

Abstract
As structural genomics efforts succeed in solving protein structures with novel folds, the number of proteins with known structures but unknown functions increases. Although experimental assays can determine the functions of some of these molecules, they can be expensive and time consuming. Computational approaches can assist in identifying potential functions of these molecules. Possible functions can be predicted based on sequence similarity, genomic context, expression patterns, structure similarity, and combinations of these. We investigated whether simulations of protein dynamics can expose functional sites that are not apparent to the structure-based function prediction methods in static crystal structures. Focusing on Ca2+ binding, we coupled a machine learning tool that recognizes functional sites, FEATURE, with Molecular Dynamics (MD) simulations. Treating molecules as dynamic entities can improve the ability of structure-based function prediction methods to annotate possible functional sites.
1. Introduction
The problem of function prediction is central to bioinformatics. Recently, the number of approaches dealing with solving this problem has increased dramatically. Some methods use interaction data collected from genomic and microarray experiments [1]. Sequence based approaches use sequence conservation through analysis of related sequences [2]. Some methods recognize sequence motifs, compiled into databases such as PROSITE [3] and PRINTS [4], and analyze their patterns of co-occurrence in the related sequences. There are also aggregate approaches that apply several methods at once to examine a given structure [5, 6].
Structural genomics efforts specifically attempt to solve the structures of proteins with novel folds [7, 8]. As such, there is a pressing need for reliable structure-based function prediction methods. These methods rely on conserved geometric context of sites of interest. They range from global, identifying possible substrate binding pockets, to local, concentrating on the particular atoms coordinating ligand binding [9-12]. Because three-dimensional (3D) environment is more conserved than sequence, these methods may have more success when sequence similarity is too low to detect 1D motifs or even overall similarity.
Methods that identify calcium (Ca2+) binding sites in protein structures have had variable success. These methods employed artificial neural networks [13], graph theory and geometric similarity [14], bond-valence calculations [15, 16] and distribution of distances between Cα atoms of residues [17]. We have previously described FEATURE [18], a machine learning algorithm which employs Bayesian scoring scheme, and its ability to identify Ca2+ binding sites [19]. FEATURE uses models built by examination of local physico-chemical environments to predict whether a site of interest has a potential for a particular function of interest. The chief advantages of FEATURE are its generality, extending to many types of sites [18], and its focus on 3D environments which allows it to recognize divergent binding sites, without depending on sequence or structure similarity.
Until now, FEATURE has only been applied to static structures. However, protein molecules are dynamic and examining their behavior over time may improve the performance of structure-based function prediction methods. Molecular Dynamics (MD) uses physical principles to simulate the motion of protein molecules, and has been applied for many purposes, including structure refinement, drug docking, protein engineering, and protein folding [8,9]. We propose a novel application of MD simulations: generating structural diversity in order to improve our ability to detect functional sites. For a single protein, there can now be many structural examples that can reveal its functions. In order to test this idea, we asked whether MD simulations coupled with FEATURE analysis could reproduce and further improve performance of FEATURE alone. We present our preliminary results on a single protein, parvalbumin β.
2. Methods
2.1 Structures
From the Protein Data Bank we obtained two structures for parvalbumin β (10) (PDB IDs 1B9A and 1B8C). Since we were only interested in the monomeric protein structures and associated ions we used only the coordinates of the structure and Ca2+ atoms for 1B9A (HOLO) and the coordinates of the first monomer and associated Mg2+ atom for 1B8C (APO).
2.2 Molecular Dynamics Simulations
Using software suit GROMACS version 3.3.1 [20], we set up two simulation systems - one for each structure. In case of 1B9A, the structure was solvated in 4,411 simple point charge (SPC) [21] water molecules. 1B8C structure was solvated in 4,479 SPC water molecules. The solvent buffer zone comprised 1nm. Addition of one Na+ atom and three Na+ atoms neutralized the 1B9A and 1B8C systems, respectively.
Each simulation started with a 200-step energy minimization run using the steepest descent algorithm and was followed by a 10 picosecond (ps) simulation with harmonic position restraints applied to all protein atoms to allow relaxation of the solvent molecules and added ions. The use of LINCS [22] algorithm and GROMOS96 [23] force field (GROMOS96 43a1) allowed for a 2 femtosecond (fs) integration time step. The systems were coupled to external temperature baths [24] of 300K with a coupling constant tauT = 0.1ps separately for the protein and the solvent with added ions. Electrostatic and Van der Waals interactions and neighborlist cut-offs were set at 1nm. Finally, each of the systems underwent free dynamic simulations for 1 nanosecond (ns), at constant temperature, as above, and constant pressure, kept at 1 bar by weak coupling to pressure baths [24] with tauP = 0.5ps. We obtained snapshots of the simulations every 2.5ps, 401 total for each simulation, to examine the generated structures further with FEATURE.
RMS fluctuations per residue were calculated by averaging the atomic RMS fluctuations of each residue, as determined by GROMACS.
2.3 FEATURE Scanning
Using FEATURE [18] version 1.9 and Ca2+ binding site model [19] we analyzed original PDB structures and those generated by the simulations in two ways: using a 6 × 6 × 6 Å3 grid with point density of 1Å (local grid) placed directly over the Ca2+ binding sites, and a grid that encompassed the whole protein with point density of 1Å (global grid). The local grid was centered on an equivalent atom in each of the structures obtained from the simulations, identified as the closest atom to the Ca2+ of interest in the HOLO PDB structure file. For both 1B9A and 1B8C structures this atom was defined as ASP90 OD1675 for EF loop binding site and PHE57 O407 for CD loop binding site. At each grid point, FEATURE generated a score, representing the likelihood of there being a potential Ca2+ binding site centered at that point. The threshold of the Ca2+ binding model is 50, therefore, any point scoring 50 or above is considered a plausible center for a Ca2+ binding site. From the local grid scanning, only the coordinates of the highest scoring point were kept for further visualization and analysis. Coordinates of all the points scoring above model threshold in the global grid scanning were kept for further visualization.
For negative controls, local grids were centered on two points located far away from the true Ca2+ binding sites in the HOLO structure with the following criteria: the atom density of the environment near the points and the distance of the points from the surface were comparable to the local grid centers of the true Ca2+ binding sites. These points were LEU15 N93 and ASP24 C295. Coordinates for the top scoring points were kept, just as in the local grid scanning of Ca2+ binding sites.
2.4 Viewing of the Structures
Visual Molecular Dynamics (VMD) [25] allowed visual analysis of simulation trajectories and illustrations of the structures and results of FEATURE scanning. Molecular images were generated using Tahyon Parallel/Multiprocess Ray Tracer System (Fig. 1,2,4) [26, 27].
Figure 1.

Visited structural conformations over the course of the MD simulations for the HOLO and APO systems. Grey balls represent the first and the last atoms in each of the chains. Greater flexibility of the HOLO structure is noticeable.
Figure 2.

FEATURE scores profiles of local grid analysis and associated structures. 1ns Simulation:  . Original FEATURE Score:
. Original FEATURE Score:  . Model Threshold:
. Model Threshold:  . a) 1B9A EF loop binding site. b) 1B9A ASP24C295, negative control. c) 1B8C EF loop binding site, positive control. d) 1B8C CD loop binding site, which was abolished by mutations. In the structures, location of the binding site is made obvious by the close association of oxygens, black balls, whether terminal ones from ASPs and GLUs or main-chains ones, near the highest-scoring points, white balls. 1) 1B9A EF loop binding site in the original static structure, which scores ∼77. 2) 1B9A EF loop binding site at 445ps, which scores ∼20. 3) 1B8C EF loop binding site at 615ps, which scores ∼112. 4) 1B9A CD loop binding site at 184ps, which scores ∼52. 5) 1B8C CD loop binding site, abolished by the mutations, at 187.5ps, which scores ∼13.5. 6) 1B9A negative control centered on ASP24C295 at 805ps, which scores ∼13.5.
. a) 1B9A EF loop binding site. b) 1B9A ASP24C295, negative control. c) 1B8C EF loop binding site, positive control. d) 1B8C CD loop binding site, which was abolished by mutations. In the structures, location of the binding site is made obvious by the close association of oxygens, black balls, whether terminal ones from ASPs and GLUs or main-chains ones, near the highest-scoring points, white balls. 1) 1B9A EF loop binding site in the original static structure, which scores ∼77. 2) 1B9A EF loop binding site at 445ps, which scores ∼20. 3) 1B8C EF loop binding site at 615ps, which scores ∼112. 4) 1B9A CD loop binding site at 184ps, which scores ∼52. 5) 1B8C CD loop binding site, abolished by the mutations, at 187.5ps, which scores ∼13.5. 6) 1B9A negative control centered on ASP24C295 at 805ps, which scores ∼13.5.
Figure 4.
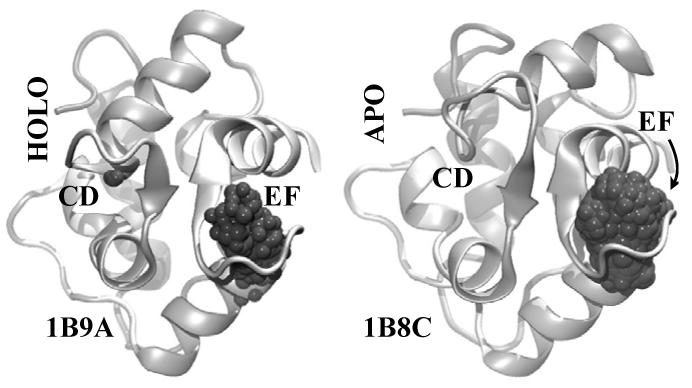
Structures of 1B9A, HOLO, and 1B8C, APO, analyzed by global grid scanning. All of the structures generated by the simulations were aligned for each molecule, and global backbone RMSD was minimized. Shown are the initial structures used in the 1ns simulation in a backbone cartoon representation, as well all of the points that scored over 50 during the simulation as grey balls.
3. Results
3.1 Molecular Dynamics Simulations
On Dual Core AMD Opteron™ Processor 880, 2.4 GHz, MD simulations of the HOLO system took 8.698 hours and of the APO system took 8.768 hours. Analysis of potential and kinetic energies of the systems over the course of the simulations revealed that both systems were stable throughout. Backbone root mean square deviation (RMSD) to the original crystal structure continued to increase slowly over the course of the simulation for 1B9A, reaching a maximum value of 4.5Å at the end of 1ns. In the case of 1B8C, backbone RMSD to the original crystal structure seemed to stabilize around 2.5Å towards the end of 1ns. Average RMSD between the consecutive structures generated by the simulations every 2.5ps was ∼0.5Å for both systems, demonstrating that the systems were evolving slowly over time.
3.2 FEATURE Scanning
On Dual Core AMD Opteron™ Processor 880, 2.4 GHz, local grid analysis took 7.117 minutes for HOLO and 7.167 minutes for APO, while global grid analysis took 3.225 hours for HOLO and 3.942 hours for APO. With the local grid scanning approach, points located in the vicinity of the Ca2+ binding sites present in the original HOLO and original APO structures, respectively, scored above the model threshold of 50. HOLO EF loop and APO EF loop binding sites showed persistent Ca2+ binding conformations over the course of MD simulation, obtaining scores of 50 or greater at several different time points. The local grid scan detected the Ca2+ binding signal in the HOLO CD loop binding site in the beginning of the simulation, but over time the signal was lost. At numerous time points, some conformations achieved higher scores than the original starting structures. Negative controls had scores clustered around zero, never exceeding fifteen, while showing similar extent of structural diversity and sampling (Fig. 2).
FEATURE correctly identified all of the Ca2+ binding sites expected in the HOLO and APO structures in the global grid scanning analysis approach. Figure 4 shows all the points which scored over the model threshold of 50 in the structures at all of the examined time points of the simulations. The structures from all the time points were aligned globally to minimize the total RMSD among them and only the starting structures of the HOLO and APO 1ns simulations are shown.
4. Discussion
Parvalbumin β is a small, 10-12kDa member of EF-hand Ca2+ binding proteins. Structurally, it consists of 108 residues, which form six α-helices and intermittent loops. There are two Ca2+ binding sites in wild type parvalbumin β: one located in the loop between helices C and D (CD loop binding site) and one located in the loop between helices E and F (EF loop binding site). Several wild type and mutant structures exist for parvalbumin β in the Protein Data Bank (PDB). We used two structures taken from PDB entries 1B9A and 1B8C [28]. These two structures are not of wild type parvalbumin, but share two mutations: F102W and D51A. The first mutation allows experimental monitoring of metalion presence by following the magnitude of tryptophan fluorescence upon metal binding. This mutation did not affect metal binding properties of the molecule. The second mutation was introduced to study the cooperative effects of the CD loop on the EF loop binding site. Interestingly, this mutation did not change the binding properties of the EF loop site, but severely decreased Ca2+ affinity for the CD loop site. Nevertheless, Ca2+ binding was observed in both of these sites in the original 1B9A structure [28].
There are several differences between the 1B9A and 1B8C structures. Firstly, 1B9A crystallized with Ca2+ present in the binding sites (HOLO), while 1B8C crystallized without Ca2+ in the binding sites (APO). Secondly, the 1B8C original structure contains a third mutation, E101D, introduced to study the role of the glutamic acid at the last coordinating position of the Ca2+ binding loop. This mutation stabilizes EF loop, since the side chain of aspartate is shorter and less mobile than the side chain of glutamate. This mutation had similar effects on the two Ca2+ binding sites in the molecule, as measured experimentally. The Ca2+ affinity of EF loop site was reduced 10-fold. The affinity of the CD loop site was also reduced, such that in combination with the D51A mutation, Ca2+ binding was no longer observed at this site, even when crystallization media did contain Ca2+ [28]. Therefore, while HOLO structure retains the two Ca2+ binding sites present in the wild type parvalbumin, only one Ca2+ binding site remains in the APO structure.
MD simulations provided conformations alternative to the original HOLO and APO structures, allowing to generate conclusions on the basis of a set of snapshot structures for HOLO and APO systems, rather than on a single static structure. The structures generated by the 1ns simulation seemed to be sufficiently diverse for the purposes of this study. The backbone RMS deviations over 1ns were no more than 4.5 Å, and were sufficient to find conformations that achieved a varied range of scores. Figure 1 presents the structural diversity sampled by the MD simulations, as given by structures created by the simulations at the 401 analyzed time points.
FEATURE examines the local environment of functional sites, compares it to a set of non-sites, and builds a model to identify the same functional sites in other structures. Using a Ca2+ binding site model [19], FEATURE correctly identified all of the Ca2+ binding sites present and expected in the original static HOLO and APO structures. Although the original training set for this Ca2+ binding site model contained other parvalbumin structures, these formed a small proportion of the total number of structures used to create the general model. Therefore, this model does not specifically bias results towards Ca2+ binding sites of parvalbumin. As such, this HOLO-APO pair formed a good test case for determining whether simulations of molecular dynamics can be applied to structure based function prediction.
The coupling of FEATURE scanning to structures generated by the MD simulations is promising. First, using local grid scanning, we observed local structural conformations in the vicinity of Ca2+ binding sites that were both high scoring and low scoring for Ca2+ binding (Fig. 2). Such dynamic FEATURE score profiles may be useful in assessing the presence, stability and persistence of Ca2+ binding sites. For the HOLO structures, local grid scanning revealed that EF loop binding site was persistent and stable (Fig. 2a). The scores repeatedly surpassed the model threshold of 50. CD loop showed good Ca2+ binding site signal in the beginning of the simulation, but over time that signal was lost, due to structural rearrangements within the loop. The APO structures yielded similar results. The EF loop binding site exhibited stability and persistence even more than in the HOLO case (Fig. 2c). In essence, this result could be considered a positive control: an environment created and shown experimentally to be more stable obtained higher FEATURE scores. On the other hand, the CD loop binding site did not at any point in the simulation attain favorable Ca2+ binding conformations (Fig. 2d). This site, in essence, could be considered as a negative control, since based on experimental evidence, there was no Ca2+ binding site to be found in the CD loop of the APO structure.
In order to understand how the E101D mutation present in the APO molecule stimulates a difference in structural behaviors of the HOLO and APO systems, we examined RMS fluctuations per residue over the course of the two simulations (Fig. 3). The two grey rectangles on the Residue Number axis mark where the two Ca2+ binding sites are in the sequence. The small lightning bolt symbol points to the location of the concerned mutation. It is interesting to note that the change from glutamine to aspartate at position 101 dampens dramatically not only the flexibility of the EF loop surrounding this residue, as expected, but also the flexibility of the CD loop and its immediate upstream surroundings. Such a reduction in the possible motion space may be the reason why CD loop binding site is abolished in the APO molecule - it may require greater structural fluctuations, such as the ones observed in the HOLO case, to form properly.
Figure 3.
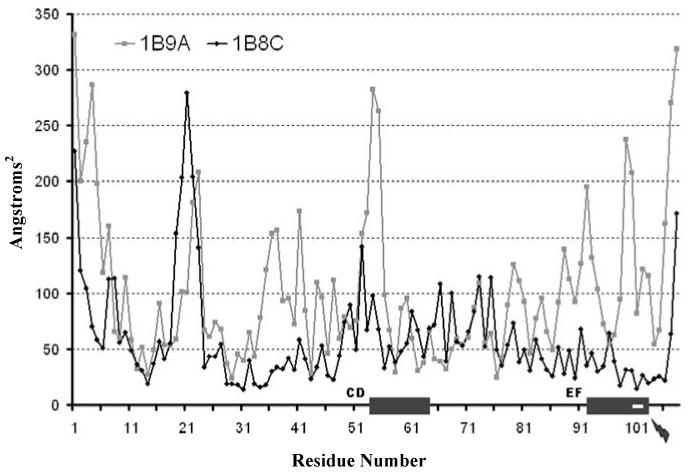
RMS fluctuations per residue over the course of the simulations for both HOLO and APO systems. Grey boxes along the Residue Number axis depict location of CD loop and EF loop Ca2+ biding sites. White rectangle in the EF loop grey box as well as the small lightning bolt depict the position of the mutation that abolishes CD loop binding site in the APO structure.
In order to explore further the potential of MD simulations coupled with FEATURE analysis to discern true positive Ca2+ binding sites, we performed local grid scanning analysis centered on two other points in the HOLO structure. These points were chosen to contain in their environment the same number of atoms as FEATURE sees at the centers of the local grids in the true Ca2+ binding sites in HOLO and APO structures. Additionally, these points resided as close to the surface of the protein, as the centers of the grids chosen for the true Ca2+ binding sites, and far away from the true Ca2+ binding sites, to avoid aberrant influences. FEATURE score profiles of these negative sites showed that the frequency and magnitude of the structural changes distinguished by FEATURE were comparable to the magnitude observed in the true Ca2+ binding sites. However, the scores at these sites were much lower (Fig. 2b). The scores observed for LEU15 N93 site were even lower than the ones observed for ASP24C295. As such, the FEATURE scores profiles of the negative controls underscored the lack of Ca2+ binding site in the CD loop of 1B8C (Fig. 2d).
Global grid analysis confirmed further the results observed with the local grid scanning (Fig. 4). Over the course of the simulations, FEATURE recognized only the points in the vicinity of the true sites as favorable centers for Ca2+ binding. Thus, EF loop and CD loop of 1B9A and EF loop of 1B8C were correctly identified as Ca2+ binding sites. Since only the points at the true Ca2+ binding sites scored over the model threshold, the rest of the points encompassing the whole protein could be thought of as correctly predicted negative controls.
The profiles of FEATURE scores obtained by local grid analysis revealed that some of the conformations generated during the simulations achieved scores that were higher than in the original crystal structures. These structures offer intriguing alternative local conformations that the protein may adopt in order to facilitate Ca2+ binding. These conformations may be worth studying to investigate shared features that contribute to the details of Ca2+ binding. In addition, the ability of MD to create alternative conformations, in which the score at the Ca2+ binding site is significantly different from the score of the crystal structure, indicates that it should be able to discover Ca2+ binding sites in simulations of proteins whose Ca2+ sites happen not to be sufficiently well-formed in their crystalline state. In fact, even if FEATURE were not to recognize Ca2+ binding sites in the original structures, correct assignment of Ca2+ binding sites would have been possible for parvalbumin β based on the HOLO and APO conformations and associated FEATURE score distributions generated by the simulations. Furthermore, using the global grid approach, it was possible to identify de novo potential Ca2+ binding sites, when a structure of the molecule without bound Ca2+ was used. Lacking knowledge of the HOLO structure, APO structure would have been correctly annotated to have one Ca2+ binding site based on our results.
It is interesting to note that the FEATURE scores change significantly even across relatively small time steps, thus indicating that it is very sensitive to the detailed position of atoms during the simulation. This is a good sign that FEATURE will be sensitive to small conformational changes that might affect the ability to bind calcium. Based on this, we are optimistic that MD will be able to sample conformational diversity sufficiently to improve performance of FEATURE when it misses false negative sites in crystal structures that are within 2 - 4Å of local atom RMSD from conformations that show the missed function. This estimate is based on the amount of conformational change seen in our simulations and the associated variation in the FEATURE scores. It is also reassuring that the true negative control sites never achieved scores close to the range sampled by the true positives.
An intriguing question emerges from our results: is there a correlation between the frequency of high scores and binding affinity of the site? A study involving a larger set of diverse Ca2+ binding proteins is necessary to pursue the answer to this. In addition, we only simulated protein dynamics for 1ns. It is likely that with longer simulations, other conformations would be visited that would broaden the distribution of scores to reach both higher and lower than the scores of the conformations sampled in 1ns. We are keen to explore the conformational diversity at different lengths of MD simulations and to asses the value of longer simulations for function recognition purposes.
The results of this study are limited to a single protein and a single function, and thus they can not be generalized to all proteins and functions. Experiments are underway to explore further the potential of MD to improve FEATURE predictions of Ca2+ binding sites using more HOLO - APO pairs. These include examples of sites that FEATURE can not identify by itself as Ca2+ binding. Furthermore, we plan to examine functions other than Ca2+ binding, as well as couple together different methods to sample conformational space and to identify functional sites in 3D structures, that are less expensive computationally and thus can be applied to larger datasets..
Coupling MD simulations with FEATURE showed potential in being able to allow better annotation of novel structures with unknown function and of structures where functions have been already assigned.
5. Acknowledgements
This work was supported by Stanford Genome Training Grant (NIH 5 T32 HG00044) and FEATURE Grant (NIH LM-05652) and the Simbios National Center for Biomedical Computing (http://simbios.stanford.edu/, NIH GM072970). FEATURE is available online at http://feature.stanford.edu/ and http://simtk.org.
Contributor Information
DARIYA S. GLAZER, Department of Genetics, Stanford University, 318 Campus Drive Clark Center S240 Stanford, CA 94305, USA
RANDALL J. RADMER, SIMBIOS National Center, 318 Campus Drive Clark Center S231 Stanford, CA 94305, USA
RUSS B. ALTMAN, Departments of Bioengineering and Genetics, Stanford University 318 Campus Drive Clark Center S170, Stanford, CA 94305, USA
6 References
- 1.Walker MG, Volkmuth W, Spinzak E. Genome Research. 1999;9:1198. doi: 10.1101/gr.9.12.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sjölander K. Bioinformatics. 2004;20:170. doi: 10.1093/bioinformatics/bth021. [DOI] [PubMed] [Google Scholar]
- 3.Hulo N, Sigrist C, Saux VL, Langendijk-Genevaux P, Bordoli L, Gattiker A, Castro ED, Bucher P, Bairoch A. Nucleic Acids Research. 2004;32:D134. doi: 10.1093/nar/gkh044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attwood T, Bradley P, Flower D, Gaulton A, Maudling N, Mitchell A, Moulton G, Nordle A, Paine K, Taylor P, Uddin A, Zygouri C. Nucleid Acids Research. 2003;31:400. doi: 10.1093/nar/gkg030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laskowski RA, Watson JD, Thornton JM. Nucleic Acids Research. 2005;33(Web Server issues):W89. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pal D, Eisenberg D. Structure. 2005;13:1. doi: 10.1016/j.str.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Chandonia JM, Brenner SE. Science. 2006;311(5795):347. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- 8.Terwilliger TC. Nature Structural and Molecular Biology. 2004;11(4):296. doi: 10.1038/nsmb0404-296. [DOI] [PubMed] [Google Scholar]
- 9.Holm L, Sander C. Journal of Molecular Biology. 1993;233:123. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 10.Fetrow JS, Godzik A, Skolnick J. Journal of Molecular Biology. 1998;282:703. doi: 10.1006/jmbi.1998.2061. [DOI] [PubMed] [Google Scholar]
- 11.Laskowski RA, Watson JD, Thornton JM. Journal of Molecular Biology. 2005;351:614. doi: 10.1016/j.jmb.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 12.Krissinel E, Henrick K. Acta Crystallographica. 2004;D(60):2256. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 13.Sodhi JS, Bryson K, McGuffin LJ, Ward JJ, Wernisch L, Jones DT. Journal of Molecular Biology. 2004;342(307-320):307. doi: 10.1016/j.jmb.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 14.Deng H, Chen G, Yang W, Yang JJ. Proteins: Structure, Function, and Bioinformatics. 2006;64(34-42):34. doi: 10.1002/prot.20973. [DOI] [PubMed] [Google Scholar]
- 15.Müller P, Köpke S, Sheldrick GM. Acta Crystallographica. 2003;D59:32. doi: 10.1107/s0907444902018000. [DOI] [PubMed] [Google Scholar]
- 16.Nayal M, Cera ED. Proc. Natl. Acad. Sci. USA. 1994;91:817. doi: 10.1073/pnas.91.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asaoka T, Ando T, Meguro T, Yamato I. Chem-Bio Informatics Journal. 2003;3(3):96. [Google Scholar]
- 18.Wei L, Altman RB. PSB: Pacific Symposium of Biocomputing. Maui, HI: 1998. Recognizing Protein Binding Sites Using Statistical Descritions of Their 3d Environments; p. 497. [PubMed] [Google Scholar]
- 19.Wei L, Altman RB. Journal of Bioinformatics and Computational Biology. 2003;1(1):119. doi: 10.1142/s0219720003000150. [DOI] [PubMed] [Google Scholar]
- 20.Lindahl E, Hess B, van der Spoel D. Journal of Molecular Modeling. 2001;7:306. [Google Scholar]
- 21.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. In: Intermolecular Forces. Pullman B, editor. D. Reidel Publishing Company; Dordrecht, The Netherlands: 1981. p. 331. [Google Scholar]
- 22.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. Journal of Computational Chemistry. 1997;18:1463. [Google Scholar]
- 23.van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, Scott WRP, Tironi IG. Biomolecular Simulation: The Gromos96 Manual and User Guide. Hochschulverlag AG an der ETH Zürich; Zürich, Switzerland: 1996. [Google Scholar]
- 24.Berendsen HJC, Postma JPM, van Gunsteren WF, Nola AD, Haak JR. Journal of Chemical Physics. 1984;81(8):3684. [Google Scholar]
- 25.Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics. 1996;14(1):33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 26.Stone J. Computer Science Department. University of Missouri; Rolla: 1998. An Efficient Library for Parallel Ray Tracing and Animation. [Google Scholar]
- 27.Frishman D, Argos P. Proteins: Structure, Function and Genetics. 1995;23:566. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 28.Cates MS, Barry MB, Ho EL, Li Q, Potter JD, George N, Phillips J. Structure. 1999;7:1269. doi: 10.1016/s0969-2126(00)80060-x. [DOI] [PubMed] [Google Scholar]