterol and disrupt the lipid rafts disperses the α7-nAChR clusters. The treatments also disperse the EphB2R clusters (Fig. 3). Strikingly, conditions that activate the EphB2Rs provides resistance against the dispersal both for EphB2Rs and for α7-nAChRs. This was found by treating the neurons with an EphB2R ligand, ephrinB1-Fc, together with anti-Fc Ab to cluster the ligand. This procedure has been used previously to activate EphB2Rs (Dalva et al., 2000). (Because the ligand activates other EphBRs as well, herein we refer to ephrinB1 effects as mediated by EphBRs as a class.) With E14 CG neurons, the treatment prevented dispersal of the α7-nAChR clusters both after dispersal of lipid rafts (Fig. 3A) and after spine collapse (Fig. 3B). The latrunculin A did indeed collapse the F-actin, as shown by co-staining with phalloidin for F-actin. Notably ephrinB1-Fc/Ab did not prevent the MβCD treatment from dispersing the Src family member(s) associated with the lipid raft (Fig. 3C). Staining with cholera toxin for the ganglioside GM1 confirmed that the MβCD treatment disrupted the lipid rafts. Taken together, the results suggest a specific, though not direct, interaction of EphBRs with α7-nAChRs on the neurons.
Figure 3.
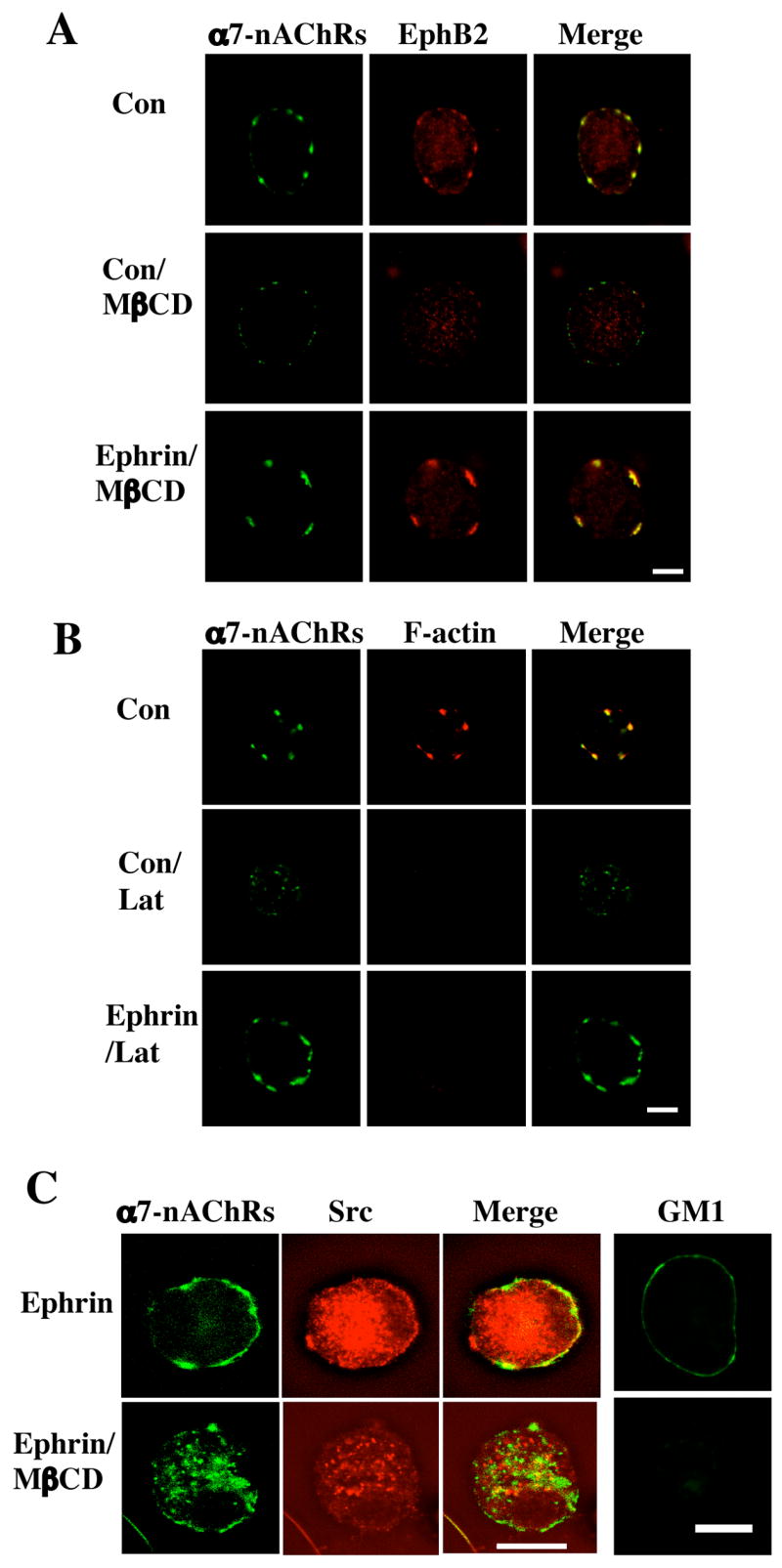
Retention of α7-nAChRs by ephrinB1/EphB2Rs after spine collapse or lipid raft dispersal. Freshly dissociated E14 CG neurons were either treated with ephrinB1-Fc/Ab to activate EphBRs (Ephrin) or with Fc/Ab as a control (Con) and then treated with methyl-βcyclodextrin (MβCD) to extract cholesterol and disperse lipid rafts or with latrunculin A (Lat) to collapse F-actin and induce spine retraction. The cells were then labeled for α7-nAChRs (left; green) and either (A) EphB2Rs, (B) F-actin, or (C) Src (middle; red), and the images merged (right; yellow). Alternatively, the cells were immunostained with cholera toxin for the ganglioside GM1, confirming that MβCD treatment dispersed the lipid rafts (C, far right). Scale bar: 10 μm. EphrinB1-Fc/Ab protected both EphB2Rs and α7-nAChRs from dispersal after spine retraction or raft disruption; it did not protect Src from dispersal.
EphrinB1 Protection of Functional α7-nAChRs
Nicotine-induced whole-cell currents were analyzed to determine whether the ephrinB1-Fc/Ab treatment protected the functional response of α7-nAChRs as it did the distribution following lipid raft dispersal. The rapidly decaying component of the nicotine-induced whole-cell response can be attributed to α7-nAChRs on chick CG neurons while the slowly decaying component of the response is produced by α3*-nAChRs (Zhang et al., 1994; Liu and Berg, 1999). Patch-clamp analysis of freshly dissociated E14 CG neurons showed that EphBR activation by ephrinB1-Fc/Ab had no significant effect on either the α7-nAChR component or the α3*-nAChR component of nicotine-evoked currents (Fig. 4). Similarly, no differences were found in the decay constants (tau values) for the fast and slow components when compared for control and ephrin treatment conditions (data not shown). The ephrin treatment did, however, provide partial protection against loss of the whole-cell nicotinic response following cholesterol extraction and lipid raft dispersal by MβCD. The was specific for the α7-nAChR component (Fig. 4).
Figure 4.

Effects of ephrinB1 treatment on nicotine-induced whole-cell currents. (A) Representative traces of nicotine-induced whole-cell responses in E14 neurons after treatment with Fc/Ab as a control (Fc, left) or with ephrinB1-Fc/Ab (EphrinB, right) and then incubated in culture medium (Con, top) or extracted with MβCD to extract cholesterol and disperse lipid rafts (MβCD, bottom). (B) Quantification of the peak currents, normalized for capacitance to correct for differences in cell size. (C) Quantification of the peak current components contributed by α7-nAChRs (left) and α3*-nAChRs (right), separated based on decay rates (fast vs. slow, respectively). Values represent the mean ± SEM of 12–18 cells. Asterisks, p < 0.05. The ephrinB1 treatment does not significantly enhance the nicotine-induced whole-current but does partially protect the rapidly decaying α7-nAChR component of the response against MβCD-induced loss.
EphBR Modulation of α7-nAChR downstream signaling
Nicotinic stimulation of α7-nAChRs activates calcium-dependent intracellular pathways, including ones capable of activating the transcription factor CREB and altering gene expression (Chang and Berg, 2001; Hu et al., 2002). We find that treating E14 CG neurons with ephrinB1-Fc/Ab specifically enhances the ability of α7-nAChRs to activate CREB. This was assessed by monitoring the appearance of phospho-CREB (pCREB) in the nucleus, sustained at least 20 min after termination of the nicotinic stimulation (long-term pCREB). Long-term pCREB in CG neurons under these conditions correlates with changes in gene expression (Chang and Berg, 2001).
In CG neurons, the ability of α7-nAChRs to produce a sustained activation of CREB depends on voltage-gated calcium channels failing to deliver significant calcium to the neurons. For this reason, experiments designed to reveal long-term pCREB require the presence of blockers such as cadmium or nifedipine being present along with the nicotine (Chang and Berg, 2001). Treating E14 neurons with ephrinB1-Fc/Ab significantly increased the proportion of cells displaying long-term pCREB after nicotine/cadmium treatment, and all of the increase was prevented by blocking α7-nAChRs with βbungarotoxin (αBgt) during the exposure to nicotine (Fig. 5). Treating neurons with thapsigargin to deplete internal calcium stores did reduce the baseline amount of nicotinic-induced pCREB but did not affect the increase induced by the ephrinB1 treatment. (Internal calcium stores are apparently required for the α3*-nAChR component of the effect but not the α7-nAChR component.) The ephrinB1-mediated increase did appear to depend, however, on a receptor tyrosine kinase (blocked by K252a; also by lavendustin A, not shown), a Src family member (blocked by SU6656; also by PP2 but not PP3 as a negative control, not shown), PI3 kinase (blocked by Ly294002), and protein kinase A (PKA, blocked by KT5720). The dependence on a receptor tyrosine kinase, PI3 kinase, and PKA was confined to the ephrinB1/EphBR-mediated increase in nicotine-induced pCREB. The dependence on a Src family member appeared to be more general, decreasing pCREB levels below that seen with nicotine in the absence of EphB2R activation (Fig. 5).
Figure 5.
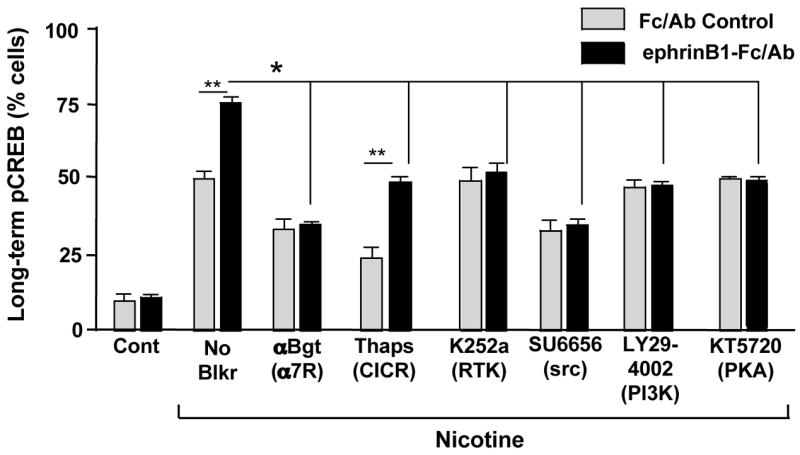
EphrinB1 enhancement of α7-nAChR-induced long-lasting pCREB. E14 CG neurons were treated with Fc/Ab (Con) or ephrinB1-Fc/Ab (Ephrin) and the indicated compounds followed by 10 μM nicotine (Nic) for 5 min in the presence of 200 μM cadmium to block voltage-gated calcium channels, and then incubated an additional 20 min in the absence of nicotine before fixing, staining, and immunostaining for nuclear pCREB. Values represent the mean ± SEM for the proportion of labeled cells. Asterisk, p < 0.05; double asterisk, p < 0.01; multi-line comparisons: group members compared to “No Blkr”. EhprinB1 treatment increased the proportion of cells showing long-term pCREB, and the increase was entirely attributable to α7-nAChRs (blocked by αBgt) and dependent on a tyrosine receptor kinase (blocked by K252a), a Src family member (blocked by SU6656; also blocked by PP2 but not PP3, not shown), PI3 kinase (blocked by LY294002) and PKA (blocked by KT5720) but not on calcium release from internal stores (not blocked by thapsigargin).
Intermediates Executing EphrinB1/EphBR Enhancement of α7-nAChR Signaling
Analysis of short-term pCREB formation yielded the same pattern of EphBR/α7-nAChR effects and was used to further dissect the mechanism. In these assays nuclear pCREB is assessed immediately after stimulating with nicotine in the presence of cadmium. The cadmium is included to prevent contributions for voltage-gated calcium channels to short-term pCREB (Chang and Berg, 2001). Nicotine treatment again caused a substantial increase in the proportion of E14 neurons displaying pCREB, and the proportion was increased by ephrinB1-Fc/Ab treatment to activate EphBRs (Fig. 6A). Removal of calcium or blockade of CaMKII with KN93 (but not KN92 as a negative control; not shown) completely prevented nicotine-induced pCREB, including the portion representing the ephrin/EphBR-induced increase. Inhibition of MAPK was without effect (data not shown); MAPK acts downstream of CaMKII in generating nicotine-induced long-term pCREB and is not required for short-term pCREB (Chang and Berg, 2001). As found for long-term pCREB, the ephrin/EphBR enhancement of nicotine-induced short-term pCREB was completely blocked by blockers of receptor tyrosine kinases (K252a), Src family members (SU6656), PI3 kinase (Ly294002), and PKA (KT5720). None of these kinases were required, however, for the baseline short-term pCREB formation induced by nicotinic stimulation, including the contributions of both α7-nAChRs and α3*-nAChRs. Thus the blockers reduced the pCREB incidence to that seen in the absence of ephrin treatment, while the specific α7-nAChR antagonist αBgt not only eliminated the ephrin-enhanced portion of pCREB but also the portion attributable to α7-nAChRs in the absence of ephrinB1/EphR activation (Fig. 6A). The remaining nicotine-induced pCREB seen in the presence of αBgt is the result of α3*-nAChR activation (Chang and Berg, 2003).
Figure 6.

Enzyme pathway mediating the ephrinB1 enhancement of nicotine-induced pCREB. E14 neurons were treated as in Fig. 5, including the addition of cadmium to block voltage-gated calcium channels, except that the 20 min rinse was omitted so that short-term pCREB could be assessed. Values represent the mean ± SEM for the proportion of labeled cells (n = 200–300 cells per condition from each of 3–6 separate experiments). Asterisk, p < 0.05; double asterisk, p < 0.01; multi-line comparisons: group members compared to “No Blkr”. (A) EphrinB1-Fc/Ab treatment increased the proportion of cells with short-term pCREB, acting specifically through α7-nAChRs (stimulated by nicotine; blocked by αBgt), and the effect depended on a receptor tyrosine kinase, a Src family member, PI3 kinase, and PKA. None of these kinases were required for short-term pCREB induced by nicotine via either α3*-nAChRs or α7-nAChRs in the absence of ephrinB1 treatment. CaMKII/IV was required for both α7- and α3*-nAChR-mediated pCREB (blocked by KN93 but not by control KN92). (B) Combining the PKA agonist 8Br-cAMP (8Br) with either SU6656 or LY294002 demonstrated that both the Src family member and PI3 kinase act upstream of PKA in mediating ephrinB1 enhancement of α7-nAChR-produced pCREB. The receptor or kinase being blocked is indicated in parentheses.
We then examined the sequence of kinase contributions to the ephrin/EphBR effect on the α7-nAChR component of short-term pCREB formation. An agonist of PKA, 8-bromo-cyclic-AMP (8Br-cAMP), replicated the ephrin/EphBR effect on pCREB and was completely blocked by αBgt, demonstrating that it acted through α7-nAChRs (Fig. 6B). Importantly 8Br-cAMP was able to override the inhibitory effects of both SU6656 and LY294002, indicating that PKA acted downstream from the Src family member and PI3K. In contrast, 8Br-cAMP was unable to override the inhibition produced by a blocker of CaMKII, consistent with CaMKII being needed for all nicotine-induced pCREB. The results indicate that (1) the 8Br-cAMP effect was confined to the same portion affected by ephrinB1 treatment, (2) the ephrinB1/EphBR complex specifically enhanced the ability of α7-nAChRs to induce pCREB, and (3) the enhancement employed a receptor tyrosine kinase, a Src family member, PI3 kinase, and PKA, with PKA being most distal in the pathway.
PKA is unlikely to exert its effects on pCREB levels by altering the current response of α7-nAChRs. Treating freshly dissociated E14 CG neurons with 8Br-cAMP for 2 hours did not change the mean peak current (180 ± 15 vs. 180 ± 27 pA/pF) or mean rise time (8.4 ± 0.2 vs. 9.2 ± 1.7 ms) of the nicotine-induced whole-cell response for 8Br-cAMP-treated vs control cells, respectively (mean ± SEM; n = 8 cells in each case). Nor did 8Br-cAMP treatment alter the mean rate of decay of the rapidly decaying component attributable to α7-nAChRs (tau values of 54 ± 10 vs. 48 ± 8 ms for 8Br-cAMP vs control; mean ± SEM, n = 11 and 14 cells, respectively). The results indicate that the ephrin/EphBR effect on α7-nAChR signaling is exerted at a site subsequent to current flow through the receptor.
Discussion
We show here for the first time that EphBRs participate in nicotinic signaling pathways, co-distributing with α7-nAChRs and enhancing their function. The ephrinB1/EphBR interaction with α7-nAChRs does not markedly affect receptor currents, and the receptors do not appear to interact directly. Nonetheless, ephrinB1/EphBR interactions partially protect α7-nAChRs against dispersal following spine retraction or lipid raft disruption. Functionally, ephrinB1/EphBR interactions enhance α7-nAChR signaling uniquely to induce nuclear pCREB formation. The kinases implicated in this enhancement – a receptor tyrosine kinase, a Src family member, PI3 kinase, and PKA – are not needed for basal level induction of short-term pCREB by α7-nAChRs but are needed for the eprinB1-induced increase, consistent with a specific action. The timing of EphB2R expression in the CG suggests that the role of the ephrinB1/EphBR complex in nicotinic pathways may primarily be one of regulating development.
The anti-EphB2R Ab used here revealed EphB2Rs on CG neurons and indicated both their relative abundance during development and their co-distribution with α7-nAChRs. The effects of ephrinB1-Fc/Ab treatment, however, need not be confined to EphB2Rs but would also activate other EphBRs if present on the neurons. For this reason, reference is consistently made to the broad family of EphBRs when interpreting ephrinB1 effects on α7-nAChR signaling. The requirement for a receptor tyrosine kinase, implicated by the effects of K252a, may reflect that of an EphBR. Consistent with this is the observation that no receptor tyrosine kinase activity is needed for either the short- or long-term generation of pCREB by either α7-nAChRs or α3*-nAChRs in the absence of ephrinB1 treatment. The Src family member, implicated by the effects of the specific Src inhibitor SU6656, also acts specifically to augment α7-nAChR-induced short-term pCREB. In the long-term assay, the Src family member reduces pCREB levels below that seen in the absence of ephrinB1 treatment, indicating that it has additional targets in this case. The blocker used, SU6656 (and PP2; data not shown) does not discriminate among Src family members, so the results do not identify the specific candidate. A likely source, however, is the Src family member(s) shown here to be embedded in the lipid rafts on CG neurons and thereby having close proximity. Direct tyrosine phosphorylation has been reported to depress α7-nAChR function (Charpantier et al., 2005) while indirect effects have been reported to decrease α7-nAChR number (Cho et al., 2005). Neither effect is likely to explain the ephrinB1/EphBR-mediated enhancement of α7-nAChR-induced pCREB. Both the receptor tyrosine kinase and Src family member act indirectly here, i.e. through PKA, to achieve their effects on α7-nAChRs, and the effect is an increase rather than a decrease in α7-nAChR effectiveness.
PKA represented the most distal kinase in the pathway shown here to mediate the enhancement of nicotine-induced pCREB. Use of 8Br-cAMP to circumvent the pathway and activate PKA directly did mimic ephrinB1 treatment, but the target substrate of PKA has yet to be identified. Incubating E14 CG neurons with 8Br-cAMP triggers phosphorylation of α7-nAChRs (Z. Liu and D. Berg, unpublished results), but the phosphorylation does not appear to alter the electrophysiological response of the receptors. An alternative possibility is that ephrinB1 treatment increases the amount or persistence of intracellular calcium resulting from α7-nAChR activation. Imaging experiments to detect calcium elevations, however, failed to show any reproducible change in the α7-nAChR response following ephrin treatment (Z. Liu and D. Berg, unpublished results). In view of this, likely alternatives to consider are that PKA phosphorylation of key substrates results in more efficient sequestration or functionality of rate-limiting components needed to transduce the α7-nAChR effect.
EphB2Rs are unlikely to associate directly with α7-nAChRs, given the inability to co-immunoprecipitate the two from detergent-solubilized CG extracts. There is a clear physical link, however, demonstrated by the ability of ephrinB1 treatment to restrain both EphB2Rs and α7-nAChRs following spine collapse and lipid raft dispersal. One candidate for the link would be the PDZ-containing scaffold protein PICK1. PICK1 has recently been shown to bind to α7-nAChRs (Baer et al., 2007), and EphB2Rs are thought to bind PICK1 as well (Torres et al., 1998). PICK1 is usually not found concentrated at synapses, however, and in the case of AMPA receptors is thought to participate in trafficking both to and from the surface, rather than synaptic stabilization (Lu and Ziff, 2005; Cao et al., 2007). Nonetheless, the PDZ binding domains of EphB2Rs, as well as other binding motifs in the receptors, suggest numerous possible indirect links to α7-nAChRs.
The peak of EphB2R expression in the CG, normalized for ganglionic protein, coincides with a time (E10-E14) during which important developmental changes occur in the ganglion. All of the neurons are innervated by E7, but naturally-occurring neuronal die-off is largely confined to the period E8-E14 and removes half of the neurons in the embryonic ganglion (Landmesser and Pilar, 1974). Recently it has been shown that α7-nAChRs play a determinant role in this die-off; blockade of α7-nAChRs in vivo enables many neurons to survive that otherwise would have died (Hruska and Nishi, 2007). E8-E14 is also a critical time during which the chloride gradient reverses developmentally in CG neurons, allowing GABA to become inhibitory in the ganglion, rather than excitatory as is the case initially. Both α7-nAChRs and α3*-nAChRs contribute to this developmental switch (Liu et al., 2006). The ability of the ephrinB1/EphBR complex to interact with α7-nAChRs and enhance their downstream signaling at this time could have important consequences for these developmental transitions.
Experimental Methods
Western Blots
Western blot analyses were performed on freshly dissected and detergent-solubilized E8-E18 CGs as previously described (Conroy et al., 2003). The blots were probed with an anti-chick EphB2R Ab kindly provided by Dr. Elena Pasquale (Burnham Institute; La Jolla, CA). In addition to the affinity purification initially described (Holash and Pasquale, 1995), the EphB2R Abs used here were purified further by cross-absorption with a GST-fusion protein containing the relevant region of the EphB4R (equivalent to the EphB2R portion used as antigen) to eliminate any cross-reaction (E. Pasquale, personal communication). Values were normalized to the amount of ganglionic protein (Chiappinelli and Giacobini, 1978) and to the number of neurons (Landmesser and Pilar, 1974). Co-immunoprecipitations were performed as described (Conroy et al., 2003).
Cell Preparations
Dissociated E10-18 CG neurons were prepared and maintained for 1–4 hours on glass coverslips lacking lysed fibroblasts as previously described for E14 CG neurons (Conroy et al., 2003).
Fluorescent Labeling and Immunostaining
Fluorescence imaging with deconvolving software was used to examine dissociated E14 CG neurons stained for α7-nAChRs with either Alexa488-αBgt or rhodamine-αBgt (both at 100 nM from Molecular Probes, Eugene, OR) prior to fixation as previously described (Conroy et al,. 2003; Liu et al., 2005, 2006). Co-staining with a primary Ab was carried out by fixing the labeled cells in 2–4% PFA for 20–30 min at room temperature and incubating in PBS containing 0.1% Triton X-100 with 5% normal donkey serum and either anti-Src Ab (Src2 polyclonal Ab, 1:500 dilution, Santa Cruz Biotech, Santa Cruz, CA) for 2 hrs at room temperature, or anti-chick EphB2R Ab (1:1000 dilution; see above for source/purification) at 4ºC overnight. Bound Ab was detected by incubating the labeled neurons with cy3-conjugated donkey anti-rabbit polyclonal Ab (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1–2 hrs at room temperature as secondary Ab. Co-staining for F-actin was achieved by incubating fixed cells with rhodamine-phalloidin (1:2000 dilution, Molecular Probes) for 1 hr at 4ºC. In some cases fixed neurons were labeled for α7-nAChRs by incubating with goat anti-α7-nAChR Ab (Santa Cruz; 1:1000 overnight at room temperature) as previously described (Conroy et al., 2003). To stain for the ganglioside GM1, cells were lightly fixed with 0.1% PFA for 20 min, then incubated with fitc-conjugated cholera toxin (1:500 dilution) for 1 hr followed by fixation with 2% PFA for 20 min at room temperature.
For ephrinB1-Fc/Ab treatment, the neurons were incubated 45 min at 37ºC in media containing ephrinB1-Fc peptide (R&D Systems, Minneapolis, MN), together with anti-Fc Ab (Jackson ImmunoResearch Laboratories). The peptide and Ab were mixed at concentrations of 125 μg/ml and 12.5 μg/ml, respectively, and incubated 30 min at room temperature before adding to neurons at a final dilution of 1:250. Controls substituted the Fc fragment for the ephrinB1-Fc.
To remove cholesterol and disrupt lipid rafts on the cell, neurons were treated with 10 mM MβCD at 37°C in media for 45 min. To collapse F-actin and induce spine retraction, cells were treated with 4 μg/ml latrunculin A for 2 hrs. When ephrinB1-Fc/Ab or Fc/Ab treatment was applied, it preceded incubation with the MβCD or latrunculin A.
Electrophysiological Analyses
Freshly dissociated E14 CG neurons were examined for whole-cell currents induced by rapid application of nicotine, and analyzed as previously described (Zhang et al., 1994; Liu and Berg, 1999; Nai et al., 2003). Cells were perfused (1 ml/min) at room temperature with recording solution containing (in mM) 145 NaCl, 5.3 KCl, 0.8 MgSO4, 5.4 CaCl2, 5.6 Glucose, 5 HEPES, supplemented with 10% heat-inactivated horse serum, pH 7.35. Patch pipettes (2 –4 Omega;M ) were filled with intracellular solution containing (in mM) 145.6 CsCl, 1.2CaCl2, 2.0 EGTA, 15.4 glucose, and 5.0 HEPES, pH 7.35. Pipettes were visually guided to the surface of individual neurons, and a giga-ohm seal was formed. Nicotine (20 μM) was rapidly applied using a multibarrel large-bore drug applicator (Zhang et. al., 1994). All voltage-clamp experiments were performed with an Axopatch 200A and pClamp 9 software (Molecular Devices Corp., Sunnyvale, CA). Data were filtered at 2 kHz, sampled at 5 kHz, and pipette capacitance was minimized. All experiments were performed within 5 hours of plating. Analysis of currents was performed using Clampfit 9 software (Molecular Devices Corp.), and statistical analysis was performed using Excel software (Microsoft Corp., Redmond, WA). Incubation with Fc + Fc Ab as a negative control and with ephrinB1-Fc + Fc Ab was as described above. Incubation with 100 μM 8Br-cAMP was for 1 hr.
pCREB Assay
Nicotine-induced short- and long-term pCREB was assayed with freshly dissociated E14 CG neurons as previously described with values representing the proportion of neurons having nuclear pCREB staining (Chang and Berg, 2001; Triana-Baltzer et al., 2006). Nicotine was applied at 10 μM. Cadmium (200 μM) was included in the assays to block voltage-gated calcium channels, preventing their contributions to pCREB formation and degradation. Treatment of cultures with ephrinB1-Fc + Fc antibody (vs controls in which Fc replaced the ephrinB1-Fc) were carried out as previously described for L1-Fc (Triana-Baltzer et al., 2006). Other drug treatments were as follows: K252a (200 nM), KT5720 (2 μM), SU6656 (1 μM), and 8Br-cAMP (100 μM) were all applied for 1 hr prior to nicotine treatment. Thapsigargin (1 μM) was applied for 15 min and αBgt (100 nM) for 30 min prior to nicotine treatment. All other compounds were applied at 10 μM for 1 hr prior to nicotine treatment. When cells were co-incubated with 8Br-cAMP and blockers, the blockers were added first at the concentrations and durations indicated above.
Statistics
Values represent the mean ± SEM of results from the indicated number of experiments. Statistical differences between two means were determined using the two-tailed Student’s t test; comparisons among more than two were determined by one-way ANOVA with Bonferroni post-hoc tests.
Materials
White leghorn chick embryos were obtained locally and maintained in a humidified incubator. Latrunculin A was purchased from Invitrogen (Carlsbad, CA), and KN93, KN92, PP2, PP3, LY294002, and KT5720 were purchased from EMD Chemicals (San Diego, CA). All other reagents were purchased from Sigma (St. Louis, MO) unless otherwise indicated.
Acknowledgments
Grant support was provided by the National Institutes of Health Grants NS12601 and NS35469 and by a Young Investigator grant from the Tobacco-Related Disease Research Program (15KT-0157 to Z.L.). We thank Elena Pasquale (Burnham Institute; La Jolla, CA) for generous gifts of anti-chick EphB2R Ab, and Xiao-Yun Wang for expert technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baer K, Burli T, Huh K-H, Wiesner A, Erb-Vogtli S, Gockeritz-Dujmovic D, Moransard M, Nishimune A, Rees MI, Henley JM, Fritschy J-M, Fuhrer C. PICK1 interacts with α7 neuronal nicotinic receptors and controls their clustering. Mol Cell Neurosci. 2007;35:339–355. doi: 10.1016/j.mcn.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers-Thiéry A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal a7 nicotinic receptor. Proc Natl Acad Sci (USA) 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruses JL, Chauvet N, Rutishauser U. Membrane lipid rafts are necessary for the maintenance of the α7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J Neurosci. 2001;21:504–512. doi: 10.1523/JNEUROSCI.21-02-00504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Xu J, Shen C, Huganir RL, Xia J. PICK1-ICA69 heteromeric BAR domain complex regulates synaptic targeting and surface expression of AMPA receptors. J Neurosci. 2007;27:12945–12956. doi: 10.1523/JNEUROSCI.2040-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Berg DK. Dependence of circuit function on nicotinic acetylcholine receptors containing a7 subunits. J Neurosci. 1999;19:3701–3710. doi: 10.1523/JNEUROSCI.19-10-03701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron. 2001;32:855–865. doi: 10.1016/s0896-6273(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Charpantier E, Wiesner A, Huh K-H, Ogier R, Hoda J-C, Allaman G, Raggenbass M, Feuerbach D, Bertrand D, Fuhrer C. α7 Neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and src-family kinases. J Neurosci. 2005;25:9836–9849. doi: 10.1523/JNEUROSCI.3497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli VA, Giacobini E. Time course of appearance of a-bungarotoxin binding sites during development of chick ciliary ganglion and iris. Neurochem Res. 1978;3:465–478. doi: 10.1007/BF00966328. [DOI] [PubMed] [Google Scholar]
- Cho C-H, Song W, Leitzell K, Teo E, Meleth AD, Quick MW, Lester RA. Rapid upregulation of α7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J Neurosci. 2005;25:3712–3723. doi: 10.1523/JNEUROSCI.5389-03.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggan JS, Bartol TM, Esquenazi E, Stiles JR, Lamont S, Martone ME, Berg DK, Ellisman MH, Sejnowski TJ. Ectopic neurotransmitter release at a neuronal synapse. Science. 2005;309:446–451. doi: 10.1126/science.1108239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy WG, Liu Z, Nai Q, Coggan J, Berg DK. PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron. 2003;38:759–771. doi: 10.1016/s0896-6273(03)00324-6. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Nai Q, Ross B, Naughton G, Berg DK. Postsynaptic neuroligin enhances presynaptic inputs at neuronal nicotinic synapses. Dev Biol. 2007;307:79–91. doi: 10.1016/j.ydbio.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- Dryer S. Functional development of the parasympathetic neurons of the avian ciliary ganglion: a classic model system for the study of neuronal differentiation and development. Prog Neurobiol. 1994;43:281–322. doi: 10.1016/0301-0082(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Farias GG, Valles AS, Colombres M, Godoy JA, Toledo EM, Lukas RJ, Barrantes FJ, Inestrosa NC. Wnt-7a induces presynaptic co-localization of α7-nicotinic acetylcholine receptors and Adenomatous Polyposis Coli in hippocampal neurons. J Neurosci. 2007;27:5313–5325. doi: 10.1523/JNEUROSCI.3934-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holash JA, Pasquale EB. Polarized expression of the receptor protein tyrosine kinase Cek5 in the developing avian visual system. Dev Biol. 1995;172:683–693. doi: 10.1006/dbio.1995.8039. [DOI] [PubMed] [Google Scholar]
- Hruska M, Nishi R. Cell-autonomous inhibition of α7-containing nicotinic acetylcholine receptors prevents death of parasympathetic neurons during development. J Neurosci. 2007;27:11501–11509. doi: 10.1523/JNEUROSCI.3057-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu Q-s, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and non-glutamatergic pathways. Mol Cell Neurosci. 2002;21:616–625. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- Jacob MH, Berg DK, Lindstrom JM. Shared antigenic determinant between the Electrophorus acetylcholine receptor and a synaptic component on chicken ciliary ganglion neurons. Proc Natl Acad Sci (USA) 1984;81:3223–3227. doi: 10.1073/pnas.81.10.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser MS, McClelland AC, Hughes EG, Dalva MB. Intracellular and trans-synaptic regulation of glutamatergic synaptogenesis by EphB receptors. J Neurosci. 2006;26:12152–12164. doi: 10.1523/JNEUROSCI.3072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser L, Pilar G. Synaptic transmission and cell death during normal ganglionic development. J Physiol. 1974;241:737–749. doi: 10.1113/jphysiol.1974.sp010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q-s, Berg DK. Actin filaments and the opposing actions of CaM kinase II and calcineurin in regulating α7-containing nicotinic receptors on chick ciliary ganglion neurons. J Neurosci. 1999;19:10280–10288. doi: 10.1523/JNEUROSCI.19-23-10280.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Tearle AW, Nai Q, Berg DK. Rapid activity-driven SNARE-dependent trafficking of nicotinic receptors. J Neurosci. 2005;25:1159–1168. doi: 10.1523/JNEUROSCI.3953-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. 2006;314:1610–1613. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Nai Q, McIntosh JM, Margiotta JF. Relating neuronal nicotinic acetylcholine receptor subtypes defined by subunit composition and channel function. Mol Pharmacol. 2003;63:311–324. doi: 10.1124/mol.63.2.311. [DOI] [PubMed] [Google Scholar]
- Ross BS, Conroy WG. Capabilities of neurexins in the chick ciliary ganglion. Dev Neurobiol. 2008 doi: 10.1002/dneu.20598. in press. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadichem J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain a7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Martone ME, Yamada N, Ellisman MH, Berg DK. Neuronal acetylcholine receptors with α7 subunits are concentrated on somatic spines for synaptic signaling in embryonic chick ciliary ganglia. J Neurosci. 1999;19:692–704. doi: 10.1523/JNEUROSCI.19-02-00692.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Chang KT, Ellisman MH, Berg DK. Synaptically driven calcium transients via nicotinic receptors on somatic spines. J Neurosci. 2001;21:771–781. doi: 10.1523/JNEUROSCI.21-03-00771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Esquenazi E, Yamada N, Ellisman MH, Berg DK. Ultrastructure of a somatic spine mat for nicotinic signaling in neurons. J Neurosci. 2002;22:748–756. doi: 10.1523/JNEUROSCI.22-03-00748.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science. 2002;295:491–495. doi: 10.1126/science.1065983. [DOI] [PubMed] [Google Scholar]
- Temburni MK, Rosenberg MM, Pathak N, McConnell R, Jacob MH. Neuronal nicotinic synapse assembly requires the adenomatous polyposis coli tumor suppressor protein. J Neurosci. 2004;24:6776–6784. doi: 10.1523/JNEUROSCI.1826-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R, Firestein BL, Dong H, Staudinger J, Olson EN, Huganir RL, Bredt DS, Gale NW, Yancopoulos GD. PDZ proteins bind, cluster, and synaptically colocalize with Eph receptors and their ephrin ligands. Neuron. 1998;21:1453–1463. doi: 10.1016/s0896-6273(00)80663-7. [DOI] [PubMed] [Google Scholar]
- Triana-Baltzer GB, Liu Z, Berg DK. Pre- and postsynaptic actions of L1-CAM in nicotinic pathways. Mol Cell Neurosci. 2006;33:214–226. doi: 10.1016/j.mcn.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Ullian EM, McIntosh JM, Sargent PB. Rapid synaptic transmission in the avian ciliary ganglion is mediated by two distinct classes of nicotinic receptors. J Neurosci. 1997;17:7210–7219. doi: 10.1523/JNEUROSCI.17-19-07210.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Pasquale EB. Eph receptors in the adult brain. Curr Op Neurobiol. 2004;14:288–296. doi: 10.1016/j.conb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Williams BM, Tamburni MK, Schwartz Levey M, Bertrand S, Bertrand D, Jacob MH. The long internal loop of the α3 subunit targets nAChRs to subdomains within individual synapses on neurons in vivo. Nat Neurosci. 1998;1:557–562. doi: 10.1038/2792. [DOI] [PubMed] [Google Scholar]
- Wilson Horch HL, Sargent PB. Perisynaptic surface distribution of multiple classes of nicotinic acetylcholine receptors on neurons in the chicken ciliary ganglion. J Neurosci. 1995;15:7778–7795. doi: 10.1523/JNEUROSCI.15-12-07778.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z-w, Vijayaraghavan S, Berg DK. Neuronal acetylcholine receptors that bind a-bungarotoxin with high affinity function as ligand-gated ion channels. Neuron. 1994;12:167–177. doi: 10.1016/0896-6273(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Zhang Z-w, Coggan JS, Berg DK. Synaptic currents generated by neuronal acetylcholine receptors sensitive to a-bungarotoxin. Neuron. 1996;17:1231–1240. doi: 10.1016/s0896-6273(00)80253-6. [DOI] [PubMed] [Google Scholar]