

Abstract
Bovine herpesvirus 1 (BHV-1), a member of the alpha-herpesvirinae subfamily, causes significant losses to the cattle industry. BHV-1 establishes latency in trigeminal ganglionic sensory neurons, but periodically reactivates from latency. Previous studies suggested that infection with BHV-1 induced novel morphological changes in rabbit skin cells (RS) versus bovine kidney cells (MDBK). Consequently, we hypothesized that viral infection led to a novel form of cell death in RS cells compared to MDBK cells. To test this hypothesis, we examined the levels of apoptosis in these cell types following infection with BHV-1. Infection of RS, but not MDBK, cells leads to high levels of apoptosis compared to mock infected cells. Previous studies indicated that a BHV-1 recombinant virus that does not express the bICP0 protein grows poorly in permissive cells and induces a persistent-like infection. This suggested that bICP0 played an important role in regulating cell death following infection. To test this hypothesis, we compared the levels of apoptosis in cells infected with the bICP0 null mutant versus viral strains that expressed bICP0. The bICP0 null mutant induces low levels of apoptosis in RS or MDBK cells. When MDBK cells are treated with UV light prior to infection, bICP0 expressing viral strains, but not the bICP0 null mutant, inhibited UV induced apoptosis. Infection of MDBK cells with the bICP0 null mutant, leads to an accumulation of autophagosomes that are not detected following infection with bICP0 expressing viruses. These studies suggest that the bICP0 null mutant induces autophagy in MDBK cells, and bICP0 protein expression mediates cell type specific cytotoxicity.
Keywords: bovine herpesvirus type 1, apoptosis, autophagy, bICP0
INTRODUCTION
Infection of cattle with bovine herpesvirus 1 (BHV-1) leads to respiratory disorders, conjunctivitis, genital infections, encephalitis, abortions and a multi-systemic fatal disease in neonates. Following acute infection, BHV-1 establishes latency in sensory neurons within trigeminal or dorsal root ganglia (Jones, 1998; Jones, 2003). Reactivation from latency is stimulated by dexamethasone treatment or increases in natural corticosteroids resulting in virus shedding and spread to susceptible hosts. Although the main site of BHV-1 latency is sensory neurons, BHV-1 DNA is consistently detected in tonsils (Perez et al., 2005; Winkler et al., 2000), peripheral blood cells (Fuchs et al., 1999), lymph nodes, and spleen (Mweene et al., 1996) of latently infected calves.
Like other alpha-herpesvirinae subfamily members, viral gene expression is regulated temporally in three distinct phases: immediate-early (IE), early (E) or late (L). The bICP0 protein is encoded by IE transcription unit 1 (IEtu1) (Wirth et al., 1992), and is expressed constitutively during productive infection because it has an IE and E promoter that is activated by bICP0 (Fraefel et al., 1994). When BHV-1 DNA is transfected into permissive cells, plaque formation is inefficient. Co-transfection of BHV-1 DNA with bICP0 significantly increases the number of plaques and decreases the time prior to plaque formation (Geiser and Jones, 2003; Geiser et al., 2002; Inman et al., 2001b) indicating bICP0 stimulates productive infection. The ability of bICP0 to associate with chromatin remodeling enzymes, histone deacetylase 1 (HDAC1) and p300, stimulates viral gene expression and productive infection (Zhang and Jones, 2001; Zhang et al., 2006). Since bICP0 stimulates productive infection, the finding that a bICP0 null mutant did not produce well-defined plaques or grow efficiently in cultured cells was expected (Geiser et al., 2005).
In cultured cells or in infected calves, BHV-1 induces apoptosis (Devireddy and Jones, 1999; Lovato et al., 2003; Winkler et al., 1999). In the absence of viral gene expression, bICP0 indirectly induces caspase 3 activation and apoptosis (Henderson et al., 2004b). The LR gene, which is antisense with respect to bICP0, can inhibit apoptosis in transfected or infected cells (Ciacci-Zanella et al., 1999; Henderson et al., 2004a), trigeminal ganglia (Lovato et al., 2003), but not tonsils of infected calves (Perez et al., 2005). In addition, the UL14 gene inhibits apoptosis in transiently transfected cells (Marfe et al., 2006). It is unlikely that these three viral genes are the only BHV-1 genes that regulate apoptosis during productive infection or the latency-reactivation from latency because numerous HSV-1 genes have been identified that regulate apoptosis (Blaho and Aubert, 2001).
Although our previous studies indicated that bICP0 is not required for productive infection, bICP0 did have a profound effect on viral growth and the outcome of infection (Geiser et al., 2005). Consequently, we compared the cytotoxicity that occurs following infection with the bICP0 null mutant versus a bICP0 expressing virus strain. Following infection of rabbit skin (RS) cells with BHV-1, high levels of apoptosis were detected. In contrast, non-apoptotic cell death was the predominant form of cell death observed in bovine kidney (MDBK) cells following infection. In RS or MDBK cells, the bICP0 null mutant is not as cytotoxic as the bICP0-rescued virus. When MDBK cells are infected with the bICP0 null mutant, many of the cells contained numerous autophagosomes in the cytoplasm relative to mock infected cells or cells infected with the bICP0-resuced virus. In summary, these results demonstrated that BHV-1 kills cells in a cell type specific fashion, and bICP0 protein expression regulates cell death as well as virus growth.
RESULTS
Wt BHV-1 induces high levels of apoptosis in RS cells
Previous studies suggested that following infection of MDBK (bovine kidney) or RS (rabbit skin) cells with BHV-1 there were significant differences in the morphology of these cells. For example, infected RS cells appeared to contain extensive condensed chromatin relative to infected MDBK cells (data not shown). Consequently, we hypothesized that BHV-1 infection of RS cells led to higher levels of apoptosis relative to infection of MDBK cells. To compare BHV-1 induced cell death in different cell types, apoptotic DNA was examined by agarose gel electrophoresis following infection with MDBK or RS cells. Following infection of RS cells with wt BHV-1, a DNA ladder was present at 16 h after infection, which is indicative of extensive apoptosis (Figure 1). In mock-infected RS cells, the DNA ladder was not observed. When MDBK cells were infected with BHV-1, the DNA ladder was not readily observed at any time point examined. However, at 24 h after infection high molecular weight DNA (>10 Kb) was readily detectable in the gel suggesting that DNA damage had occurred. As expected, UV treatment of MDBK or RS cells led to apoptosis as judged by the appearance of DNA laddering.
Figure 1. Induction of DNA laddering following infection with BHV-1 or UV light treatment.
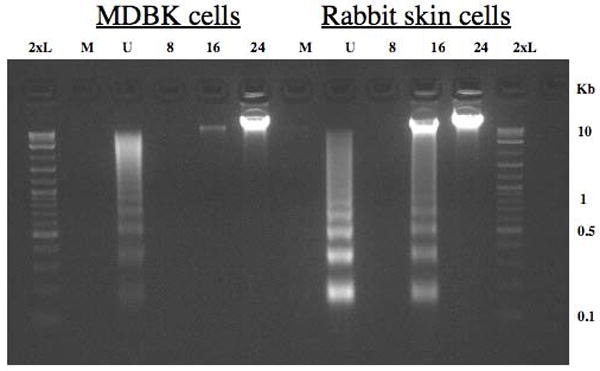
RS or MDBK cells were mock infected (M), UV treated (U), or infected with wt BHV-1 (MOI = 1) for 8, 16, or 24 hours. UV treated cells were exposed to UV light from a trans-illuminator for 30 sec (312 nm), fresh media was added to each well, and cultures were incubated for 4 h at 37 °C. After virus inoculation, cells were incubated for 1 h at 37°C, monolayers were rinsed twice with CMF-PBS, and EMEM supplemented with 5% FCS was added to each well. DNA was electrophoresed through a 2% agarose gel in 1x TAE. A 2-log molecular weight DNA marker (New England Biolabs) (2xL) was used as a DNA marker.
Comparison of DNA laddering following infection with a bICP0 null mutant virus or the bICP0 rescued virus
The bICP0 null mutant has decreased infectivity compared to bICP0 expressing viruses, does not produce well defined plaques, and is able to persist for several passages in bovine cells without killing all of the cells (Geiser et al., 2005). Consequently, we compared the bICP0 mutant to wt BHV-1 to better understand the role that efficient productive infection plays in virus induced cells death. For these studies, RS cells were infected with the bICP0 null mutant or the bICP0-rescued virus (MOI=1) and the appearance of DNA ladders monitored by agarose gel electrophoresis (Figure 2). We also included the LR mutant strain (lane L) in this study because previous studies demonstrated that the LR gene inhibits apoptosis in transfected cells, trigeminal ganglia, but not tonsils (Ciacci-Zanella et al., 1999; Henderson et al., 2004a; Lovato et al., 2003; Perez et al., 2005). Relative to mock infected RS cells (lane M), there was an increase in apoptotic DNA laddering following infection with the bICP0-rescued virus (lane R), the LR mutant virus (lane L), or UV light (lane U). Reduced DNA laddering was consistently observed when RS cells were infected with the bICP0 null mutant (lane N).
Figure 2. Induction of DNA laddering following infection with the bICP0 null mutant.
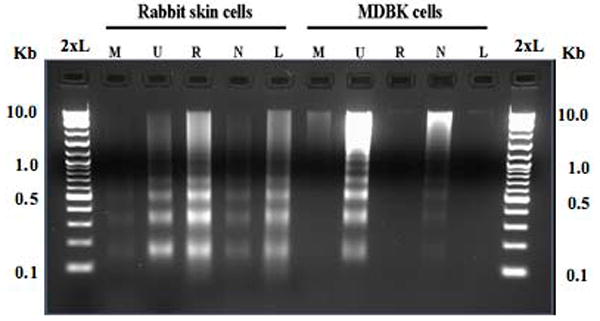
RS or MDBK cells were mock infected (M), UV treated (U), or infected with the bICP0-rescued virus (R), bICP0 null mutant (N), or the LR mutant (L). A MOI = 1 was used for all viruses. After virus inoculation, cells were incubated for 1 h at 37°C, monolayers were rinsed twice with CMF-PBS, and EMEM supplemented with 5% FCS was added to each well. Two days after plating, media was removed from mock-infected RS or MDBK cells, and these cells were exposed to UV light as described in Figure 1. At 48 h after infection, cells were collected and small molecular weight DNA prepared as described in materials and methods. DNA was electrophoresed along with a 2-log molecular weight DNA marked (New England Biolabs) through a 2% agarose gel in 1x TAE (2xL).
To compare the results obtained with RS cells, MDBK cells were infected with the respective viruses (Figure 2). Relative to mock infected MDBK cells (lane M), there was an increase in apoptotic laddering following UV treatment, which was expected (lane U). Following infection with the bICP0 null mutant, we observed a slight increase in levels of apoptotic DNA laddering (Figure 2, lane N), and/or a smear of low molecular weight DNA (for example, see Figure 4, lane N). DNA laddering was not readily detected when MDBK cells were infected with the bICP0-rescued virus (lane R) or the LR mutant (lane L), which was consistent with the results obtained in Figure 1.
Figure 4. The bICP0 null mutant does not protect cells from UV-induced DNA laddering.

MDBK cells were mock-infected (M), or infected with the bICP0-rescued virus (R), bICP0 null mutant (N), or the LR mutant (L). A MOI = 1 was used for all virus infections. After virus inoculation, cells were incubated for 1 h at 37°C, monolayers were rinsed twice with CMF-PBS, and EMEM supplemented with 5% FCS was added. At 4 h after infection, media was removed from 1 well of mock or viral infected MDBK cells. These cells were exposed to UV light (U) as described in Figure 1. Cells were collected and small molecular weight DNA was prepared as described in materials and methods. The DNA was eletrophoresed along with 2-log molecular weight DNA maker (New England Biolabs) (2xL) through a 2% agarose gel in 1x TAE.
To confirm and extend the results obtained from examining apoptotic DNA laddering in agaraose gels, we measured the sub G0/G1 DNA content by FACS analysis. Mock infected or infected cells were fixed in ethanol, stained with PI, and then analyzed. Following infection of MDBK cells with wt BHV-1, most of the cells were floating at 24 h after infection. In contrast, it took 2-3 days for this to occur in RS cells. Consequently, we examined RS cells at later times after infection. At 16 or 48 h after infection, a statistically significant difference in the percentage of cells containing sub G0/G1 DNA content was seen following bICP0-rescued virus infection of MDBK cells compared to mock infected (Figure 3A). At 48 h after infection, a higher percentage of cells with sub G0/G1 DNA content were detected when MDBK cells were infected with the bICP0 null mutant, however these values were not statistically significant (P > 0.05). The finding that slightly higher levels of sub G0/G1 DNA content was observed in MDBK cells following infection with the bICP0-rescued virus suggested that non-apoptotic death was occurring, which resulted in DNA degradation that was not typical DNA laddering observed during apoptosis. Similar results were also observed when wt BHV-1 was compared to the bICP0-rescued virus (data not shown). With respect to RS cells, a statistically significant increase in sub G0/G1 DNA was observed following infection with the bICP0-rescued virus at 4 dpi. Following infection of RS cells with the bICP0 null mutant virus, enhanced levels of cells containing sub G0/G1 DNA was detected at 4 dpi, however the values were not statistically significant compared to mock infected cells. When RS cells were cultured, an increase in the levels of sub G0/G1 DNA occurred as a function of time.
Figure 3. Examination of membrane permeability and DNA content following BHV-1 infection.
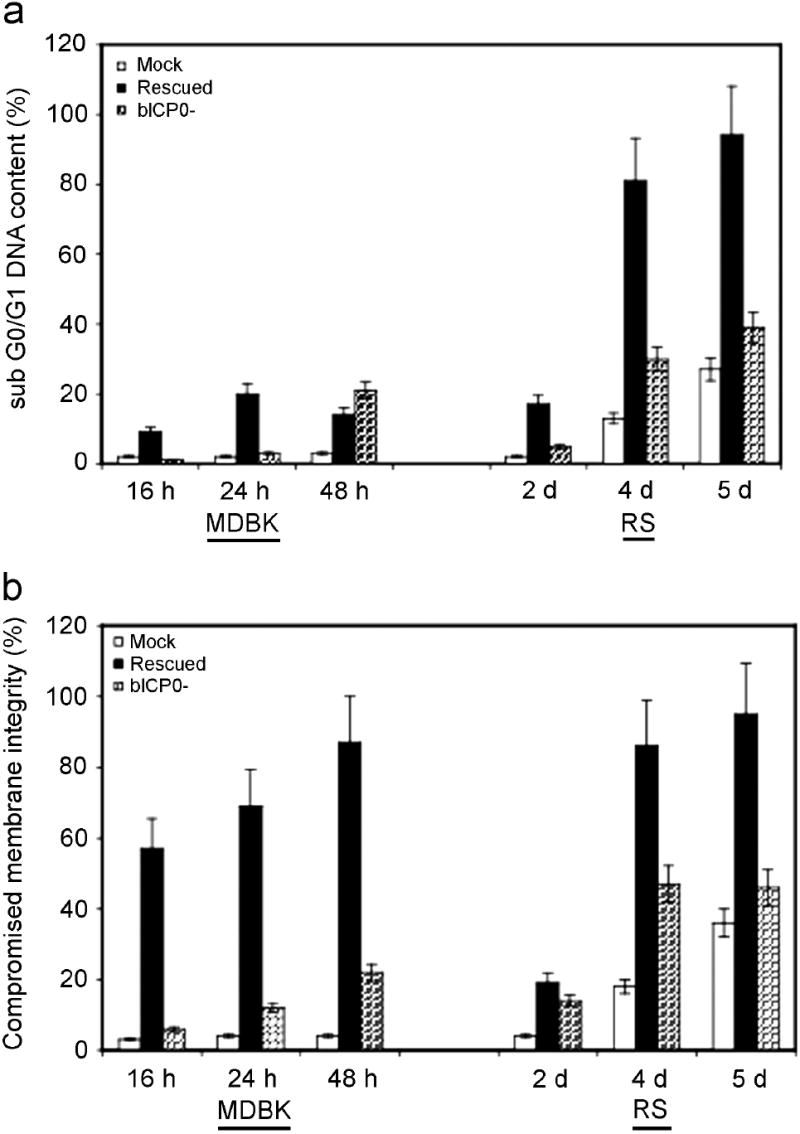
MDBK or RS cells were mock infected (M), or infected with the bICP0 null mutant (N) or the bICP0-rescued virus. A MOI = 1 was used for all viruses. At various times following infection, cells were collected, stained, and approximately 2 × 104 viable cell events were analyzed by flow cytometry as described in materials and methods. The data presented are the average and standard deviation of two experiments and summarize the percent of the total cells that stained for altered membrane integrity. The data sets for RS cells at 2 and 5 dpi represent a single experiment. A Tukey-Kramer multiple comparisons post-ANOVA test demonstrated that the differences between MDBK cells infected with the bICP0 null mutant at 16 h after infection, and bICP0-rescued virus infected cells at 24 and 48 h after infection were significantly different compared to mock infected cells at the same time points (P < 0.01, 0.02, and 0.001, respectively). Although bICP0-rescued virus infected MDBK cells at 16 h after infection appeared to have a higher percentage of cells with compromised membrane integrity compared to mock infected cells, there was not a significant difference (P > 0.05). Rabbit skin cells infected with the bICP0 null mutant and the bICP0-rescued virus at 4 dpi were significantly different compared to mock infected cells on the same day (P < 0.01, and 0.002, respectively). Panel A: Determination of DNA content. Panel B: Determination of membrane permeability.
We also compared membrane integrity in cells infected with the bICP0 null mutant versus the bICP0-rescued virus. One method to distinguish cells that are undergoing apoptosis or necrosis is to evaluate cell membrane integrity following infection. If the cell membrane is intact, propidium iodide does not enter the cell. At various times after infection (16, 24, or 48 h after infection for MDBK cells or 2, 4, or 5 d after infection for RS cells), cells were collected, and a portion of the cell population was stained briefly with propidium iodide (PI). Flow cytometry analysis of PI staining intensity was then determined (Figure 3B). At 24 and 48 h after infection, a statistically significant difference in membrane integrity was observed when MDBK cells were infected with the bICP0-rescued virus versus mock-infected cells or cells infected with the bICP0 null mutant. The number of cells with compromised membranes in MDBK cells was also significantly different when infected with the bICP0 null mutant for 16, but not 24 or 48 h after infection. When RS cells, were infected with the bICP0 null or the bICP0-rescued virus, statistically significant differences in membrane integrity were also detected at 4 dpi. As expected, the bICP0-rescued virus had a higher percentage of cells with compromised integrity compared to the bICP0 null mutant at 2, 4, or 5 dpi. Collectively, these studies confirmed that BHV-1 induced apoptosis in RS cells, and that expression of bICP0 had a profound effect on how cultured cells die after infection. These results also indicated that most of the infected MDBK cells underwent DNA damage that was not due to apoptosis, which suggested these cells died as a result of necrosis.
bICP0 expressing viral strains inhibit UV induced apoptosis in MDBK cells
HSV-1, another alpha-herpesvirinae sub-family member, can induce or inhibit apoptosis by cell-type-dependent mechanisms (Galvan and Roizman, 1998). When cells are treated with pro-apoptotic chemicals, HSV-1 efficiently inhibits apoptosis indicating that the viral genome contains several anti-apoptotic genes. To test whether BHV-1 can inhibit apoptotic induction by pro-apoptotic stimuli, MDBK cells were treated with UV light to initiate the apoptotic response, and then these cells were infected. Since the bICP0-rescued virus and LR mutant virus did not contain detectable levels of DNA laddering in MDBK cells, we compared the ability of these bICP0 expressing viruses to inhibit UV-induced DNA laddering relative to the bICP0 mutant. Cells were mock infected or pre-infected with the respective virus strain for 4 h prior to UV treatment, and then DNA laddering analyzed (Figure 4). Cells infected with the bICP0 rescued virus (lane RU) or LR mutant (lane LU) had reduced levels of UV-induced DNA laddering compared to mock infected cells that were UV-treated (U). In contrast, the bICP0 null mutant appeared to enhance UV induced apoptosis (lane NU). As expected, DNA laddering was not detected in mock infected MDBK cells not treated with UV light (M), or when infected with the bICP0 rescued virus (R) or the LR mutant virus (L). A smear of low molecular weight DNA was detected in cells infected with the bICP0 null mutant (N), which was seen in varying degrees in all of the studies. In summary, bICP0-expressing viruses inhibited UV induced apoptosis in MDBK cells or RS cells (data not shown).
Analysis of infected MDBK cells by electron microscopy
We next examined the cytopathic effects that BHV-1 has on MDBK cells following virus infection by transmission electron microscopy (TEM). MDBK cells were used for these studies because results derived from Figures 1-4 suggested that following infection with the bICP0 null mutant, a novel form of cell death occurred that was, in large part, non-apoptotic. Cells infected with the bICP0 null mutant had increased numbers of cytoplasmic vacuoles (autophagosomes) (Figure 5a, arrows) compared to mock-infected cells (Figure 5c), or following infection with bICP0 expressing strains (Figure 5b and 5d). bICP0 expressing virus strains (Figure 5b and 5d) also exhibited marginated chromatin, nuclear reorganization, and contained structures that resembled viral replication centers (Figure 5b and 5d, arrowheads). In contrast, these changes were not evident in mock-infected cells (Figure 3c) or in cells infected with the bICP0 null mutant (Figure 5a).
Figure 5. MDBK cells infected with the bICP0 null mutant exhibit increased the numbers of cytoplasmic vacuoles.
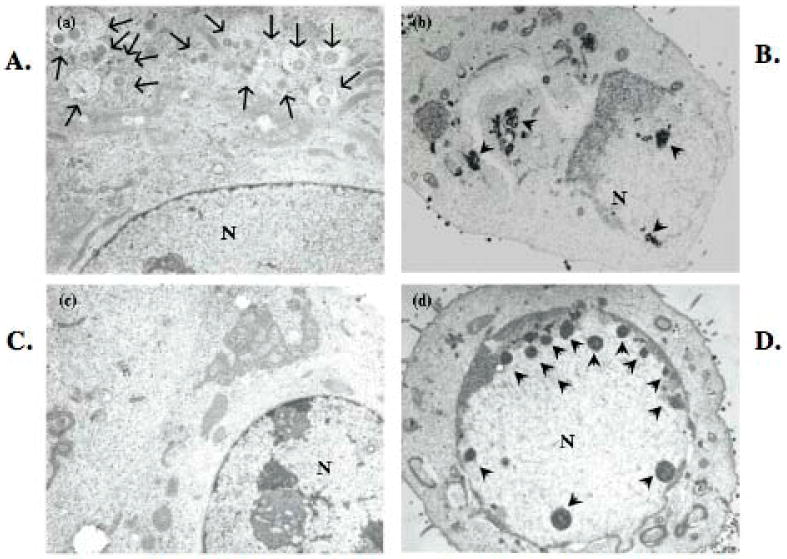
MDBK cells were infected with the bICP0 null mutant (21,000 X magnification) (a) or wt BHV-1 (18,000 X magnification) (b), were mock-infected (18,000 X magnification) (c) or were infected with the bICP0-rescued virus (15,000 X magnification) (d). Virus infection was performed with MOI=1. After incubation for 1 h at 37°C, monolayers were rinsed twice with CMF-PBS, fresh media was added to each plate and incubated for 24 h at 37°C. Cells were fixed with 2.5% glutaraldehyde for 2 hr and ultra-thin sections prepared. Sections were fixed in 1% osmium tetroxide (OsO4) for 1 h, dehydrated in graded ethanol, and embedded in Epon 812 mixture. The ultrathin sections of cells were stained with 2% uranyl acetate and examined under a Hitachi H7500 transmission electron microscope (TEM). Arrows denote autophagosomes present in the bICP0 null mutant infected cells. Arrowheads denote viral replication centers in wt or bICP0-rescued virus infected cells. The nuclei are denoted by the N.
DISCUSSION & CONCLUSIONS
These studies provided evidence that BHV-1 induced high levels of apoptosis in RS, but not MDBK, cells suggesting cell type specific factors mediate viral induced apoptosis. Furthermore, BHV-1 strains that expressed bICP0 inhibited UV induced apoptosis in RS or MDBK cells. HSV-1, another alpha-herpesvirinae family member, also induces or inhibits apoptosis by cell-type-dependent mechanisms (Galvan and Roizman, 1998), in part, because it encodes several anti-apoptotic factors (Blaho and Aubert, 2001). Interestingly, these studies have also demonstrated that HSV-1 can inhibit apoptosis in cells that were previously treated with chemicals to induce apoptosis, which confirms that HSV-1 encodes several anti-apoptotic genes. These demonstrate that BHV-1 also has the potential to induce or inhibit apoptosis, and this occurs in a cell type dependent fashion. The ability of BHV-1 to induce apoptosis (Lovato et al., 2003; Perez et al., 2005; Winkler et al., 1999) and non-apoptotic forms of programmed cell death in infected calves is likely to mediate certain aspects of pathogenesis.
Productive infection of RS, but not MDBK, cells led to a high frequency of apoptosis. We have observed apoptosis in various tissues of infected calves (Lovato et al., 2003; Perez et al., 2005; Winkler et al., 1999; Winkler et al., 2002); thus it was not surprising to find that BHV-1 induced high levels of apoptosis in RS cells. However, it was unexpected that productive infection did not lead to high levels of apoptosis in MDBK cells. Although one could argue that MDBK cells were not susceptible to apoptosis, this was not the case because MDBK cells undergo apoptosis following UV light treatment. BHV-1 encodes at least two genes with anti-apoptotic activity; the LR gene (Ciacci-Zanella et al., 1999; Henderson et al., 2004a; Lovato et al., 2003), and UL14 (Marfe et al., 2006). In the context of productively infected RS cells, the LR gene did not play a major role in inducing or inhibiting apoptosis, which is consistent with previous studies demonstrating it has no effect on the frequency of apoptosis in tonsils of infected calves (Perez et al., 2005). Consequently, these studies suggested that: 1) a viral gene induced apoptosis in RS, but not MDBK cells; 2) a viral encoded anti-apoptotic gene does not function in RS cells; and/or 3) cell type specific interactions occurred between viral and cellular proteins that regulated the mechanism of cell death.
In general, four types of cell death exist in eukaryotes: apoptosis, autophagy, oncosis, and pyroptosis (Fink and Cookson, 2005). Regardless of the signal that initiates programmed cell death, membrane disruption can lead to necrosis. FACS analysis demonstrated that membrane disruption occurred in RS or MDBK cells following infection with BHV-1, but apoptosis did not occur frequently in cultures of infected MDBK cells. Consequently, these studies suggested that necrosis was the predominant type of cell death in infected MDBK cells. In contrast, when MDBK cells were infected with the bICP0 null mutant virus many cells contained numerous autophagosomes suggesting these cells were undergoing autophagy. The hallmarks of authophagy include formation of cytoplasmic autophagosomes leading to degradation of cytoplasmic components in an intact cell, and slight chromatin condensation (Burch, 2001). Although autophagy appears to occur upstream or parallel to apoptosis (Akdemir et al., 2006), independent studies have also concluded that autophagy is crucial for cell survival in starving cells (Baehrecke, 2006). A previous study found that the bICP0 null mutant persists in infected MDBK cells, and that a subset of infected cells continued to grow and divide (Geiser et al., 2005). It is tempting to speculate that formation of autophagosomes in MDBK cells is due to autophagy, which promoted cell survival following infection with the bICP0 null mutant.
Expression of the bICP0 gene was important for cytotoxicity in RS or MDBK cells indicating that bICP0 expression directly or indirectly induced cell death (apoptosis or necrosis respectively). The bICP0 null mutant does not express detectable levels of bICPO protein, but does express detectable levels of RNA (Geiser et al., 2005) indicating that bICP0 protein expression was necessary for the effects observed in this study. In the absence of other viral genes, bICP0 protein expression indirectly activates caspase 3, thus increasing the frequency of apoptosis (Henderson et al., 2004b). bICP0 also activates viral gene expression and productive infection, which consequently promotes cell death. In contrast to these two observations, two bICP0 expressing strains (wt or the bICP0 rescued virus and the LR mutant virus) inhibited UV induced apoptosis in MDBK cells. The bICP0 null mutant had no effect on UV induced apoptosis. The most logical explanation for these observations is that during the course of productive infection bICP0 activated expression of viral encoded anti-apoptotic genes, which consequently interfered with ongoing apoptosis. At the same time, bICP0 promoted productive infection, which also led to cell death. It was also possible that when BHV-1 infected a cell undergoing apoptosis, the viral anti-apoptotic genes are expressed earlier and/or at higher levels. In contrast to bICP0, a recent study concluded that HSV-1 ICP0 RNA expression is an apoptotic stimulus following infection of human epithelial cells (Sanfilippo and Blaho, 2006). Studies designed to examine the mechanism by which bICP0 protein expression regulates cell death in certain cell types, but under certain conditions inhibited apoptosis, is in progress.
MATERIALS AND METHODS
Cells and Virus
Madin–Darby bovine kidney (MDBK) cells and rabbit skin cells (RS) were plated at a density of 5×105 cells per 100 mm2 plastic dish in Earle’s modified Eagle’s medium supplemented with 5% fetal bovine serum (FBS), penicillin (10 U/ml) and streptomycin (100 μg/ml). MDBK and RS cells were split in a 1:8 ratio every 3-4 days.
The Cooper strain of BHV-1 (wt virus) was obtained from the National Veterinary Services Laboratory, Animal and Plant Health Inspection Services, Ames, IA, USA. The bICP0 null mutant contains mutations at the 5’ end of bICP0, including the initiating methionine that prevents bICP0 protein expression (Geiser et al., 2005). The bICP0-rescued virus behaves like wt BHV-1.
The LR mutant virus was constructed by replacing wt (Cooper strain) LR gene sequences with an oligonucleotide that contain a unique EcoRI restriction site and three stop codons that is designed to block protein synthesis in each reading frame (Inman et al., 2001a). Viral stocks were prepared by infecting MDBK cells at an MOI of 0.01 from plaque-purified virus and subsequently titrating on MDBK cells.
The bICP0 null mutant contains mutations in the first in frame ATG of bICP0, a C→A substitution down stream of the first in frame ATG (Geiser et al. 2005). As a result of these mutations, bICP0 protein expression is not detectable in infected cells. DNA prepared from the bCIP0 null mutant virus and a plasmid that contains the bICP0 gene was cotransfected into bovine cells to generate the bICP0-rescued virus. The resulting bICP0-rescued virus produces wt plaques and has identical properties as wt BHV-1. For further details of the bICP0 null mutant virus and the bICP0-rescued virus, see Geiser et al. (2005).
Flow cytometry
Cell cycle analysis was performed according to standard techniques. MDBK or RS cells were infected with the designated virus. As negative controls, mock-infected cells were used. At the indicated times following infection, adherent and non-adherent cells were collected in a 15 ml conical tube. Cells were pelleted by centrifugation at 4° C, 300 × g for 5 min. The cell pellet was suspended in CMF-PBS at a density of 1×106 cells/ml. Propidium iodide (PI) viability staining was conducted on a fraction of cells as described previously (Sasaki et al., 1987). Immediately before flow-cytometry analysis, PI staining solution was added to the cell suspension to yield a final concentration of 50 ug/ml PI.
The remaining cell suspension was prepared for PI stained DNA content analysis. Cells were suspended at a concentration of 2×106 cells/ml in 70% ethanol, and incubated at -20° C for 15 min. The cell suspension was pelleted, washed in PBS, and suspended at 2×106 cells/ml in 1× PI working solution (40 μg/mL PI, 100 μg/mL ribonuclease A in CMF-PBS), incubated at 37° C in the dark for 30 minutes, and then incubated at 4° C in the dark for 12-16 h.
Flow cytometry analysis was performed for PI fluorescence (488-nm argon ion laser, 493-536 nm dye excitation, 617-639 nm dye emission; 585/42 nm filter) of individual cells with a flow cytometer (FACScan; Becton Dickinson Immunocytometry Systems, San Jose, CA). Cells that were not stained with PI were used to set gates, and set up photomultiplier tube voltages and amperage gain. Approximately 2 × 104 viable or cycling cell events were acquired at the low flow rate and analyzed using the Cell Quest program. To exclude cell debris, cells were gated using standard forward and side scatter properties.
Apoptotic DNA laddering assay
Fragmented DNA from virus infected and UV-treated cells was analyzed using a modified DNA laddering assay (Jin et al., 2004). For virus-induced apoptotic laddering, 1 to 2×106 cells was seeded into 6 well plates. For virus protection from UV-induced apoptosis, 3 × 105 MDBK cells were seeded into 6 well plates, and allowed to reach confluence by incubating for 3 days prior to infection. Cells were collected by scraping adherent cells into the spent media, and then centrifuged at 4° C, 2000 × g, for 10 min. Cells were suspended in 400 uL of hypotonic buffer and incubated at 20° C for 10 min. Samples were pelleted by centrifuging at 4° C, 10,000 × g for 10 min. to remove nuclei. The supernatant was mixed with 2 ml of Qiagen miniprep kit (#27106) PB buffer (#19066), and loaded onto a miniprep column (#1018398). Columns were washed with 750 ul of PE buffer. PB and PE buffers were supplied by Qiagen. Following an additional spin to remove residual wash buffer, 100 ul of 70°C TE was added to the column to elute DNA. The DNA solution was concentrated under vacuum to 15 ul, suspended in DNA loading buffer, and separated through a 2% agarose gel in 1 × TAE.
Electron microscopy
At 24 h after infection, cells were fixed with 2.5% glutaraldehyde for 2 h and ultra-thin sections prepared. Sections were fixed in 1% osmium tetroxide (OsO4) for 1 h, dehydrated in graded ethanol, and embedded in Epon 812 mixture. Sections were stained with 2% uranyl acetate and examined under a Hitachi H7500 transmission electron microscope (TEM).
Acknowledgments
This work was supported by two USDA grants (2005-01554, 2006-01627) and, in part, by a Public Health Service grants P20RR15635. VG was partially supported by a NIH Ruth L. Kirschstein fellowship (T32 AI060547).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akdemir F, Farkas R, Chen P, Juhasz G, Medved’ova L, Saas M, Wang L, Wang X, Chittaranjan S, Gorski SM, Rodriguez A, Abrams JM. Autophagy occurs upstream or parallel to the apoptosome during histolytic cell death. Development. 2006;133:1457–1465. doi: 10.1242/dev.02332. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Molec Cell Biol. 2006;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Blaho JA, Aubert M. Modulation of apoptosis during herpes simplex virus infection in human cells. Microbes and Infection. 2001;3:1–8. doi: 10.1016/s1286-4579(01)01444-7. [DOI] [PubMed] [Google Scholar]
- Burch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death and Differentiation. 2001;8:569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- Ciacci-Zanella J, Stone M, Henderson G, Jones C. The latency-related gene of bovine herpesvirus 1 inhibits programmed cell death. J Virol. 1999;73(12):9734–40. doi: 10.1128/jvi.73.12.9734-9740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devireddy LR, Jones CJ. Activation of caspases and p53 by bovine herpesvirus 1 infection results in programmed cell death and efficient virus release. J Virol. 1999;73(5):3778–88. doi: 10.1128/jvi.73.5.3778-3788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying cells. Infect Immun. 2005;73:197–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraefel C, Zeng J, Choffat Y, Engels M, Schwyzer M, Ackermann M. Identification and zinc dependence of the bovine herpesvirus 1 transactivator protein BICP0. J Virol. 1994;68(5):3154–62. doi: 10.1128/jvi.68.5.3154-3162.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs M, Hubert P, Detterer J, Rziha HJ. Detection of bovine herpesvirus type 1 in blood from naturally infected cattle by using a sensitive PCR that discriminates between wild-type virus and virus lacking glycoprotein E. J Clin Microbiol. 1999;37(8):2498–507. doi: 10.1128/jcm.37.8.2498-2507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Roizman B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell- type-dependent manner. Proc Natl Acad Sci U S A. 1998;95(7):3931–6. doi: 10.1073/pnas.95.7.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser V, Jones C. Stimulation of bovine herpesvirus 1 productive infection by the adneovirus E1A gene and a cell cycle regulatory gene, E2F-4. J Gen Virol. 2003;84:929–938. doi: 10.1099/vir.0.18915-0. [DOI] [PubMed] [Google Scholar]
- Geiser V, Inman M, Zhang Y, Jones C. The latency related (LR) gene of bovine herpes virus 1 (BHV-1) can inhibit the ability of bICP0 to activate productive infection. J Gen Virol. 2002;83:2965–2971. doi: 10.1099/0022-1317-83-12-2965. [DOI] [PubMed] [Google Scholar]
- Geiser V, Zhang Y, Jones C. Characterization of a BHV-1 strain that does not express the major regulatory protein, bICP0. J Gen Virol. 2005;86:1987–1996. doi: 10.1099/vir.0.80921-0. [DOI] [PubMed] [Google Scholar]
- Henderson G, Perng G-C, Nesburn A, Wechsler S, Jones C. The latency related gene of bovine herpesvirus 1 can suppress caspase 3 and caspase 9 during productive infection. J Neurovirol. 2004a;10:64–70. doi: 10.1080/13550280490261716. [DOI] [PubMed] [Google Scholar]
- Henderson G, Zhang Y, Inman M, Jones D, Jones C. The infected cell protein 0 encoded by bovine herpes virus 1 (bICP0) can activate caspase 3 when over-expressed in transfected cells. J Gen Virol. 2004b;85:3511–3516. doi: 10.1099/vir.0.80371-0. [DOI] [PubMed] [Google Scholar]
- Inman M, Lovato L, Doster A, Jones C. A mutation in the latency-related gene of bovine herpesvirus 1 leads to impaired ocular shedding in acutely infected calves. J Virol. 2001a;75:8507–8515. doi: 10.1128/JVI.75.18.8507-8515.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman M, Zhang Y, Geiser V, Jones C. The zinc ring finger in the bICP0 protein encoded by bovine herpes virus-1 mediates toxicity and activates productive infection. J Gen Virol. 2001b;82:483–492. doi: 10.1099/0022-1317-82-3-483. [DOI] [PubMed] [Google Scholar]
- Jin L, Perng G-C, Brick DJ, Naito J, Nesburn AB, Jones C, Wechsler SL. Methods for detecting the HSV-1 LAT anti-apoptosis activity in virus infected tissue culture cells. J Virol Meth. 2004;118:9–13. doi: 10.1016/j.jviromet.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Jones C. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv Virus Res. 1998;51:81–133. doi: 10.1016/s0065-3527(08)60784-8. [DOI] [PubMed] [Google Scholar]
- Jones C. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin Micro Rev. 2003;16:79–95. doi: 10.1128/CMR.16.1.79-95.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovato L, Inman M, Henderson G, Doster A, Jones C. Infection of cattle with a bovine herpesvirus 1 (BHV-1) strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J Virol. 2003;77:4848–4857. doi: 10.1128/JVI.77.8.4848-4857.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfe G, De Martino L, Filomeni G, Di Stefano C, Giganti MG, Pagnini U, Napoletano F, Iovane G, Circolo MR, Salimei Sinibaldi P. Degenerate PCR method for identification of an antiapoptotic gene in BHV-1. J Cell Biochem. 2006;97:813–823. doi: 10.1002/jcb.20636. [DOI] [PubMed] [Google Scholar]
- Mweene AS, Okazaki K, Kida H. Detection of viral genome in non-neural tissues of cattle experimentally infected with bovine herpesvirus 1. Jpn J Vet Res. 1996;44(3):165–74. [PubMed] [Google Scholar]
- Perez S, Inman M, Doster A, Jones C. Latency-realated gene encoded by bovine herpesvirus 1 promotes virus growth and reactivation from latency in tonsils of infected calves. J Clin Micro. 2005;43:393–401. doi: 10.1128/JCM.43.1.393-401.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanfilippo CM, Blaho JA. ICP0 gene expression is a herpes simplex virus type 1 apoptotic trigger. J Virol. 2006;80:6810–6821. doi: 10.1128/JVI.00334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki DT, Dumas SE, Engelman EG. Discrimination of viable and non-viable cells using propidium iodide in two color immunofluorescence. Cytometry. 1987;8:413–420. doi: 10.1002/cyto.990080411. [DOI] [PubMed] [Google Scholar]
- Winkler MT, Doster A, Jones C. Bovine herpesvirus 1 can infect CD4(+) T lymphocytes and induce programmed cell death during acute infection of cattle. J Virol. 1999;73(10):8657–68. doi: 10.1128/jvi.73.10.8657-8668.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler MT, Doster A, Sur JH, Jones C. Analysis of bovine trigeminal ganglia following infection with bovine herpesvirus 1. Vet Microbiol. 2002;86(12):139–55. doi: 10.1016/s0378-1135(01)00498-9. [DOI] [PubMed] [Google Scholar]
- Winkler MTC, Doster A, Jones C. Persistence and reactivation of bovine herpesvirus 1 in the tonsil of latently infected calves. J Virol. 2000;74:5337–5346. doi: 10.1128/jvi.74.11.5337-5346.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth UV, Fraefel C, Vogt B, Vlcek C, Paces V, Schwyzer M. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3’ coterminal and encode a putative zinc finger transactivator protein. J Virol. 1992;66(5):2763–72. doi: 10.1128/jvi.66.5.2763-2772.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jones C. The bovine herpesvirus 1 immediate-early protein (bICP0) associates with histone deacetylase 1 to activate transcription. J Virol. 2001;75(20):9571–9578. doi: 10.1128/JVI.75.20.9571-9578.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiang Y, Zhou J, Jones C. The bovine herpes virus 1 (BHV-1) immediate early protein (bICP0) interacts with the histone acetyltransferase p300, and these interactions correlate with stimulation of gC promoter activity. J Gen Virol. 2006;87:1843–1851. doi: 10.1099/vir.0.81766-0. [DOI] [PubMed] [Google Scholar]