

Abstract
The HIV-1 envelope glycoprotein gp41 undergoes a sequence of extensive conformational changes while participating in the fusion of the virus with the host cell. Since the discovery of its postfusion conformation, the structure and function of the protease-resistant six-helix bundle (6-HB) have been the subject of extensive investigation. In this work, we describe additional determinants (S528–Q540 and W666–N677) in the fusion peptide proximal region (FP-PR) and the membrane proximal external region (MPER) that stabilize the six-helix bundle and are involved in the interaction of T-20 (FUZEON, an anti-HIV-1 fusion inhibitor drug) with the gp41 FP-PR. Circular dichroism and sedimentation equilibrium measurements indicate that the 1:1 mixture of N′ and C′ peptides comprising residues A541–T569 and I635–K665 from the gp41 first and second helical repeats, HR1 and HR2, respectively, fail to form a stable six-helix bundle. Triglutamic acid and triarginine tags were added to these N′ and C′ peptides, respectively, at the termini distant from the FP-PR and the MPER to alter their pI and increase their solubility at pH 3.5. The tagged HR1 and HR2 peptides were elongated by addition of residues S528–Q540 from the FP-PR and residues W666–N677 from the MPER, respectively. A 1:1 complex of the elongated peptides formed a stable six-helix bundle which melted at 60 °C. These results underscore the importance of a detailed high-resolution characterization of MPER interactions, the results of which may improve our understanding of the structure–function relationship of gp41 and its role in HIV-1 fusion.
The human immunodeficiency virus type 1 (HIV-1)1 envelope protein is initially produced as a precursor glycoprotein gp160, which is proteolytically cleaved into two subunits. The resulting surface subunit (gp120) and transmembrane subunit (gp41) remain noncovalently associated and oligomerize as trimers on the surface of the virion. The transmembrane subunit gp41 mediates fusion between HIV-1 and its target cells (1). After the penetration of the gp41 fusion peptide into the membrane of the target cell, gp41 undergoes structural rearrangements into a fusion-active conformation, which is believed to form the six-helix bundle (6-HB) by bringing the viral and cellular membranes together in a hairpin conformation (see Figure 1). It has been shown that the free energy released when gp41 undergoes this conversion is directly and immediately used for pore formation by the merging of the viral and target cell membranes (2). The 6-HB, revealed by crystallographic studies of the gp41 core (3–5), is thought to be the gp41 conformation in the late stages of the fusion process. This conformation involves a trimer of gp41 molecules and consists of the C-terminal helical segments of three gp41 molecules (C′ heptad repeat, HR2) packed onto a trimer of helices made of N-terminal segments (N′ heptad repeat, HR1) of gp41 (3–5). To date, structural knowledge of HIV-1 gp41 is limited to the protease-resistant core domain (3–5) of the 6-HB which includes residues A541–K588 of the N-segment and residues W628–K665 of the C-segment. However, there is currently no structural information about segments preceding and following this core in the 6-HB state of gp41, that is, the fusion peptide proximal region of residues S528–Q540 (FP-PR) and the membrane proximal external region of residues W666–N677 (MPER), respectively (Figure 1).
Figure 1.
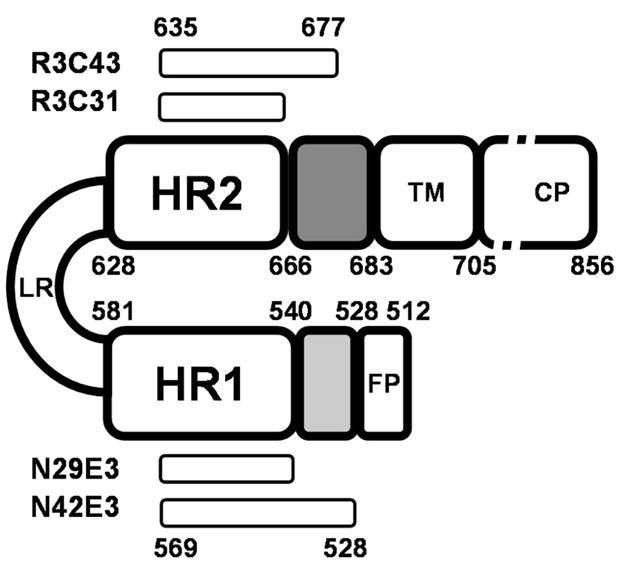
Schematic representation of HIV-1 gp41. Internal regions are specified by abbreviations: FP, fusion peptide; HR1, N′ heptad repeat 1; LR, loop region; HR2, C′ heptad repeat 2; TM, transmembrane region; CP, cytoplasmic tail. The FP-PR and the membrane proximal external region are colored light and dark gray, respectively. Bars represent sequence location of key peptides used in this study. The residues are numbered according to their position in gp160 of HIV-1HXB2.
Previous studies revealed that the segment comprising residues E657–W678 of gp41 includes the epitopes for the two rare anti-gp41 broadly neutralizing antibodies 2F5 and 4E10 and represents the only neutralizing determinant of this protein (6). The X-ray structure of the two antibodies in complex with their respective gp41 peptides (both epitopes are not present in previously published 6-HB structures) revealed the conformation of these two epitopes (extended for 2F5 and α-helical preceded by a short 310-helix and extended structure at the N′ terminus for 4E10) corresponding to the prefusion, prehairpin state of gp41, before the formation of the 6-HB (7, 8).
The C-terminal region (residues W666–K683) of the gp41 ectodomain preceding the transmembrane domain (TM) is rich in tryptophan residues that are conserved in lentiviruses (9) and is crucial for the pathogenesis of HIV-1. Deletion of residues L660–W670 (LLELDKWASLW) resulted in partial dissociation of the oligomeric structure of gp41, abolished fusion, and decreased the extent of gp160 precursor cleavage (10). Mutation of the first three tryptophan residues (W666, W670, and W672) sufficed to eliminate viral fusion (9). Like C34 (W628–L661) (4), an overlapping peptide corresponding to residues Y638–F673 of gp41, termed T-20, was found to inhibit the fusion of the virus with the target cell and is currently being used as an Federal Drug Administration-approved entry inhibitor for the treatment of HIV-1-infected individuals (11, 12). In accordance with the studies cited above, truncation of the last 13 residues of T-20 or mutation of two tryptophans and a phenylalanine at the C-terminal segment of T-20 reduced its activity by more than 5 orders of magnitude (13).
The segment that includes the first 28 residues of T-20 and is comprised of gp41 residues W628–K665 was found to be involved in formation of the 6-HB (3). Therefore, it has been suggested that T-20 inhibits the formation of the 6-HB by interacting with the trimer formed by the N-terminal helices. However, T-20 C-terminal residues W666–F673 were not included in any of the structures of the gp41 6-HB, and there are no conclusive data on the interactions of this C-terminal membrane proximal domain with other regions of gp41, or with gp120. Furthermore, it has been found that unlike C′ peptide C34 (W628–L661), T-20 does not form a stable 6-HB with either the N36 peptide (S546–L581) (14) or the N46 peptide (T536–L581) and may form insoluble nonhelical complexes or aggregates (15). Moreover, it has been found that T-20 interacts with targets on gp120 (14). Thus, the exact role and structure of the last eight residues of T-20 in preventing viral fusion remains to be determined. To shed light on the contribution of the C-terminal segment of T-20 to its antiviral activity, and to study possible interactions of the segment of residues W666–N677 with the N-terminal segment of gp41, we searched for N′ and C′ gp41 peptides that form a stable 6-HB. These peptides were designed to contain the entire segment between the fusion peptide and HR1 as the N-terminal component, and the entire T-20 sequence within the C-terminal component (for the definition of the various gp41 peptides, see Table 1). All the peptides described in this paper, except the peptides corresponding to core segments N29E3 (A541–T569–E3) and R3C31 (R3–I635–K665), include two domains. These are shortened core segments (A541–T569 or I635–K665) (3–5), together with the FP-PR and the MPER, respectively, which are the focus of this work (Figure 1). The N42E3 peptide, with a triglutamic acid solubility tag at its C-terminus, and the R3C43 peptide, with a triarginine solubility tag at its N-terminus, were found to form a stable 6-HB. N′ and C′ gp41 peptides that contain only the A541–T569 core segment linked to the triglutamic acid solubility tag (N29E3) and I635–K665 core segment linked to the triarginine solubility tag (R3C31) did not form a stable 6-HB, clearly indicating that the S528–Q540 N-terminal segment and the W666–N677 C-terminal segment interact with each other and that this interaction contributes to the stabilization of the 6-HB formed by gp41.
Table 1.
Amino Acid Sequences of gp41 Peptidesa
| Peptide | Gp41 position | Sequence – N′ Peptides | M.W |
|---|---|---|---|
| N49a | S546-L581 | 541-ARQLLSGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARILAVERYLKDQ-590 | 5807.7 |
| N52 | S528-R579 | 528-STMGAASMTLTVQARQLLSGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQAR-579 | 5757.7 |
| N47 | S528-K574 | 528-STMGAASMTLTVQARQLLSGIVQQQNNLLRAIEAQQHLLQLTVWGIK-574 | 5161.0 |
| N42 | S528-T569 | 528-STMGAASMTLTVQARQLLSGIVQQQNNLLRAIEAQQHLLQLT-569 | 4577.3 |
| N42E3 | S528-T569-EEE | 528-STMGAASMTLTVQARQLLSGIVQQQNNLLRAIEAQQHLLQLT-569-EEE | 4964.6 |
| N38 | S528-L565 | 528-STMGAASMTLTVQARQLLSGIVQQQNNLLRAIEAQQHL-565 | 4121.7 |
| N29E3 | A541-T569-EEE | 541-ARQLLSGIVQQQNNLLRAIEAQQHLLQLT-569-EEE | 3685.1 |
| Peptide | Gp41 position | Sequence – C′ Peptides | |
| C42a | N624-K665 | 624-NHTTWMEWDREINNYTSLIHSUEESQNQQEKNEQELLELDK-665 | 5187.6 |
| R3C49R3 | RRR-I635-K683-RRR | RRR-635-INNYTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWFNITNWLWYIK683RRR | 7061.9 |
| C48 | E630-NB77 | 630-EWDREINNYTSUHSLIEESQNQQEKNEQELLELDKWASLWNWFNITN-677 | 5950.4 |
| R3C43 | RRR-I635-N677 | RRR-635-INNYTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWFNITN-677 | 5703.2 |
| C43 | I635-N677 | 635-INNYTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWFNITN-677 | 5234.7 |
| C42 | NB36-N677 | 638-NNYTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWFNITN-677 | 5121.5 |
| R3C31 | RRR-I635-K665 | RRR-635-INNYTSLIHSLIEESQNQQEKNEQELLELDK-665 | 4169.6 |
Amino acid numbering (in subscript) is according to the gp160 sequence of the HXB2 strain. Amino acids participating in core formation are underlined. Solubility tags are in italics.
Core peptides studied by Weissenhorn et al. (3).
EXPERIMENTAL PROCEDURES
Peptide Synthesis
Peptide N47 was synthesized on a preloaded Fmoc-Lys(Boc)-Wang resin with 0.6 mmol/g substitution at 0.05 mmol scale using an Applied Biosystems Inc. model 433A synthesizer. The coupling strategy was FastMoc chemistry. Double coupling was carried out for each residue using HBTU/HOBt activation, and capping was accomplished with acetic anhydride in the presence of DIEA. After the completion of chain assembly, the resin was treated with a solution of 0.75 g of phenol, 0.5 mL of thioanisole, 0.25 mL of EDT, 0.5 mL of water, and 10 mL of TFA. The reaction was carried out at room temperature for 3 h. The reaction mixture was filtered, and the filtrate was concentrated to a small volume on a rotator evaporator. The crude peptide was precipitated by addition of ethyl ether and purified by high-pressure liquid chromatography (HPLC) (Thermo Separation Products) using a C4 reverse-phase preparative column (GraceVydac). All synthetic peptides were purified to >98% homogeneity and had the expected mass as determined using electrospray ionization mass spectrometry.
Vector Construction
The DNA encoding the TrpΔLE polypeptide (16) with a His tag at the N-terminus was inserted between the NdeI and BamHI sites of pET24a (Novagen, Darmstadt, Germany) and used as a starting point to construct fusion proteins containing various segments of gp41. The coding DNA for either the N-segment(S528–R579) or the C-segment (E630–N677) of the HIV-1 gp41 extra-cellular domain, strain HXB2, including two TAA stop codons, was attached to the DNA encoding the TrpΔLE polypeptide sequence using HindIII and BamHI restriction sites. A hydroxylamine cleavage site, Asn-Gly (17), solubility tags, and stop codons in all constructs were created using a Quik-Change XL site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Expression of the Fusion Peptide
Escherichia coli BL21 or Rosetta DE3 cells were transformed by standard heat-shock transformation with plasmids encoding the expression of gp41 C′ and N′ peptides, respectively. Cells were grown overnight in 5 mL of LB medium at 37 °C with shaking at 225 rpm and then diluted 200-fold into 1 L of culture medium in 5 L flasks. After dilution, cells were grown until the culture reached an OD595 of 0.6–0.7, and then expression was induced by the addition of 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) (Fermentas), followed by incubation for 16 h at 37 °C with shaking at 225 rpm. Cells were harvested by centrifugation (8000 rpm for 30 min), suspended in buffer A [50 mM Tris (pH 7.8), 0.5 mM EDTA, and 50 mM NaCl], and sonicated while being cooled by ice (five 1 min pulses at 70% amplitude, VibraCell, Sonics & Materials, Inc.). Inclusion bodies were collected by centrifugation (13000 rpm for 30 min at 4 °C), washed four times with buffer A, and stored at −20 °C until cleavage.
Protein Cleavage
To release the gp41 peptides from TrpΔLE carrier protein (16), we chose hydroxylamine cleavage (17), since there are no naturally occurring asparagine-glycine (NG) sequences in our gp41 peptides and since the N′ peptides contain two methionine residues that prevented the use of cyanogen bromide cleavage. Inclusion bodies were cleaved in modified cleavage buffer [6 M guanidine-HCl, 0.25 M hydroxylamine-HCl, and 50 mM free methionine (pH 9)] at 45 °C for 6 h. Cleavage was stopped by cooling and adding formic acid until the pH reached 7.
Peptide Purification
Cleavage samples were loaded on a 10 mL HisTrap column (GE Healthcare, Buckinghamshire, England) immediately after the cleavage reaction [loading buffer, 6 M guanidine-HCl, 10 mM imidazole, and 50 mM Na2HPO4 (pH 7.5)]. The flow-through solution containing the gp41 peptides was dialyzed twice (1 kDa molecular mass cutoff, Spectra/Por 6, Spectrum Laboratories, Inc.) against 50 mM NH4HCO3 (Fluka) in deionized Milli-Q water and then lyophilized. The bound carrier protein was eluted from the column by buffer containing 6 M guanidine-HCl, 0.8 M imidazole, 0.5 M NaCl, and 50 mM buffer acetate (pH 4) (elution buffer). The lyophilized crude gp41 peptides were resuspended in 40% HPLC buffer B (buffer A, 99.9% H2O and 0.1% TFA; buffer B, 74.9% acetonitrile, 25% H2O, and 0.1% TFA) and purified twice by high-pressure liquid chromatography (HPLC) (Thermo Separation Products) using C4 reverse-phase preparative and analytical columns (GraceVydac). All gp41 peptides eluted between 45 and 55% buffer B using a gradient from 45 to 55% buffer B over 120 min. Peptides were purified to >99% purity, and the molecular mass was verified by MALDI/TOF and electrospray ionization mass spectroscopy.
Circular Dichroism Spectroscopy
Circular dichroism (CD) spectra were recorded using an AVIV Instruments Inc. (model 202) spectrometer equipped with a thermoelectric temperature controller. Spectra were recorded using 30 μM samples at 4 °C in 1 mm quartz cells in 1 nm steps from 190 to 260 nm, a 1.5 nm bandwidth, and an averaging time of 4 s/step. Data points were averaged over triplicate determinations. Peptides were dissolved in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)]. After the spectrum of the buffer was subtracted, raw ellipticity values were converted to mean residue ellipticity using standard methods. Thermal stability measurements were performed at 222 nm with 2 °C steps from 4 to 94 °C and back to 4 °C, and an averaging time of 30 s/step. Apparent thermal melting points were calculated with a Boltzman sigmoid fit (Origin version 7.0, OriginLab Corp.).
Kd Determination
Peptide binding measurements were carried out on a PC1 photon counting spectrofluorometer (ISS Inc., Champaign, IL). C′ peptides (R3C31 or R3C43) were mixed with their respective N′ peptides (N29E3 or N42E3) at different N′/C′ molar ratios and then diluted in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)] to 2 μM in a total sample volume of 250 μL. The excitation wavelength was set to 276 nm to allow excitation of aromatic residues (18), and emission spectra were recorded at 290–450 nm at 25 °C (18). The differences between the emission intensity of the N′ and C′ peptides’ mixtures and that of the free C′ peptide at 308 nm (for R3C31 which contains only one tyrosine and neither tryptophan nor phenylalanine residues) or at 350 nm (for R3C43 which contains one tyrosine, three tryptophans, and one phenylalanine) at varying N′ peptide/C′ peptide molar ratios were fitted to the following equation (19):
where ΔFi is the difference between the fluorescence of the peptide mixture and that of the unbound C′ peptide, ΔFmax is the fluorescence difference in the case of a 50-fold molar concentration of the N′ peptide with respect to the C′ peptide, [C]0 is the constant C′ peptide concentration, [N]i is the variable N′ peptide concentration, and Kd is the N′–C′ dissociation constant.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were performed on a Beckman Optima XL-A analytical ultracentrifuge (Beckman, Coulter, CA) at 4 °C unless otherwise indicated. Six-channel cells were used with an An60 titanium rotor. The cells were scanned using a step size of 0.001 cm, and data points were averaged over 10 replicates. Solutions of 10–30 μM peptides were prepared in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)]. Samples were initially scanned at 3000 rpm to identify appropriate wavelengths for data collection. The absorbance obtained at 3000 rpm was compared to the absorbance at higher rpm to ensure that the peptides remained in solution over the course of the experiment. Data were collected at different rotation speeds and wavelengths according to the sample’s absorbance and the expected molecular mass. Weight-averaged molecular masses were obtained by averaging the molecular masses from all data sets after reaching equilibrium. The apparent molecular mass was calculated by the formula M = [2RT/(1 − νρ)ω2][d(ln Abs)/dr2] (20), where M is the solute molar weight (in grams per mole), R is the gas constant in grams per square centimeter per square minute, T is the temperature in kelvin, 1 − νρ = 0.27 (ν is the partial specific volume in milliliter per gram; ρ is the density in grams per milliliter) (21), ω is the angular velocity of the rotor in revolutions per minute, and d(ln Abs)/dr2 is the first derivative of ln Abs with respect to r2, where Abs is the absorbance of the solute (OD) at r which is the radial distance from the axis of rotation. Different rotor speeds were tested to achieve a good fit of the data to probable multimerization states (i.e., monomers, dimers, etc.). Equilibrium was reached after an overnight run and verified by overlay and subtraction of at least two successive scans.
RESULTS
Matching the HR1 Peptide to NN-T-20-NITN (C42)
To examine whether a peptide that contains most of the MPER and the entire T-20 sequence, but only part of the core segment, can form a 6-HB similar to the one formed by core peptides C34 and N36 (4), we followed the interactions of C42 with different N′ peptides. We used the T-20 peptide analogue C42 (NN-T-20-NITN) (Table 1) in these experiments because of its improved solubility at neutral or slightly basic pH in comparison with T-20 which is poorly soluble (21). Three N′ peptides, N47, N42, and N38 (Table 1), were synthesized or expressed and then purified and mixed with C42. Complex formation was followed by measuring the CD spectrum of the free peptides and their mixtures in a 1:1 molar ratio. The theoretical isoelectric points (pI) of the N′ peptides are distributed between 10.5 and 12. However, the pI of C42 is 4.1. The solubilities of the N′ peptides alone are relatively high at acidic pH (up to 2 mM at pH <4). Despite the improvement obtained by the addition of the NN and NITN gp41 segments at the N- and C-termini of T-20, C42 had a solubility of only 0.1 mM at pH >7 and was not soluble at acidic pH. The large differences in the pIs and solubilities at the same pH resulted in technical problems during their simultaneous dissolution in the same buffer. To avoid precipitation of mixtures of the N′ and C′ peptides, we prepared dilute solutions of each of the peptides and incubated them together. The first N′ peptide examined was N47. According to the X-ray structure of the gp41 core (4), this peptide aligns well with C42 with only five residues of its C-terminal end that are part of the HR1 core left unmatched with opposing residues of the HR2 core. Following incubation with the C42 peptide, the sample severely precipitated over a broad pH range (pH 3.5–8), and the CD spectrum of the mixture could not be measured. The next peptide examined was N42. The gp41 core segment of this peptide should match perfectly with the core segment of C42. Only slight precipitation was observed following incubation of N42 with C42 (at pH <4). To better define the minimal N′ peptide that can interact with C42, the next complex examined contained the N38 peptide in which four more HR1 residues were omitted from the C-terminus of the peptide. Similar to the case of N42, the mixture of N38 and C42 slightly precipitated out of solution, making mean residue ellipticity conversions inaccurate.
Secondary Structure and Stability Analyses of HR1–Peptide Complexes with C42
Circular dichroism (CD) spectroscopy was used to analyze the secondary structure of the N42–C42 and N38–C42 complexes. Figure 2A shows that while N42 and C42 alone do not exhibit a high content of α-helices as judged by the relative intensities of the 208 and 222 nm minima, the N42–C42 mixture in a 1:1 molar ratio had a CD spectrum typical of an α-helix, with two almost equal-intensity minima at 208 and 222 nm. Similar changes were observed for the N38–C42 mixture (Figure 2B). Due to the slight precipitation in these solutions, no attempt was made to calculate the mean residue ellipticities of the mixtures. However, the change in shape of the CD curve of the peptides upon mixing the peptides in a 1:1 molar ratio suggests the formation of an α-helix in the complex. The thermal unfolding of the complexes was examined by CD to determine their stabilities. As shown in Figure 2C, while the N38–C42 complex had a melting temperature of 32 °C, the melting temperature of the N42–C42 complex was 53 °C. The CD measurements suggest that the additional four-residue segment (566LQLT569) in the N42 peptide contributed to the significant increase in complex stability. We hypothesize this is due to interaction with the N-terminal segment of C42. Notably, residues 566LQLT569 are the first residues of the hydrophobic cavity formed by the N′ peptides (22). The lower stability of the N38–C42 complex in comparison with that of the N42–C42 complex could be explained by the absence of a hydrophobic interaction of Y638 with 566LQLT569 which is absent in the shorter N′ peptide complex.
Figure 2.

Circular dichroism of untagged peptides. Spectra were collected at a peptide concentration of 30 μM in 2.5 mM HEPES at pH 7 and 4 °C. (A) N42 (black), C42 (gray), and their 1:1 complex (dashed). (B) N38 (black), C42 (gray), and their 1:1 complex (dashed). (C) Thermal unfolding of 1:1 complexes of N38 and C42 (▼) and N42 and C42 (■). The CD signal is represented in millidegrees. The molar ellipticity was not determined because of precipitation of the peptides.
Addition of Solubility Tags
As mentioned above, the N′ peptides, which form the core onto which the C-terminal helices are packed, have pI values (pI = 10.9) drastically different from those of the C′ peptides (pI = 4.1) (Figure 3A, B). On the basis of the theoretical pI values, the N-terminal peptides should exhibit maximum solubility at acidic pH whereas the C′ peptides are expected to have maximum solubility at neutral to basic pH (Figure 3). This difference in the solubility properties of the N′ and C′ peptides limited the concentration of the complexes to at most 0.1 mM at acidic pH (Table 2). Attempts to obtain a more concentrated sample by preparing a highly diluted (10 μM) complex followed by lyophilization, SpeedVac vacuum concentration, buffer exchange via dialysis, or concentration using semipermeable membranes (VivaScience, Gloucestershire, U.K.) resulted in irreversible aggregation. To enable future structural studies of the complex between N′ and C′ peptides containing the residues S528–Q540 and W666–N677, respectively, which were not included in any of the 6-HB X-ray and NMR structures (3–5, 23), it was important to develop constructs that would form a stable and soluble complex.
Figure 3.
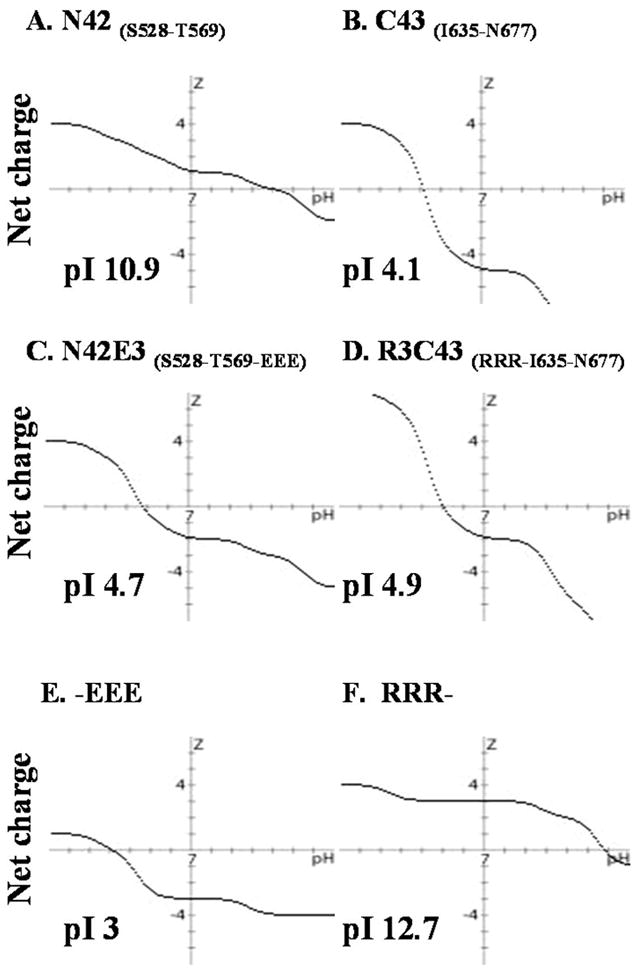
Effect of solubility tags on the overall peptide charge profile. Charge over pH plots created by Peptide property calculator, Innovagen. The pI values of each peptide or tag are indicated.
Table 2.
Summary of Biophysical Properties of N′ Peptide–C′ Peptide Mixturesa
| N′ peptide | C′ peptide | solubility (pH 3) | precipitation | Tm (°C) by CD | complex molecular mass (kDa) by AUC | theoretical (6-HB) molecular mass (kDa) |
|---|---|---|---|---|---|---|
| N38 S528–L565 | C42 N636–N677 | <100 μM | + | 32 | NDb | 27.9 |
| N42 S528–T569 | C42 N636–N677 | <100 μM | + | 53 | NDb | 28.3 |
| N47 S528–K574 | C42 N636–N677 | <100 μM | +++ | NDb | NDb | 30 |
| N52 S528–R579 | C42 N636–N677 | <100 μM | ++++ | NDb | NDb | 31.9 |
| N42 S528–T569 | R3C43 RRR-I635–N677 | >1 mM | – | 42 | NDb | 31.2 |
| N42E3 S528–T569-EEE | R3C31 RRR-I635–K665 | >1 mM | – | 34 | 8.1 ± 0.5 | 27.7 |
| N29E3 A541–T569-EEE | R3C31 RRR-I635–K665 | >1 mM | – | 38 | 11.6 ± 2 | 23.9 |
| N42E3 S528–T569-EEE | R3C49R3 RRR-I635–K683-RRR | ND | – | 45 | 20.2 ± 2 | 36.2 |
| N29E3 A541–T569-EEE | R3C49R3 RRR-I635–K683-RRR | ND | – | 42 | 11.5 ± 1 | 33.5 |
| N29E3 A541–T569-EEE | R3C43 RRR-I635–N677 | >1 mM | – | 42 | 30 ± 2 | 29.5 |
| N42E3 S528–T569-EEE | R3C43 RRR-I635–N677 | >1 mM | – | 60 | 31 ± 2 | 32.3 |
Solubility represents the maximum solubility at pH 3.2. Precipitation estimated by visual inspection. The apparent melting temperature of the helical conformation was measured by CD analysis as described in Experimental Procedures. Molecular masses were determined by analytical ultracentrifugation.
Not determined.
Residue I635 is one of the three hydrophobic residues (I635, W631, and W628) of the C core that penetrates deeply into the hydrophobic cavity formed by N core residues L565–Q577 (4, 24). To further increase the stability of the 6-HB, I635 was added to the C42 peptide. In addition, to avoid precipitation of the N′ and C′ peptides and/or their complexes, to enable quantitative measurements of the ellipticity per residue, and to obtain concentrated solutions of the complex for future NMR structural studies, we explored the possibility of adding solubility tags to the peptides. Since a basic pH is problematic for NMR measurements of peptides due to fast exchange of the amide protons with the solvent (25), we sought to optimize the solubility of both peptides at acidic pH. Previously, simian immunodeficiency virus gp41 had been studied by NMR at pH 3, providing a precedent for evaluating HIV-1 gp41 at acidic pH (26). To increase the solubility of the N′ and C′ peptides and of their complexes at acidic pH, we constructed expression vectors with a short triarginine tag attached to the C′ peptide (RRR, basic, pKa = 12.7). A triglutamic acid tag was attached to the N′ peptide (EEE, acidic, pKa = 3), to adjust the pI of each peptide to approximately 5 (Figure 3C, D). The solubility tags were added to the side of the peptide that contained the gp41 core segment (27) to prevent interference with possible interactions between the S528–Q540 and W666–N677 segments which are the major focus of this study. Clearly, the solubility tags altered the charge versus pH profile of the peptides (Figure 3), and we found that at pH 3.2 N29E3–R3C31 and N42E3–R3C43 complexes exhibited solubilities of up to 2 and 1.5 mM, respectively (Table 2).
Secondary Structure of Different gp41 Complexes with Added Solubility Tags
As mentioned in the introductory section, residues S528–Q540 and W666–N677 were not included in the structures of any gp41 core complexes that had been previously determined by X-ray crystallography or NMR (3–5). To examine whether the S528–Q540 and W666–N677 segments that correspond to the FP-PR and most of the MPER, respectively, contribute to the stability of the 6-HB, we prepared peptides N29E3 (A541–T569-EEE) and R3C31 (RRR-I635–K665) that do not include the S528–Q540 and W666–N677 segments, respectively, and compared their structures and stabilities with those of the N42E3 and R3C43 peptides and of the different possible complexes. CD spectra of peptides N29E3, R3C31, N42E3, and R3C43 and the 1:1 N29E3–R3C31, N29E3–R3C43, N42E3–R3C31, and N42E3–R3C43 mixtures reveal that R3C31 has a low fraction of helical structure while N29E3 is somewhat helical. The helical content is practically unchanged when these two peptides are mixed (Figure 4A). The R3C43 peptide with a mean residue ellipticity of −23000 deg cm2 dmol−1 at 222 nm is considerably more helical than the shorter R3C31 peptide which exhibited a mean residue ellipticity of −4000 deg cm2 dmol−1 at 222 nm (compare panels A and C of Figure 4). The experimental CD spectrum of a 1:1 mixture of N29E3 and R3C43 resembles the theoretical sum of the CD spectra of the two peptides (Figure 4C). Thus, the CD of the N29E3–R3C43 mixture does not provide an indication for interaction between the two peptides. Similarly, the CD spectra of N42E3, R3C31, and their 1:1 mixture (Figure 4B) did not provide any indication for interaction between N42E3 and R3C31. Both N42E3 and R3C43 exhibit considerable helical structure content with a mean residue ellipticity of approximately −23000 deg cm2 dmol−1. When these two peptides are mixed in a 1:1 ratio, the experimental CD spectrum differs significantly from the theoretical average of the spectra of the individual peptides and a mean residue ellipticity of −35000 deg cm2 dmol−1 is observed (Figure 4D), indicating an interaction between N42E3 and R3C43 that leads to an increase in the overall helicity of the complex. To examine whether the addition of the remaining MPER residues (679WLWYIK683) stabilize the formation of the 6-HB, we prepared the peptide R3C49R3, which includes the complete MPER sequence, and made its complexes with N42E3 and N29E3. The mixture of N42E3 and R3C49R3 exhibited slightly more helical content than their theoretical average, indicating interaction between these two peptides that stabilizes a helical conformation (Figure 4F). However, the increase in helical content was much less noticeable than with the mixture of N42E3 and R3C43, suggesting that R3C49R3 had weaker binding to N42E3 than R3C43. N29E3 and R3C49R3 hardly bind at all (Figure 4E), which is evident from the almost complete overlay of calculated average versus experimental data.
Figure 4.
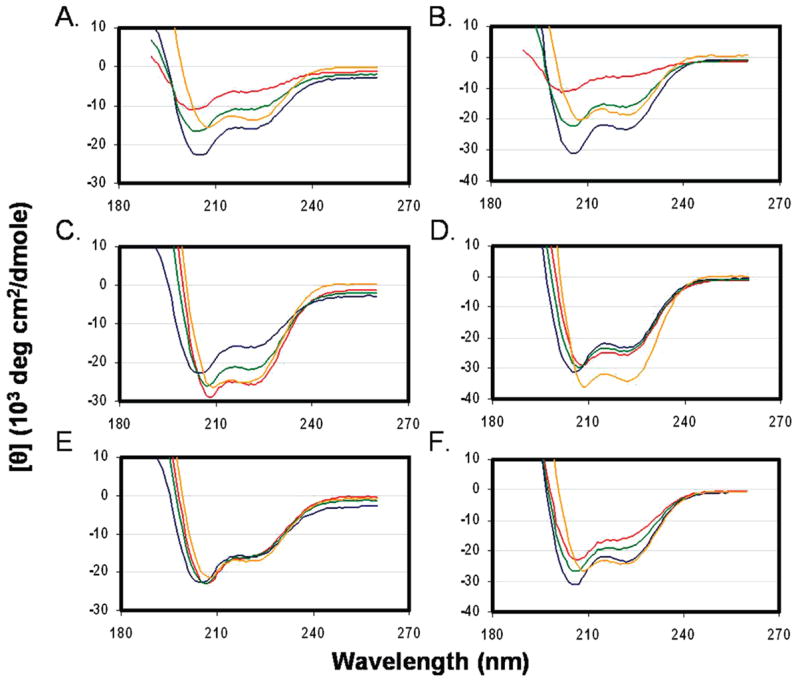
Circular dichroism spectra of tagged gp41 peptides at acidic pH. Spectra were recorded at a peptide concentration of 30 μM in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)] at 4 °C: (A) N29E3 and R3C31, (B) N42E3 and R3C31, (C) N29E3 and R3C43, (D) N42E3 and R3C43, (E) N29E3 and R3C49R3, and (F) N42E3 and R3C49R3. The traces are colored as follows: blue for N′ monomers, red for C′ monomers, orange for a mixture of N′ and C′ monomers, and green for the calculated average ellipticity of the mixture.
Thermal Stability of Different gp41 Complexes with Added Solubility Tags
The thermal stability of the conformations formed by the tagged N′ and C′ peptides and their 1:1 mixtures was examined by measuring the CD spectrum at 222 nm between 4 and 94 °C (Figure 5). Among the individual peptides, only R3C43 exhibited a sigmoidal curve indicating cooperative melting of a helical conformation (Figure 5A). The melting temperature of the helix in R3C43 was found to be 42 °C. The other individual peptides (N29E3, N42E3, R3C31, and R3C49R3) exhibited gradual melting. When the N′ and C′ peptides were mixed, except for R3C43 and the N29E3–R3C43 mixture, more sigmoidal curves were observed for the mixture of the peptides in comparison with the melting curves of the individual peptides (Figure 5B). This suggests that the melting of the mixtures was more cooperative than the melting of the individual peptides. The most stable helical structure was formed by the 1:1 mixture of N42E3 and R3C43 with a melting temperature of 60 °C (Figure 5B). This is considerably higher than the melting temperature of 42 °C observed for R3C43 alone and the temperature (~20 °C) at which N42E3 exhibited a 50% decrease in its ellipticity at 222 nm. Thus, we conclude that the melting curve of the 1:1 mixture of N42E3 and R3C43 represents that of a complex formed by these two peptides. Interestingly, a complex between N42 with R3C43 exhibited a sigmoidal melting curve with a melting temperature of 54 °C (Figure 6). The higher melting temperature of the 6-HB of the complex of the two tagged peptides suggests that additional stabilization of the 6-HB occurred probably as a result of preventing the electrostatic repulsion between the positively charged N-terminally tagged C′ peptide molecules. Moreover, a salt bridge may exist in the 6-HB between the three glutamic acid residues of the E3 tag of N42E3 and the arginine residues of the R3 tag of R3C43. Electrostatic interactions and hydrogen bonds involving side chain carboxyls can considerably lower the pKa values below 2.5 (28, 29), causing the glutamic acids to be negatively charged even under our experimental conditions (pH 3.2). Among the tagged peptides, the second most stable complex was the N42E3–R3C49R3 complex (Figure 5B), suggesting that the added MPER sequence not only did not strengthen N′–C′ interaction but also reduced the stability of the 6-HB. The next most stable complexes were the N29E3–R3C43 and N29E3–R3C49R3 complexes (Figure 5B). However, the melting curves resemble the melting curve of R3C43 alone, and therefore, the melting of N29E3, R3C43, and the 1:1 N29E3–R3C49R3 mixture measured by CD does not provide an indication of additional stability of a helical conformation obtained by formation of a complex between the peptides. Similarly, when the melting curves of N29E3 and R3C31 in a 1:1 ratio and N42E3 and R3C31 in a 1:1 ratio are compared to the melting curve of the individual peptides, except for changes in cooperativity, little evidence of complex formation was observed. Since all thermal melts were not reversible, we refer to the calculated Tm as the “apparent Tm”.
Figure 5.
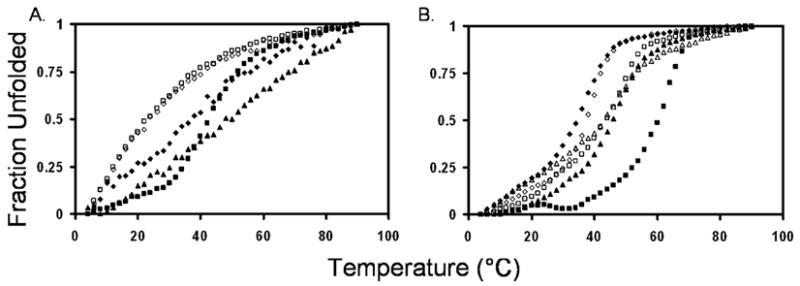
Thermal unfolding transitions of tagged gp41 peptides (left) and peptide mixtures (right) at a peptide concentration of 30 μM in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)]: (A) (□) N42E3, (◇) N29E3, (▲) R3C49R3, (■) R3C43, and (◆) R3C31 and (B) (□) N29E3 and R3C43, (■) N42E3 and R3C43, (◇) N29E3 and R3C31, (◆) N42E3 and R3C31, (△) N29E3 and R3C49R3, and (▲) N42E3 and R3C49R3. All complexes have a 1:1 molar ratio of N′ to C′ peptides.
Figure 6.
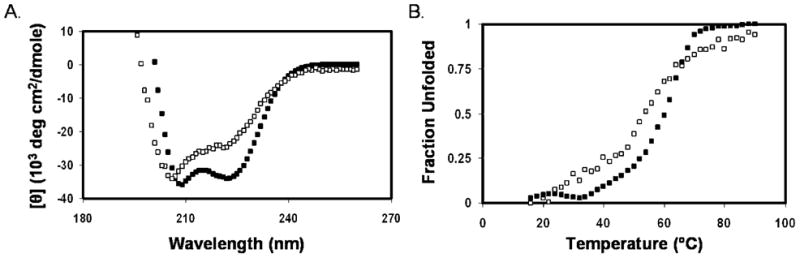
Circular dichroism spectra of gp41 peptides at acidic pH. Spectra were recorded at a peptide concentration of 30 μM in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)] at 4 °C for (A) (□) N42 and R3C43 and (■) N42E3 and R3C43. (B) Thermal unfolding transitions of (□) N42 and R3C43 and (■) N42E3 and R3C43.
Dissociation Constants of the Six-Helix Bundle Complexes
The large difference in stability between the shortened core complex (N29E3–R3C31) and the elongated complex (N42E3–R3C43), which includes most of the MPER sequence, was further investigated in an effort to assess the contribution of the interaction of the MPER with the FP-PR to the stability of the 6-HB. For this purpose, Kd measurements of the different complexes were carried out by following the change in the fluorescence intensity of the aromatic residues of the C segments upon complex formation (Figure 7) (19). While R3C31 could not be fully saturated by N29E3 even with 50-fold molar excess and a concentration of 100 μM (Kd = 113 ± 24 μM) (Figure 7), R3C43 bound N42E3 well with a Kd of 1.9 ± 0.3 μM (Figure 7) This is clear evidence that the interaction between parts of the MPER region and the matching N-terminal segment contributes to the stabilization of the 6-HB. The 2-fold increase in fluorescence intensity and the 9 nm blue shift in fluorescence maxima between the unbound and bound R3C43 suggest that aromatic residues of R3C43 moved to a more hydrophobic environment upon binding to N42E3 (30), possibly by interacting with an extension of the hydrophobic groove of the inner trimeric core created by the FP-PR (4).
Figure 7.
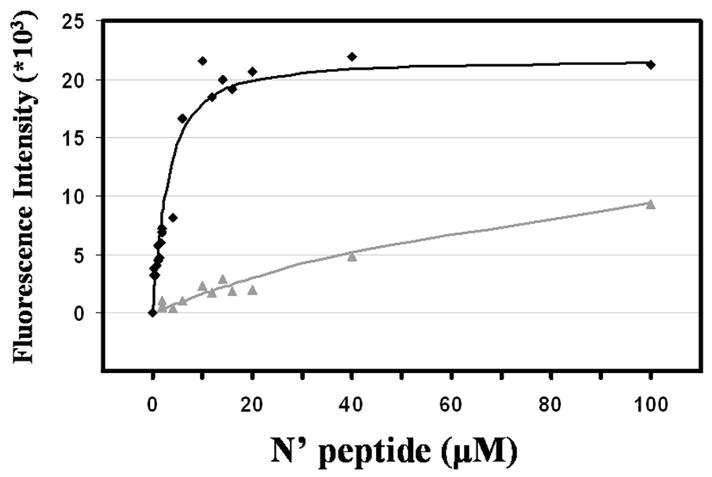
Measurement of the binding of N′ and C′ peptides using fluorescence spectroscopy. Fluorescence intensities were measured at a constant R3C43 or R3C31 concentration of 2 μM in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)] at 25 °C. N42E3 (◆) or N29E3 (▲) was added to R3C43 or R3C31, respectively, in an up to 50-fold molar excess. The fluorescence intensity scale is for R3C43 titration (R3C31 fluorescence intensity was multiplied by 10-fold for convenience).
Oligomerization State of the Mixtures of the Tagged N′ and C′ Peptides
To further characterize the extent of N′–C′ interactions, we determined the oligomerization state of various peptide mixtures using sedimentation equilibrium (31). Equimolar mixtures of different N′ and C′ gp41 peptides were tested for the ability to form a stable 6-HB. The mixtures of N29E3 and R3C31 and of N42E3 and R3C31 both at a 1:1 molar ratio revealed molecular masses of 11.6 and 8.1 kDa (Table 2), respectively, which are larger than the molecular masses of the monomeric peptides, indicating that there are some aggregates or weak interaction between the N′ and C′ peptides. However, the population of the 6-HB, if it exists at all for these complexes, is rather low. As shown in Table 2, a molecular mass of 32.1 kDa was measured for the 1:1 N29E3–R3C43 mixture, indicating the formation of a 6-HB (theoretical mass of 28.5 kDa). The molecular mass of the 1:1 N42E3–R3C43 mixture was 29.9 kDa, indicating the formation of a 6-HB by this mixture (theoretical mass of 32.3 kDa). To examine whether elongation of the R3C43 peptide by the MPER sequence 678WLWYIK683 could further stabilize the interaction between the N′ and C′ segments, the complex formed by R3C49R3 was also tested. Sedimentation equilibrium measurements clearly indicated that R3C49R3 could not form a stable complex with either N29E3 (exhibiting a molecular mass of 11.5 kDa instead of the value of 33.5 kDa expected for a 6-HB) or N42E3 (exhibiting a molecular mass of 20.2 kDa instead of 36.2 kDa). We therefore suggest that the W678–K683 segment at the C-terminus of the MPER does not interact with the FP-PR. It has been previously reported that this segment interacts with the viral membrane (32). In the absence of lipids, this segment (678WLWYIK683) may interact nonspecifically with hydrophobic residues in the N′ and C′ peptides perturbing the formation of a 6-HB.
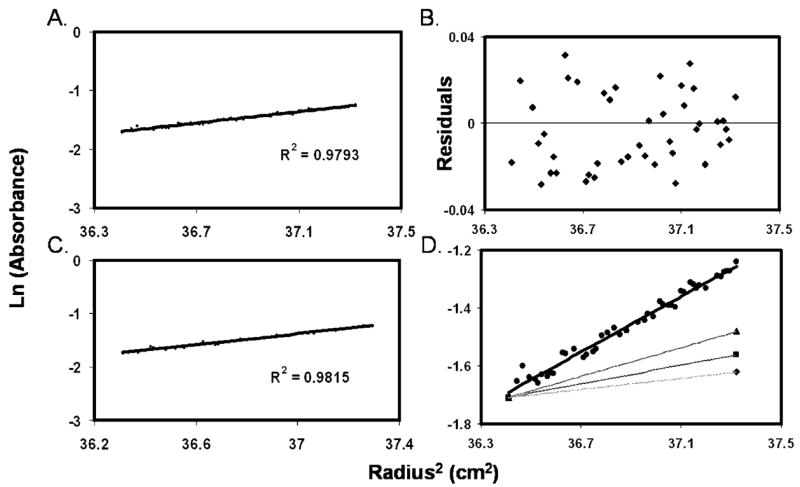
To verify that the determination of the N42E3–R3C43 6-HB molecular mass was carried out under optimal conditions, the two peptides were mixed at different molar ratios and tested for complex formation at different speeds (Figure 9A–C). Molar ratios and sample concentrations were set to reflect the Kd determined for these peptides by fluorescence measurements, and rotation speeds were adjusted for different oligomerization levels (10000, 18000, and 25000 rpm for 60, 30, and 10 kDa, respectively) according to the manufacturer’s manual. Complex formation is clearly evident, and a molecular mass of 32 ± 1 kDa is obtained when the molar ratio between R3C43 and N42E3 is 0.3 (Figure 9A) or 1 (Figure 9B). In both cases, the data fit well to a trimer-of-dimers species, reflecting complete binding of R3C43 by N42E3, even at 3 μM. When the molar ratio between R3C43 and N42E3 is 3.33 (Figure 9C), most of the R3C43 is unbound and the data deviate strongly from the theoretical linear behavior expected for a 32.3 kDa complex (Figure 9D). It should be noted that since the absorbance was measured at 280 nm it reflects only the R3C43 population in the sample (N42E3 has no aromatic residues).
Figure 9.

Sedimentation equilibrium of N42E3 and R3C43. (A–C) Different molar ratios of N42E3 (N) and R3C43 (C) were tested for 6-HB formation (20 °C) at different speeds (blue, red, and green for 10000, 18000, and 25000 rpm, respectively). (D) Data of different N′:C′ ratios (indicated) collected at 18000 rpm fitted to a 32.3 kDa complex.
DISCUSSION
Despite the importance of the MPER (E662–K683) in viral fusion (9), there is no information regarding the interaction of the MPER with the FP-PR. The MPER encompasses the entire C-terminal segment of T-20, a polypeptide whose sequence corresponds to residues 638–673 of gp41. The role of the T-20 C-terminal segment in the strong inhibition of viral fusion by T-20 had not been resolved. Previous studies proposed that unlike other C′ peptides that contain most of the HR2 segment (W628–L661) (1, 4), T-20 did not form a stable 6-HB with peptides corresponding to the N-terminal region of the gp41 heptad repeat region but rather formed an insoluble nonhelical complex or aggregates with low α-helical content (14, 15). In addition, it was found that T-20 interacted with several targets on gp120 and gp41 (14). These findings led to the suggestion that inhibition of HIV-1 fusion by T-20 resulted from interactions with multiple sites on gp41 and gp120 and that interference with the formation of the 6-HB hairpin that plays a prominent role in viral–target cell fusion was unlikely (14).
To examine the role of the T-20 C-terminal segment in fusion inhibition and whether the MPER interacts with the FP-PR, we designed N-terminal peptides that were longer at their N-terminus and shorter at their C-terminus than those previously studied and that were expected to better match the slightly extended T-20 peptide [C43 (Table 1)]. In our design, we selected an N-terminal peptide extended to S528, which encompasses the S528–T569 segment (N42). This peptide contains a relatively short region (565LLQLT569) of the hydrophobic groove formed by residues L565–Q577 of the HR1 trimer (4). Our T-20 analogue peptide (C43) includes only the first of the three HR2 hydrophobic residues (I635, W631, and W628) that interact with the hydrophobic groove formed by the HR1 trimer (4), and most of the MPER. To increase the solubility of our constructs, we added RRR and EEE tags to the termini that were distant from the regions we are investigating. Specifically, we prepared peptides N42E3 and R3C43. This allowed us to probe specific contributions of interactions between residues S528–Q540 of the FP-PR and residues W666–N677 of the MPER to the stability of hexahelical complexes formed by N′ and C-terminal peptides. Such information is not available from previous investigations (3) and is, we believe, essential for improving our understanding of the role of the MPER in viral fusion and the molecular basis for T-20 function.
Our CD and sedimentation experiments provide unequivocal evidence that at a concentration of 30 μM the 1:1 mixture of the short N′ (N29E3) and C′ (R3C31) peptides that do not include the FP-PR and the MPER, respectively, does not form a stable 6-HB. The apparent Tm (38 °C) of the 1:1 mixture of these two peptides found in the CD analysis is likely due to the melting of the helical C-terminal peptide that may be stabilized by the weakly interacting N′ peptide. Similarly, neither the CD nor the sedimentation results indicate the presence of a stable 6-HB in the 1:1 molar mixture of the long N′ peptide (N42E3) and the short C′ peptide (R3C31). Interestingly, peptides N29E3 and R3C43 formed a 6-HB as determined by sedimentation equilibrium measurements. This could be due to the increased helical content of R3C43 in comparison with R3C31 (Figure 4A–D). It is reasonable to conclude that the increase in the helical content of R3C43 compared to that of R3C31 (Figure 4) is caused by the tendency of the additional C-terminal residues (W666–N677) to assume a helical conformation. This is consistent with previous findings that showed that residues W666–N671 of gp41, as part of a short 13-residue peptide, had a strong propensity to be helical (33). The 1:1 mixture of the extended N′ (N42E3) and C′ (R3C43) peptides showed markedly enhanced minima at 208 and 222 nm and a molecular mass consistent with a 6-HB. The apparent melting point of this bundle (60 °C) was 22, 26, and 18 °C higher than those of the 1:1 N29E3–R3C31, N42E3–R3C31, and N29E3–R3C43 mixtures, respectively, and the Kd of the N42E3–R3C43 complex was 2 orders of magnitude smaller than that of the N29E3–R3C31 complex. Altogether, these data point to a significant stability increase of the 6-HB formed by the extended N′ and C′ peptides.
In an analysis of our results, it is important to pinpoint the molecular interactions that result in stabilization of the 6-HB. The solubility tags that we employed (EEE in the N′ peptide and RRR in the C′ peptide) may form attractive electrostatic interactions that would stabilize the complexes formed, even at pH 3.2 if the pKa of the glutamic acid residues of the EEE tag is shifted to a more acidic pH upon complex formation as a result of salt bridges and hydrogen bonds with the arginine residues of the RRR tag (28, 29). However, the solubility tags are the same for all four possible complexes(N29E3–R3C31, N42E3–R3C31, N29E3–R3C43, and N42E3–R3C43) and should contribute equally to their stability. Therefore, the changes in stability between the four complexes are solely attributed to the combination of the gp41 sequences used and the presence or absence of the interaction between the MPER and the FP-PR. Moreover, our observation that the apparent Tm of the N42–C42 complex, which does not have the solubility tags, is nevertheless 11 °C higher than that of the N29E3–R3C43 supports our contention that it is the interaction between the MPER which contains the entire C-terminal segment of T-20 and the matching FP-PR segment in the N′ peptide that is primarily responsible for the increased stability of the 6-HB.
In conclusion, the data presented in this report indicate that regions of the MPER (excluding 678WLWYIK683) and the C-terminal segment of T-20 can interact strongly with regions on the FP-PR of gp41. These interactions were shown to result in a highly stable 6-HB by CD, sedimentation analysis, and Kd determination. Our results indicate that it is quite reasonable that an important mode of action of T-20 is to interfere with formation of the 6-HB within gp41, thus abolishing viral fusion. The new peptides designed for this study have solubilities that make them suitable for analysis by high-resolution NMR techniques. Such analyses should pinpoint the specific residues in the MPER responsible for stabilizing the hairpin and thereby provide information that may be useful for future drug development.
Figure 8.
Sedimentation equilibrium of the 1:1 N42E3–R3C43 complex as determined by analytical ultracentrifugation at a peptide concentration of 30 μM in buffer [5% D2O and 2.2 mM deuterated formic acid (pH 3.2)]: (A) 16000 rpm and 27 °C, (B) no evident systematic deviation from the residuals, (C) 16000 rpm and 4 °C, and (D) lines of best fit for theoretical monomers (◆) [d(ln Abs)/dr2 = 0.08], dimers (■) [d(ln Abs)/dr2 = 0.16], trimers (▲) [d(ln Abs)/dr2 = 0.24], and the 6-HB (●) [d(ln Abs)/dr2 = 0.48].
Acknowledgments
We are most grateful to Prof. Amnon Horovitz and Dr. Yakov Kipnis for help in fluorescence measurements and Kd determination, to Professor Peter S. Kim for giving us the TrpΔLE vector for expressing the peptides, and to Mr. Yehezkiel Hayek for help in peptide purification.
Footnotes
This study was supported by NIH Grants GM53329 (J.A.) and GM22086 (F.N.), by the Kimmelman Center for Macromolecular Assemblies (J.A.), and by US-Israel Binational Science Foundation Grant 2003-220 to J.A. and F.N. J.A. is the Dr. Joseph and Ruth Owades Professor of Chemistry and F.N. the Leonard and Esther Kurtz Term Professor at the College of Staten Island of the City University of New York.
Abbreviations: CD, circular dichroism; DIEA, diisopropylethylamine; EDTA, ethylenediaminetetraacetic acid; EEE, Glu-Glu-Glu tag; FP-PR, fusion peptide proximal region; gp41, HIV-1 glycoprotein 41; gp120, HIV-1 glycoprotein 120; HIV-1, human immunodeficiency virus type 1; HPLC, high-pressure liquid chromatography; HR1, HIV-1 gp41 N′ heptad repeat 1; HR2, HIV-1 gp41 C′ heptad repeat 2; IPTG, isopropyl β-D-1-thiogalactopyranoside; Kd, dissociation constant; MPER, membrane proximal external region; MALDI, matrix-assisted laser desorption ionization; NMR, nuclear magnetic resonance; RRR, Arg-Arg-Arg tag; SIV, simian immunodeficiency virus; T-20, HIV fusion inhibitor corresponding to gp41 residues Y638–F673; TFA, trifluo-roacetic acid; TOF, time-of-flight; 6-HB, six-helix bundle.
References
- 1.Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 2.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151:413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 4.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 5.Tan K, Liu J, Wang J, Shen S, Lu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci USA. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zwick MB, Jensen R, Church S, Wang M, Stiegler G, Kunert R, Katinger H, Burton DR. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J Virol. 2005;79:1252–1261. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22:163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poumbourios P, el Ahmar W, McPhee DA, Kemp BE. Determinants of human immunodeficiency virus type 1 envelope glycoprotein oligomeric structure. J Virol. 1995;69:1209–1218. doi: 10.1128/jvi.69.2.1209-1218.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robertson D. US FDA approves new class of HIV therapeutics. Nat Biotechnol. 2003;21:470–471. doi: 10.1038/nbt0503-470. [DOI] [PubMed] [Google Scholar]
- 12.Este JA, Telenti A. HIV entry inhibitors. Lancet. 2007;370:81–88. doi: 10.1016/S0140-6736(07)61052-6. [DOI] [PubMed] [Google Scholar]
- 13.Lawless MK, Barney S, Guthrie KI, Bucy TB, Petteway SR, Jr, Merutka G. HIV-1 membrane fusion mechanism: Structural studies of the interactions between biologically-active peptides from gp41. Biochemistry. 1996;35:13697–13708. doi: 10.1021/bi9606962. [DOI] [PubMed] [Google Scholar]
- 14.Liu S, Lu H, Niu J, Xu Y, Wu S, Jiang S. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J Biol Chem. 2005;280:11259–11273. doi: 10.1074/jbc.M411141200. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Jing W, Cheung B, Lu H, Sun J, Yan X, Niu J, Farmar J, Wu S, Jiang S. HIV gp41 C-terminal heptad repeat contains multifunctional domains. Relation to mechanisms of action of anti-HIV peptides. J Biol Chem. 2007;282:9612–9620. doi: 10.1074/jbc.M609148200. [DOI] [PubMed] [Google Scholar]
- 16.Blacklow SC, Kim PS. Protein folding and calcium binding defects arising from familial hypercholesterolemia mutations of the LDL receptor. Nat Struct Biol. 1996;3:758–762. doi: 10.1038/nsb0996-758. [DOI] [PubMed] [Google Scholar]
- 17.Bornstein P, Balian G. Cleavage at Asn-Gly bonds with hydroxylamine. Methods Enzymol. 1977;47:132–145. doi: 10.1016/0076-6879(77)47016-2. [DOI] [PubMed] [Google Scholar]
- 18.Creighton TE. Proteins. 2. W. H. Freeman and Company; New York: 1993. [Google Scholar]
- 19.Xu W, Taylor JW. A template-assembled model of the N-peptide helix bundle from HIV-1 Gp-41 with high affinity for C-peptide. Chem Biol Drug Des. 2007;70:319–328. doi: 10.1111/j.1747-0285.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- 20.Chun PW, Kim SJ. Determination of equilibrium constants of associating protein systems. Graphical analysis for discrete and indefinite association. Biochemistry. 1970;9:1957–1961. doi: 10.1021/bi00811a016. [DOI] [PubMed] [Google Scholar]
- 21.Biron Z, Khare S, Quadt SR, Hayek Y, Naider F, Anglister J. The 2F5 epitope is helical in the HIV-1 entry inhibitor T-20. Biochemistry. 2005;44:13602–13611. doi: 10.1021/bi0509245. [DOI] [PubMed] [Google Scholar]
- 22.Chang DK, Hsu CS. Biophysical evidence of two docking sites of the carboxyl heptad repeat region within the amino heptad repeat region of gp41 of human immunodeficiency virus type 1. Antiviral Res. 2007;74:51–58. doi: 10.1016/j.antiviral.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 23.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthew JB, Richards FM. The pH dependence of hydrogen exchange in proteins. J Biol Chem. 1983;258:3039–3044. [PubMed] [Google Scholar]
- 26.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorizate M, Gomara MJ, de la Torre BG, Andreu D, Nieva JL. Membrane-transferring Sequences of the HIV-1 Gp41 Ectodomain Assemble into an Immunogenic Complex. J Mol Biol. 2006;360:45–55. doi: 10.1016/j.jmb.2006.04.056. [DOI] [PubMed] [Google Scholar]
- 28.Bosshard HR, Marti DN, Jelesarov I. Protein stabilization by salt bridges: Concepts, experimental approaches and clarification of some misunderstandings. J Mol Recognit. 2004;17:1–16. doi: 10.1002/jmr.657. [DOI] [PubMed] [Google Scholar]
- 29.Joshi MD, Hedberg A, McIntosh LP. Complete measurement of the pKa values of the carboxyl and imidazole groups in Bacillus circulans xylanase. Protein Sci. 1997;6:2667–2670. doi: 10.1002/pro.5560061224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vazquez-Ibar JL, Guan L, Svrakic M, Kaback HR. Exploiting luminescence spectroscopy to elucidate the interaction between sugar and a tryptophan residue in the lactose permease of Escherichia coli. Proc Natl Acad Sci USA. 2003;100:12706–12711. doi: 10.1073/pnas.1835645100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howlett GJ, Minton AP, Rivas G. Analytical ultracentrifugation for the study of protein association and assembly. Curr Opin Chem Biol. 2006;10:430–436. doi: 10.1016/j.cbpa.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 32.Efremov RG, Volynsky PE, Nolde DE, Vergoten G, Arseniev AS. The membrane-proximal fusion domain of HIV-1 GP41 reveals sequence-specific and fine-tuning mechanism of membrane binding. J Biomol Struct Dyn. 2007;25:195–205. doi: 10.1080/07391102.2007.10507169. [DOI] [PubMed] [Google Scholar]
- 33.Biron Z, Khare S, Samson AO, Hayek Y, Naider F, Anglister J. A Monomeric 310-Helix Is Formed in Water by a 13-Residue Peptide Representing the Neutralizing Determinant of HIV-1 on gp41. Biochemistry. 2002;41:12687–12696. doi: 10.1021/bi026261y. [DOI] [PubMed] [Google Scholar]