

Abstract
The emergence of electron tomography as a tool for three dimensional structure determination of cells and tissues has brought its own challenges for the preparation of thick sections. High pressure freezing in combination with freeze substitution provides the best method for obtaining the largest volume of well-preserved tissue. However, for deeply embedded, heterogeneous, labile tissues needing careful dissection, such as brain, the damage due to anoxia and excision before cryofixation is significant. We previously demonstrated that chemical fixation prior to high pressure freezing preserves fragile tissues and produces superior tomographic reconstructions compared to equivalent tissue preserved by chemical fixation alone. Here, we provide further characterization of the technique, comparing the ultrastructure of Flock House Virus infected DL1 insect cells that were 1) high pressure frozen without fixation, 2) high pressure frozen following fixation, and 3) conventionally prepared with aldehyde fixatives. Aldehyde fixation prior to freezing produces ultrastructural preservation superior to that obtained through chemical fixation alone that is close to that obtained when cells are fast frozen without fixation. We demonstrate using a variety of nervous system tissues, including neurons that were injected with a fluorescent dye and then photooxidized, that this technique provides excellent preservation compared to chemical fixation alone and can be extended to selectively stained material where cryofixation is impractical.
Keywords: ultrastructural preservation, macromolecular structure, electron tomography, neuroanatomy, cryofixation, synapses, virus entry
1. Introduction
High pressure freezing (HPF) is a method for freezing isolated organelles, cells and tissues up to ~200–500 µm in depth without significant ice crystal damage (Dahl and Staehelin, 1989; McDonald, 1999; Moor, 1987; Shimoni and Muller, 1998). When cells are rapidly frozen, all contents are immobilized almost immediately. These fast-freezing methods involve time scales of milliseconds and are preferable to chemical fixation methods that have time scales of seconds or minutes depending on the tissue. Two other fast freezing methods, plunge-impact freezing and propane jet freezing, freeze specimens to only ~2–5 µm and ~20–50 µm in depth, respectively. This limitation is due to the poor heat conductance of water that limits the effective thickness of freezing in spite of dramatically increasing the freezing rate. While cryo-protectants can increase the thickness of specimen preservation, these agents often give rise to their own artifacts. HPF overcomes one of the significant limitations of freezing methods, namely, the small sample of tissue that can typically be preserved. High pressures prevent the expansion of water, lower the freezing point, increase the freezing rate and reduce the crystallization rate of ice. Because the freezing is not limited by the conduction of heat from the sample by a cold source, the overall effect results in obtaining a vitrified water state rather than a damaging crystalline ice environment over a much larger area, compared to propane jet freezing or plunge-impact freezing.
With HPF, one can obtain specimens in which the preservation is optimal deep into the tissue and not restricted to surface layers or small isolated cells of limited surface-to-volume ratios. HPF freezing when used in combination with freeze-substitution methods delivers plastic embedded material that can then be examined in conventional electron microscopes without the inherent technical challenges or dose-sensitivities associated with EM analysis of vitrified specimens. HPF cryo-fixation is particularly advantageous for electron tomographic imaging where sections of 0.25–1 µm thicknesses are routinely used for creating reconstructions from tissues or cells whose diameter exceeds 2–5 µm, the limit of penetrating power obtainable with 300–400 keV electron microscopes (McDonald and Auer, 2006). However, many published studies using high pressure freezing use single cell organisms, easily accessible plant tissues, tissue culture cells or accessible mammalian tissues such as skin. The technique is less successful for large, internal tissue masses such as brain or nerve tissue that require careful dissection prior to processing. Because there is a delay before fixation commences, tissues such as brain that are acutely sensitive to anoxia require a tradeoff between artifacts that may be induced due to aldehyde fixation and the tissue and cell damage occurring upon anoxia and during dissection and removal of the tissue.
Chemical fixation has been a mainstay of electron microscopy for decades. Chemical fixation is essential in order to stabilize tissue structure against damage during dissection, sectioning, staining, photooxidation, processing, and embedding. Numerous studies over the years have documented that chemical fixation does an outstanding job of preserving molecular arrangements and tissue ultrastructure (see Hayat, 1982; Peters et al., 1991). However, chemical fixatives are known to be relatively poor at preserving, or preventing the extraction by later processing steps of some classes of molecules, e.g. sugars and lipids, and thus these procedures may change the dimensions or arrangements of certain cellular components such as the dimensions of the extracellular space (Chan et al., 1992; Chan et al., 1993; Zechmann et al., 2007). Validation of chemical fixatives for well-studied tissues occurred over many painstaking investigations comparing the ultrastructure of cells and tissues using different preservation protocols.
Previously, we developed a protocol that combined glutaraldehyde fixation with HPF and freeze substitution cocktails that both optimally preserves and stains the structures in the tissue. We examined glutaraldehyde-fixed and high pressure frozen-peripheral nerves and performed a serial section electron tomographic reconstruction of the Node of Ranvier complex (Sosinsky et al., 2005). The aldehyde fixation was necessary in order to minimize structural damage during the time the spinal root nerves were surgically removed from the mice and transferred to the closest HPF machine, which at that time was at another institution ~480 miles away. Even with chemical fixation prior to high pressure freezing and overnight shipping of the sample, we found that the structural preservation was vastly improved compared to chemical fixation alone. We hypothesized that the improved preservation was the result of minimizing extraction and distortions induced during osmification and dehydration of the tissue.
Here we provide further characterization of the hybrid fixation technique using a variety of samples and an improved protocol from our previous publication (Sosinsky et al., 2005) that provides greatly improved results for preservation of brain tissue. As a control for assessing the quality of combining fixation with high pressure freezing, we used late-stage Flock House Virus (FHV) infected DL1 insect cells, which can be easily fast frozen without fixation and thus used as a basis for comparison of different fixation methods. We found that the quality of aldehyde fixed/HPF is close to that of HPF without fixation and is superior to conventional glutaraldehyde fixation methods. In addition, we extend this technique to photoconverted material where filled hippocampal and cerebellar neurons have been selectively stained in fixed brain slices, showing that preservation is improved even in samples that are processed for selective staining prior to freezing.
2. MATERIALS AND METHODS
2.1. Tissue preparation and HPF of FHV infected DL1 cells
2.1.1. Preparation of cells
Descriptions of media and DL1 cell maintenance follow published procedures (Friesen and Rueckert, 1981). Drosophila DL1 cells in suspension media were infected with FHV at a multiplicity of infection of 5 for about 20 hours. Cells were pelleted at a speed of 500 relative centrifugal force (rcf) for 5 min.
Cell pellets were prepared three ways: (1) conventionally prepared (CAF), (2) fixed and then HPF (CAF-HPF) and (3) high pressure frozen immediately after removing from the centrifuge tube (HPF). (1) CAF: Cell pellets were conventionally prepared for electron microscopy by incubation in 2% glutaraldehyde in 100 mM cacodylate buffer initially at room temperature (~5 min) and then on ice for 30 minutes, followed by 1% osmium tetroxide in double distilled water for 1 hour and 2% UA in water overnight.
(2) CAF-HPF: The media was removed and saved. Then, cell pellets were fixed in 2% glutaraldehyde in 100 mM cacodylate buffer for 30 minutes on ice. The fixed pellet was resuspended in media and centrifuged again. Cells were loaded into the 100 µm well of a type A brass planchette (Ted Pella, Inc. Redding, CA) and fast frozen in the Bal-Tec HPM010 (Bal-Tec, Liechtenstein).
(3) HPF: Cell pellets were directly placed into brass planchettes that then were loaded in to the HPM 010 high pressure freezer and fast frozen.
2.1.2. Freeze substitution
After freezing, samples (2) and (3) were placed into a Leica EM AFS Freeze substitution (FS) machine (Leica Microsystems, Bannockburn, IL) and incubated at −90°C for 24 hours in 0.1% tannic acid in acetone. Samples were washed three times with cold acetone (cooled to −90°C) over 5 minutes, and placed in 1% OsO4 and 0.1% UA in cold acetone for 72 hours and held at −90°C. After slowly warming to room temperature at 5°C per hour, the specimens were rinsed in pure acetone three times (10 min. at room temperature). Infiltration and embedding in Durcupan resin was subsequently performed at room temperature.
2.2. Tissue preparation and HPF of nervous tissue
2.2.1. Fixation of brain and nerve tissue
A 21 day old rat was perfused with a solution of 2% of paraformaldehyde/2.5% of glutaraldehyde in 0.15 M cacodylate buffer according to the protocol described in (Giepmans et al., 2005). Brain and spinal roots were taken out and post-fixed in same fixative for 2 hrs at 4°C. For brain tissue, 100µm thick sections are kept in the fixative solution until HPF.
2.2.2. Fixation and photoconversion of Lucifer Yellow injected Purkinje neurons
This procedure follows that described in Bushong et al. (2002). A 21 day old rat was perfused with 4% paraformaformaldehyde/0.1% glutaraldehyde in PBS. The initial fixation is weaker than described above because stronger fixation makes it difficult to impale cells for dye injection and compromises membrane integrity (Belichenko and Dahlstrom, 1995; Buhl, 1993). Sections of the cerebellum and hippocampus of 100 µm in thickness were obtained by dissection and Vibratome microtomy (Vibratome, St. Louis, MO). Neurons were injected with 5% aqueous dilithium Lucifer yellow CH (LY) (Calbiochem, La Jolla, CA) and then post-fixed in 2% glutaraldehyde-PBS for 30 min at 4°C. A neuron was impaled and the dye was injected into the cell by applying a 0.5 sec negative current pulse (1 Hz) until the cell was completely filled. Several neurons were injected in each slice. These dye-injected slices were then photooxidized according to procedures described in (Deerinck et al., 1994) and recently updated in (Gaietta et al., 2006).
2.2.3. HPF of fixed brain tissue
Brain sections were cut with 1.8 mm tissue puncher. This step ensures that the proper size of brain tissue will fit into the shallow side of a 100 µm-deep well in the type A HPF brass planchette. For peripheral nerve tissue, the nerves were carefully trimmed to a proper length in order to fit into the type A freezing hats. Trimmed brain or nerve tissue was loaded into the planchettes and the well was filled with 1-hexadecene. The planchette was then covered with the flat side of brass type B planchette, quickly loaded into a freezing holder and frozen with the Bal-Tec HPM 010.
2.2.4. Freeze-substitution procedure
After freezing, the planchette sandwiches were separated under liquid nitrogen and the specimen/type A hats were placed into cryo-vials and stored under liquid nitrogen or subsequently placed into the freeze substitution device. The first substitution media was a solution of filled with freshly made 0.1% tannic acid (EM grade, from Polysciences Inc., Warrington, Pennsylvania) in acetone (EM grade from Fullam Inc., Latham, New York). After 24 hours, samples were then washed three times in cold acetone over a 2 hour period. The solution was changed to 2% osmium tetroxide in acetone for 48 hrs. The temperature was slowly raised to 20 °C. The specimens were rinsed three times at room temperature for 10 minutes in acetone. Tissues were removed from the planchettes after the last wash step. The total time for this procedure is 113 hrs.
2.2.5. Infiltration and embedding
Infiltration was conducted over 3 days followed by embedding in Durcupan ACM resin (Electron Microscopy Science Inc., Hatfield, PA). Samples were infiltrated in 30% Durcupan in acetone for 4 hours and 50% Durcupan overnight. The next day, the specimens were placed into 70% Durcupan for 4 hours, 90% over 2 hours and were placed in 100% Durcupan for overnight incubation. After two incubations in fresh 100% Durcupan, the sample was then polymerized at 60°C for 2 days.
2.2.6. Preparation of conventionally fixed and embedded samples
Vibratome sections cut at 100 µm and spinal roots were incubated in 1% osmium tetroxide in 0.15M Cacodylate buffer for 1hr on ice. After washing in the same buffer, tissue was dehydrated in a graded series of ethanol, followed by embedding in Durcupan and polymerization following our usual procedures (as in Giepmans, 2004).
2.3. Electron microscopy and tomographic reconstruction
2.3.1. Sectioning for electron microscopy
Thin (80–100 nm) sections and thick (0.5–2 µm) sections were cut using a diamond knife (Diatome) and an Ultracut E ultramicrotome (Leica Microsystems, Bannockburn, IL) and mounted on uncoated copper grids. Thin sections were imaged at 80 keV using a JEOL 1200 electron microscope (JEOL, Peabody, MA) and thick sections were imaged at 300–400 KeV using a JEOL 4000EX intermediate voltage electron microscope.
2.3.2. Electron tomography data acquisition
Gold beads were applied to the top and bottom of section prior to image acquisition. For tilt series recorded at 30,000x magnification or higher, both 5 and 10 nm gold beads were used. The 5 nm gold was applied to the bottom of the section while top was coated with 10 nm beads. Stereo pairs were collected prior to tomographic data collection to check that the tissue did not have any freezing damage. Images were collected at 400 keV using a JEOL 4000EX electron microscope at 2° increments from −60 to +60° for both single and double tilt tomographic reconstruction following standard protocols (Mastronarde, 1997).
2.3.3. Electron tomographic reconstruction
Tilt series were aligned using IMOD (Kremer et al., 1996) for fiducial alignment and data manipulations prior to back projection. Back projection was performed using the TxBr package (Lawrence et al., 2006). Surface renderings were visualized in Amira (Mercury/TGS, San Diego, CA).
2.2.4. Data deposition
Tomographic data and reconstructions as well as animations of the volumes are available upon publication in the Cell Centered Database (CCDB) (Martone et al., 2002; Martone et al., 2003). The accession numbers for these data sets are: 3649 (tomogram displayed in Fig. 4A–B) and 3684 (tomogram displayed in Fig. 4C–F). Fig. 1, Fig. 2, Fig. 3 and Fig. 5 are available in the CCDB as survey sections at high resolution quality enabling panning and zooming viewing.
Figure 1. Comparison of preparation methods for preserving ultrastructure in FHV infected DL1 cells.
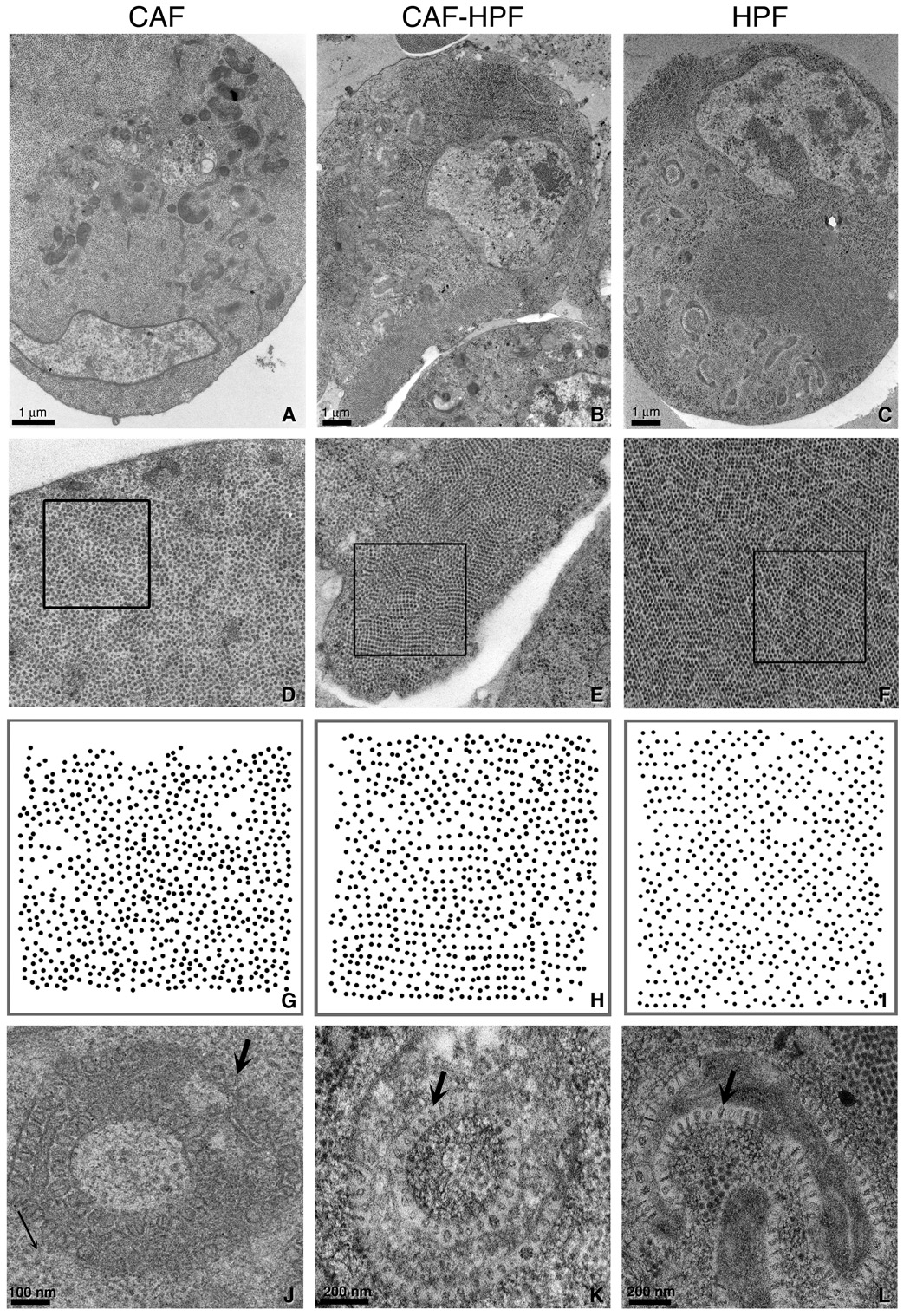
Late stage FHV infected DL1 cells prepared by conventional aldehyde fixation and embedding methods (CAF) (A, D, G, J), fixed and HPF (CAF-HPF) (B, E, H, K) and HPF alone (C, F, I, L) were examined by thin section EM. A higher magnification view of the aggregates of FHV in each DL1 cell in A (3x), B (3.5x) and C (3.5x) are shown in D, E and F respectively. The diameter of an individual FHV is 30 nm. A plot of the positions of the viruses centers in the boxed areas in D, E, and F are displayed in G, H and I, respectively and illustrating a more crystalline arrangement in H and I as compared with G. (J–L) Morphology of an appropriated mitochondrion in each preparation method. An arrow in each micrograph points to a spherule. Note how similar the mitochondrial morphologies are in the insets in K and L as compared to that in J, which appears empty and swollen.
Figure 2. Comparison of conventionally aldehyde fixed versus conventionally aldehyde fixed and high pressure frozen brain tissue.
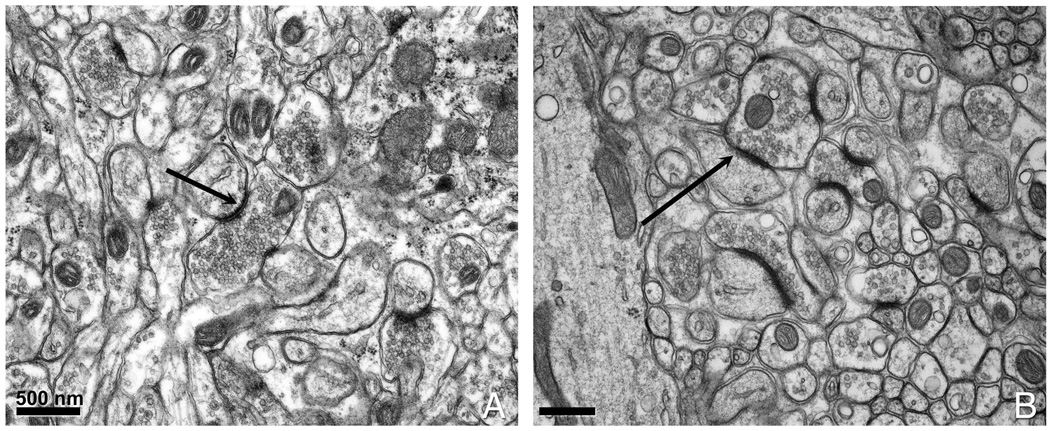
(A) Conventionally prepared cerebellar tissue. (B) Cerebellum slices that had been chemically fixed prior to HPF. The black arrows point to a postsynaptic density (PSD) that appears thicker in (B) than in (A).
Figure 3. Examples of nervous tissue prepared by a combined chemical fixation and HPF.
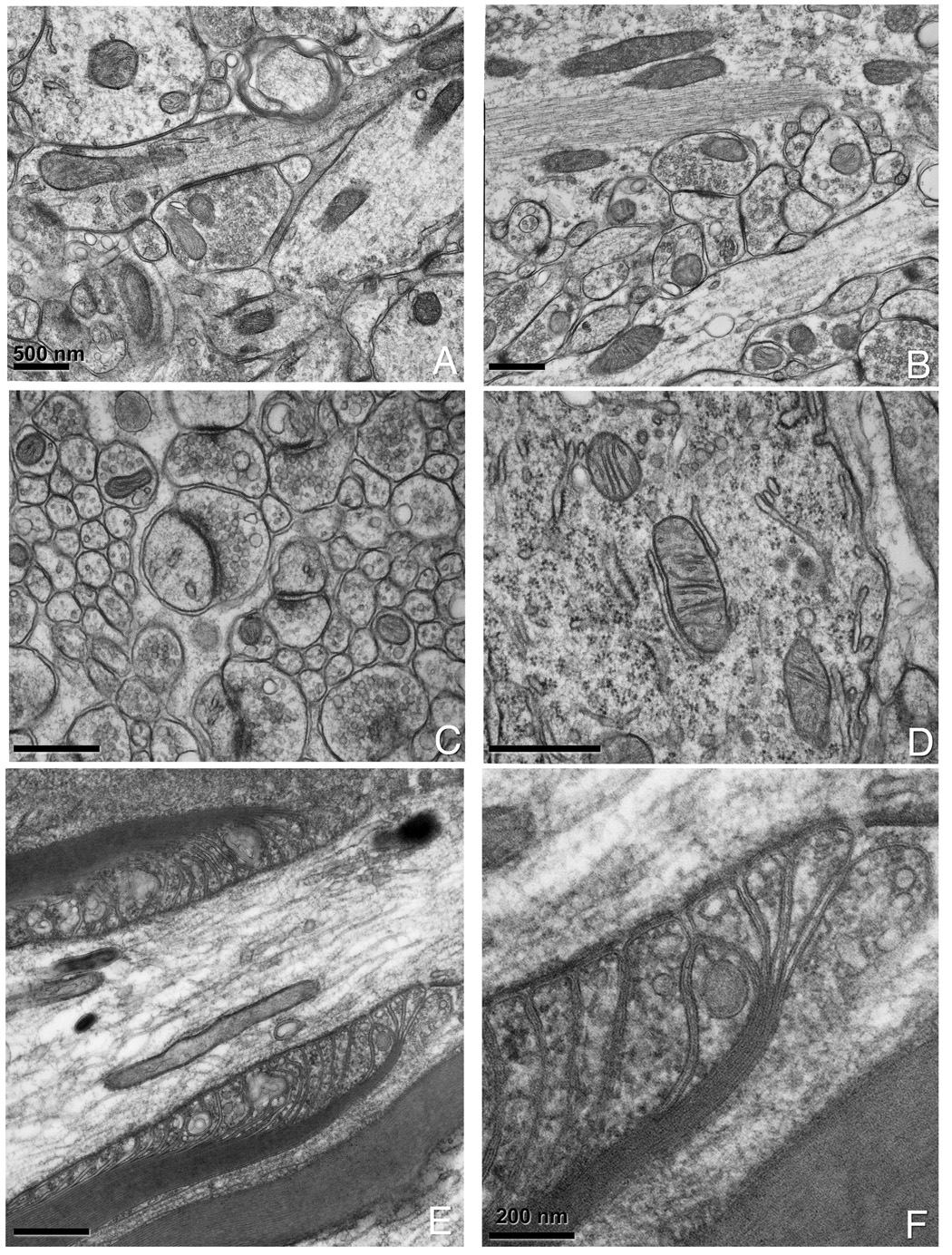
(A, B) Hippocampal slices that have been chemically fixed prior to HPF. (C) Cerebellar slices showing good preservation of synaptic vesicles and PSD and (D) mitochondria with attached smooth and rough endoplasmic reticulum. Note the ribosomes in the background of (D). (E) Spinal root prepared by combined chemical fixation and HPF. Note the smoothness of the membranes and the level of detail in the paranodal loops and axonal-glial junctions. A 3x magnification is shown in (F).
Figure 5. Thin section EM images of chemically fixed neurons that have been filled with Lucifer Yellow, photooxidized and HPF.
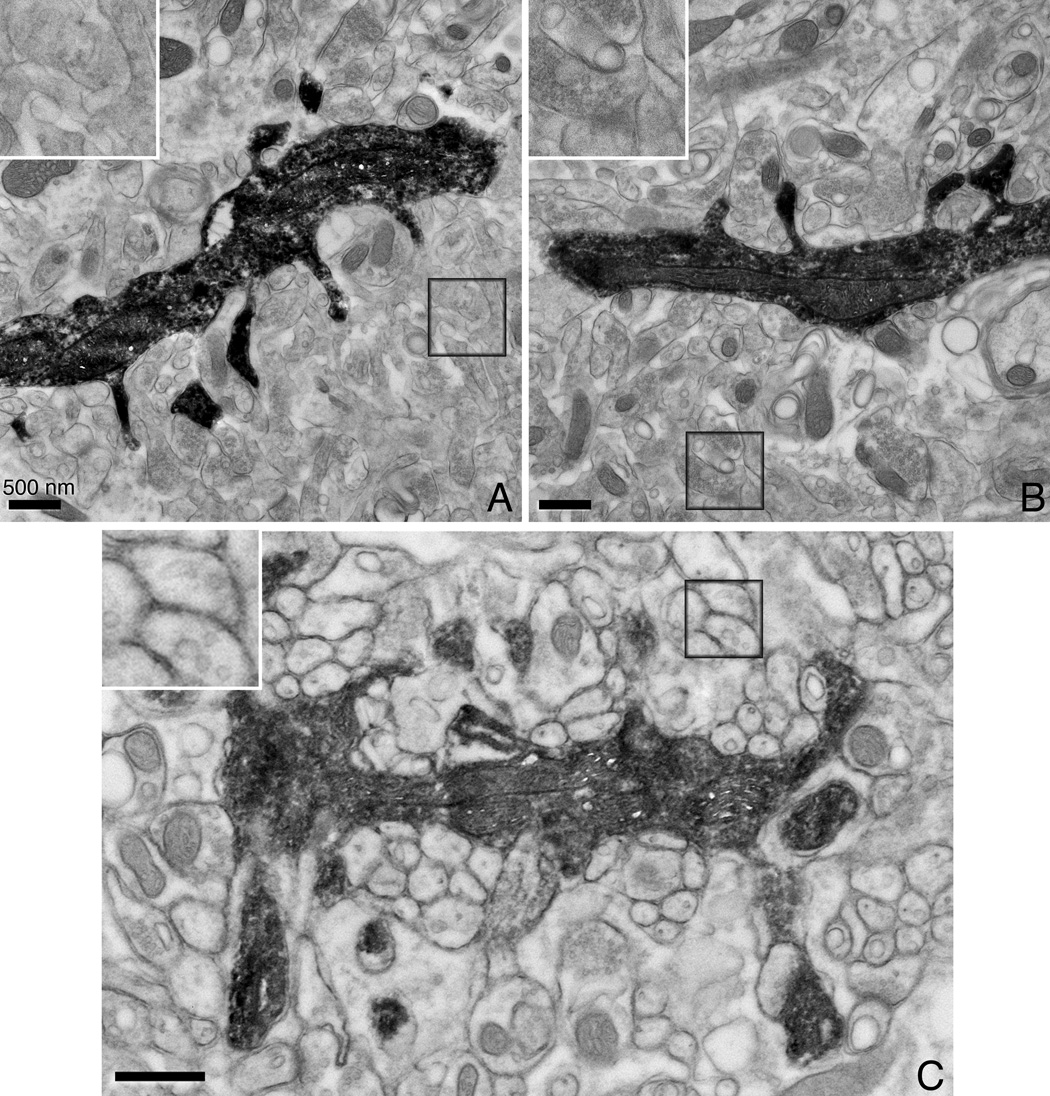
Conventionally prepared (A) and aldehyde fixed/HPF (B) hippocampal neurons. The Lucifer Yellow-DAB-osmium precipitate provides selective staining of the filled neuron with its dendrites. Note that while the ultrastructure of (A) and (B) are comparable, the delineation and preservation of the cell membranes is better in (B). (C) A photoconverted cerebellar Purkinje neuron also shows good ultrastructural preservation with chemical fixation/HPF. All scale bars correspond to 500 nm. The inset shown in each image is the boxed area shown at twice the magnification.
3. RESULTS
While HPF has been shown to be effective in cryo-fixation of samples and there is a great body of literature on aldehyde fixation techniques, combination approaches of HPF and aldehyde fixation have not been tested as rigorously with the exception of (Murk et al., 2003) and (Potrebic et al., 2003). Here, we used both a test specimen and nervous tissue to explore how glutaraldehyde fixation can be used prior to cryo-fixation with HPF.
3.1. Comparison of cryo-fixed and glutaraldehyde-fixed HPF specimens
FHV late stage infected DL1 cells represents an excellent test specimen to see if ultrastructure can be preserved with glutaraldehyde fixation as well as with cryo-fixation (“freezing from life”). In late stages of infection where the FHV has succeeded in filling the cell cytoplasm with viruses prior to release, viruses often aggregate and fill cytoplasmic areas. Whether these viruses pack into microcrystals is dependent on the specimen preparation technique used for EM (Lanman et al., 2007, submitted this issue). In addition, FHV replication takes over the cellular machinery, in particular the mitochondria and re-fashion it into a distinct morphology (Miller et al., 2001). Therefore, late stage FHV infected DL1 cells represent a good test specimen for our hybrid aldehyde fixation/HPF methods.
We prepared three specimens for this comparison. The first was conventionally prepared with aldehyde fixation (CAF: conventional aldehyde fixation) followed by osmium and uranyl staining at 4°C and dehydration in ethanol and embedding in Durcupan resin. The second and third specimens were done in parallel and only differed in that one sample was fixed with 2% glutaraldehyde prior to HPF (CAF-HPF) while the other was frozen directly (HPF). Both samples were freeze substituted. There were three hallmarks that indicated optimal preservation: paracrystalline virus arrays, preservation of uniformity in the “lollipop” structures (“spherules”) in the co-opted mitochondria and in viruses attached to the plasma membrane. Our data in Fig. 1 indicates that cryo-fixation is best, but that CAF-HPF does a very good job of preserving ultrastructural details (Fig. 1A–C), such as the paracrystals (Fig. 1D–F and Fig. 1G–I) and the unique architecture of the mitochondria (Fig. 1J–L). In CAF samples, the viruses appear to pack in close packed aggregates, however, in HPF and CAF-HPF specimens, we find that these arrays are more paracrystalline, often appearing in rows. Spherules in these optimally preserved cells also appear uniform and filled as opposed to those seen in the conventionally prepared material, which also appear larger and more swollen. In addition, viruses can be seen attached to the plasma membrane in HPF or CAF-HPF samples, but not in CAF prepared material (Lanman et al., submitted this issue) and the plasma membrane often appears ruffled in CAF samples which are usually associated with as a fixation artifact. This result implies that the majority of the degradation in ultrastructure occurs in the subsequent steps of washing and preparation of tissue for embedding rather than from fixation alone as originally suggested by Small (1981).
3.2. Protocols for optimally preserving nervous tissue
The protocol presented in the Materials and Methods section represents a refinement of initial protocols from that first presented in Sosinsky et al., (2005). It should be pointed out that there is no one HPF/FS protocol for every tissue and HPF/FS protocols must be tailored to the individual tissue in a trial and error manner. For nervous tissue, animals were perfused with 2% paraformaldehyde and 2.5% glutaraldehyde in order to maintain the tissue from anoxic deterioration. The tissue was then dissected and put into hexadecene and fast frozen in the HPF. We initially tried a combination of osmium tetroxide and uranyl acetate in pure, fresh acetone but we found precipitates within the sample due to the interaction of the uranyl acetate and the buffer used for preparation of the tissue. In addition, the contrast of structures within the tissue was sub-optimal. A freeze substitution cocktail containing 2% osmium only in acetone preserved the tissue but did not provide good enough contrast for detection of the cellular membranes in thin section electron micrographs (data not shown).
Our most successful protocol for preserving brain tissue (cerebellum, hippocampus, spinal roots) uses a freeze substitution cocktail containing 0.1% tannic acid in pure 100% acetone and incubating the tissue for 24 hours at −90°C, followed by an incubation in 2% osmium tetroxide in fresh 100% acetone for 48 hours at −90°C. The temperature was allowed to rise to 20°C at a rate of 5°C/hour. This slow warming is critical to producing specimens with little or no freezing or freeze substitution damage. The tissue is then processed for electron microscopy followed by slow infiltration and embedding, taking 3 days for infiltration of increasing concentrations of Durcupan resin prior to embedding in 100% Durcupan. This slow infiltration procedure is very important since it appears that embedding epoxy takes a much longer time to infiltrate into the biological specimen after high pressure freezing and the freeze substitution procedure.
3.3 Examples of well-preserved nervous tissue
We used three nervous system regions as test specimens for our fixation/HPF protocols: cerebellum, hippocampus and nodes of Ranvier from spinal roots. In particular, nodes of Ranvier from peripheral nerves (sciatic, saphenous and dorsal roots) are highly prone to structural damage due to freezing, mechanical instabilities, or errors that occur during the freeze substitution, more so than cerebellum or hippocampus. As shown in Fig. 2, we get improved structural preservation of cerebellar tissue with CAF-HPF (Fig. 2B) compared to CAF conventional preparation (Fig. 2A). Other benchmarks of improved specimen preparation are the uniformity and increased density in the cytoplasm and synaptic vesicles as well as smooth cell membranes. Examples of hippocampus (Fig. 3A–D) and spinal root nodes of Ranvier (Fig. 3E and F) show exquisite detail. In particular, macromolecular complexes such as the post-synaptic densities (PSDs) (Fig. 3C), ribosomes and mitochondria (Fig. 3D), paranodal loops (Fig. 3E, and at higher magnification in Fig. 3F) show complex morphologies. The membranes and axonal-glial junctions (AGJ) are extremely well delineated (Fig. 3F) and the compact myelin contains no bubbles or distortions.
3.4 EM tomography of well-preserved nervous tissue
The advantage of using HPF over other fast freezing techniques is that it can provide well-preserved thick sections for electron tomography (McDonald and Auer, 2006). Here we show two examples of tomograms from well-preserved cerebellum. Fig. 4A is a representative slice from a lower magnification tomogram showing the overall architecture of this cerebellar area. The white arrow points to an example of a PSD surrounded by synaptic vesicles. Mitochondria and synaptic vesicles are visible as well. This tomogram was volume rendered with a maximum intensity projection algorithm in the Amira package. In a second higher magnification tomogram from a different area, a synapse with a PSD and synaptic vesicles has been reconstructed. Fig. 4C and D are two slices from the volume and show the different appearance of the PSD “fuzz” on the post-synaptic side of the synapse. The PSD was threshold segmented using Amira and then, surface rendered. The 3D surface view is shown both superimposed on a volume slice (Fig. 4E) and alone (Fig. 4F). The arrow inFig. 4F points to hair-like projections that correspond to the fuzz seen in Fig. 4C.
Figure 4. Tomograms of CAF-HPF cerebellar tissue.
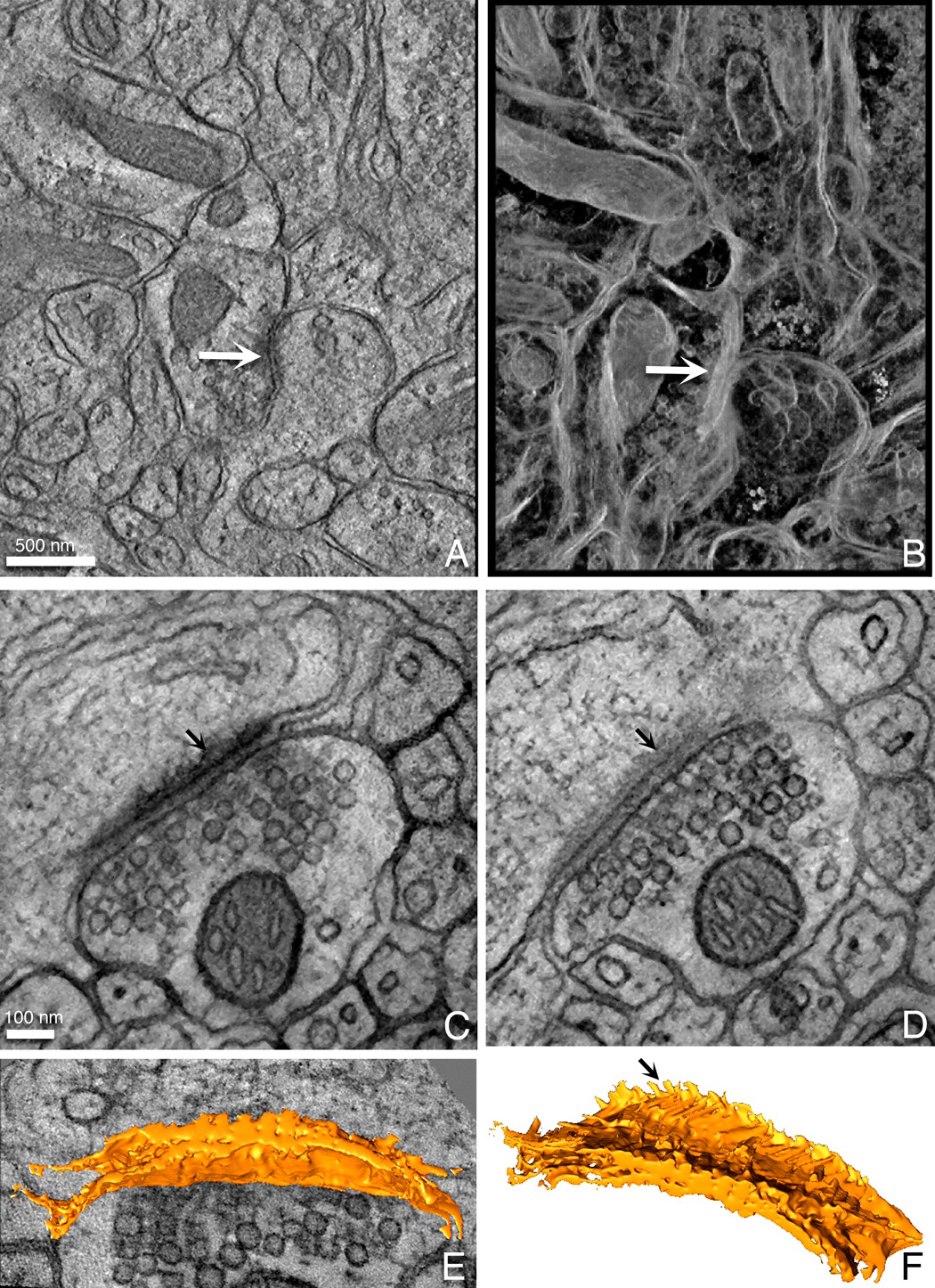
Examples of a slice (A) and a volume rendering (B) of a 0.5 µm volume of cerebellum. The yellow arrow points to a postsynaptic density (PSD) surrounded by synaptic vesicles. Slices from a higher magnification tomogram of a well-preserved PSD are shown in (C) and (D). Arrows point to the fuzzy coat of the PSD in these two slices. The PSD was semi-automatically segmented and is displayed as a surface rendering both upon a slice of the volume (E) as well as by itself (F). The arrow in (F) points to the hair-like projections that corresponds to the fuzzy coat in the slices. These datasets may be viewed in the Cell Centered Database on-line.
3.5 Extending these protocols to fluorescently labeled photoconverted samples
Fluorescence photo-oxidation is a powerful and versatile technique for LM and EM whereby the oxygen radicals generated by a fluorescent compound under illumination are used to drive the oxidation of diaminobenzidine. Oxidized DAB can be then be made electron dense through osmification. In principle, application of cryofixation methods to these labeled specimens would be highly advantageous in order to optimally preserved ultrastructure. In practice, however, cells or tissues being photo-converted cannot be fast frozen and then photo-oxidized because of two factors: first, DAB would be difficult to diffuse into the frozen tissue which would have to be maintained at low temperature to maintain the cryo-fixation and second, the ligand-fluorophore complexes need to be “locked into place” to maintain the resolution of staining. Nonetheless, we have demonstrated that aldehyde fixation used prior to rapid freezing provides very good ultrastructural preservation and we extend these protocols to photoconverted specimens.
As a proof of principle, brain slices containing either hippocampal neurons or cerebellar cortex Purkinje neurons were injected with Lucifer Yellow and then photoconverted. Purkinje neurons are some of the largest neurons in the mammalian brain, containing an elaborate dendritic arbor with a large number of dendritic spines. Slices of the hippocampus are often imaged because of its simpler and distinctive morphology. The Lucifer Yellow-DAB-osmium precipitate provides selective staining of the filled neuron with its all spiny dendrites (Fig. 5). A weaker paraformaldehyde/glutaraldehyde fixation is first performed because it is difficult to pierce a needle into strongly glutaraldehyde fixed cells. However, after dye injection a second glutaraldehyde fixation is performed prior to photoconversion. The improvement between CAF (Fig. 5A) and CAF-HPF (Fig. 5B) is not as dramatic as before, however, the preservation and delineation of the cell membranes is improved in the latter (see the 2x magnification insets of the boxed area in Fig. 5A and B). This is mostly likely due to the fact that these weaker fixed samples were not frozen directly after chemical fixation and some leaching or degradation may have occurred during the photoconversion step. Fig. 5C shows a filled and photoconverted cerebellar Purkinje neuron in which the surrounding tissue is nicely preserved. A higher magnification view of the boxed area is shown in the inset.
4. DISCUSSION
Many cells and tissues have been optimally preserved using high pressure freezing, however, in mammals, these specimens are usually limited to fairly homogeneous tissues that are easily extracted without anoxia being a significant factor. Examples of tissue that fall into this category that have been successfully cryo-fixed with HPF include rodent skin (Al-Amoudi et al., 2005; Reipert et al., 2004), cartilage (Al-Amoudi et al., 2005; Studer et al., 1995), cultured pancreatic islet cells (Marsh et al., 2004), kidney (Vanhecke et al., 2006), Xenopus laevis pituitary gland (Wang et al., 2004) and liver (Hsieh et al., 2006).
Brain tissue represents a challenging test specimen for ultrastructural preservation because the brain is a highly specialized organ with differing morphologies across its entirety. If fixatives are not used, in the time it takes to dissect and prepare tissue, tissue integrity is degraded due to anoxia and is nearly impossible to excise native brain tissue for HPF without creating structural damage during the excision. Well preserved neuronal structure has been obtained from organotypic hippocampal slice cultures that have been allowed to partially recover from excision (Frotscher et al., 2007), however, tissue cells in culture may not be the exactly same as in native tissue. Adding to the challenge of preserving brain tissue is its high water content (Schwab et al., 1997) and increased lability compared to other tissues such as heart, skin or pancreas. Epperlein et al., (2000) proposed that the high water content found in the subepidermal extracellular matrix of axolotl neural crest cells was the reason that preservation of hyaluronan networks and binding hyaluronan binding protein of was sub-optimal and problematic using HPF as compared to the sub-epidermal layer (Epperlein et al., 1997).
While aldehyde fixation has been shown to produce variable results even in conjunction with HPF (Murk et al., 2003), we demonstrate here with a test specimen of cells in suspension for which anoxia is not a consideration that the morphology with fixation followed by HPF is very similar to one that has been cryo-fixed with HPF alone. In a classic paper by Small (1981), actin networks in the leading edge of cultured fibroblasts were shown to be preserved with glutaraldehyde fixation, but were compromised or destroyed by the post-processing steps typically used for conventional electron microscopy. Other examples of hybrid fixation techniques use aldehyde fixation and cold-metal freezing for preserving anterior pituitary cells (Senda et al., 2005) or aldehyde fixation and plunge freezing for maintaining the structural integrity of reconstituted mitotic chromosomes (Konig et al., 2005). In contrast, Murk et al. (2003) demonstrated that systematic assessment by tomographic reconstruction of CAF samples versus HPF alone resulted in some shrinkage and deformation of early and late endosomes, but did not alter lysosomes even in CAF-HPF cells as compared with HPF cells. However, it should be noted that optimization of the freeze-substitution protocol is critical in obtaining superior specimens even with optimal freezing methods (Hawes et al., 2007) and that we have striven here not only to optimize the fixation methods but also the freeze substitution protocol. We show using two areas of brain, cerebellum and hippocampus, and peripheral nerves that CAF-HPF in combination with a tailored freeze substitution protocol provides very well preserved specimens for electron tomography.
In our studies, we focus on nervous tissue which does not fall into the category of being easily accessible, uniform and fairly stable. Early studies by Van Harreveld and co-workers (Vanharreveld and Crowell, 1964; Vanharreveld et al., 1965) using a cold metal mirror freezing device demonstrated that brain tissue needed to be fast frozen in 30 sec after excision from a mouse and before the onset of cardiac arrest (an ~8 minute times scale). After this time, morphological deterioration was observed and it was reported that the tissue resembled chemically fixed specimens. One of first descriptions of an HPF device was its application of cryofixation for freeze fracture electron microscopy of dissected rat cerebellar cortex and subfornical organs (Moor et al., 1980). Recent tomographic studies have shown the HPF of rat CA1 hippocampal slices produces 3D structures of synapses that were larger, had less densely packed synaptic vesicles and contained small filamentous linkers to groups of vesicles (Rostaing et al., 2006) with a network of synaptic vesicles linked together and forming a scaffold for the active zone (Siksou et al., 2007). In the latter case, the authors report that it took ~7 minutes from removal of the brain to HPF of dissected hippocampal slices. Zuber et al., (2005) used organotypic rat hippocampal slices in cryo-protectant to study synapse structure in vitreous ice sections examined at low temperature without any fixation, staining or embedding. The method provides a more near-to-native ultrastructure and produces images sometimes containing new features but the technique is difficult, requires the use of an expensive cryo-electron microscope as well as a HPF machine and is presently limited to thin sections of 70–100 nm thick. Applications to tomographic imaging of thick specimens have provided some interesting but variable results (Hsieh et al., 2006; Masich et al., 2006).
Previous successful attempts at preserving brain tissue with HPF have relied on using a commercial biopsy needle system (Vanhecke et al., 2006; Vanhecke et al., 2003) or a homemade one (Hohenberg et al., 1996; Shimoni and Muller, 1998) to quickly extract tissue without the use of chemical fixatives. This system works well primarily with the Leica unit (Vanhecke et al., 2003), but has also been used with a Bal-Tec HPF HPM 010 (Hohenberg et al., 1996). The main disadvantage with this method is that the area being extracted is hard to correlate with brain anatomy. It is important to emphasize again that correlated LM and EM provides a powerful approach for understanding functional neuroanatomy. The chemical fixation we use not only facilitates to hold tissue components in place, but also helps to mitigate any deleterious effects from anaesthetics given to animals prior to dissection (Vanhecke et al., 2003). We show using FHV infected DL1 cells that are easily cryo-fixed that a parallel sample chemically fixed and HPF has similar morphology. Therefore, we propose that chemical fixation with aldehydes is not necessarily the step where severe morphological degradation occurs, but it is during post-processing steps as shown by Small over 26 years ago.
HPF has been previously combined with immunolabeling (Donohoe et al., 2007; McDonald, 1999; Mobius et al., 2002; Monaghan et al., 1998; Nicolas et al., 1997) and in particular has been applied to brain tissue focusing on myelin glycolipids (Kirschning et al., 1998), cytoskeletal tethers in rat hippocampal PSDs (Rostaing et al., 2006; Siksou et al., 2007), kainate receptors in the monkey striatum (Kieval et al., 2001), GABA receptors in the monkey subthalamic nucleus (Galvan et al., 2004) and neurohormonal/secretory activity in neuroendocrine melanotrope cells (Calle et al., 2005; Wang et al., 2004). In several of these studies (e.g. Galvan et al., 2004; Kieval et al., 2001), cryoprotection methods were used prior to HPF for better structural preservation and to better retain antigenicity. Another study labeling serotonin 5-HT1D receptors in trigeminal and dorsal root ganglia used a similar protocol to our CAF-HPF for post-embedding immunolabel synaptic terminals in spinal cord (Potrebic et al., 2003). Immunolabeling for brain tissue is very important because it is the only way to dissect tissue complexity. For example, some types of cells are quite rare and the only way to find them in the EM is to be able to localize them at the LM, usually using a pre-embedding protocol. In these cases, correlated LM and EM analysis is critical for localizing or identifying structures in the neuropil (Martone et al., 1992).
For fluorescence photooxidation, chemical fixation prior to photoconversion is essential. Labeled or tagged molecules are dynamic within cells (Deerinck et al., 1994) and it is extremely difficult to deliver reagents into the cells since this is a diffusion-based process. In addition, heat is generated during the photooxidation process and warming and subsequent re-freezing of the tissue would likely cause ice crystal damage. Current protocols use 2–3% of glutaraldehyde as either a primary fixation, as in the case of genetically tagged fluorescent proteins, (Gaietta et al., 2002) or as a secondary fixation after immunolabeling (Deerinck et al., 1994) or small molecule labeling (Capani et al., 2001). Since specimens cannot be maintained with current light microscope stages at the low temperatures required for frozen samples and temperature dependent diffusion processes will limit DAB infiltration, cryo-fixation of photoconverted material is impractical. In this study, we present data showing that glutaraldehyde fixation in combination with HPF also improves the ultrastructure of photooxidized Lucifer Yellow injected neurons. Future improvements on our protocols will include implementation for ReAsH labeled and photoconverted cells and organelles whereby these probes allow for selective staining of proteins for correlated light and electron microscopy (Gaietta et al., 2002; Gaietta et al., 2006; Sosinsky et al., 2007). We can then place these highlighted proteins into macromolecular complexes in their cellular environments in order to fully understand their functional interactions.
Acknowledgements
We thank Joshua Brown for his help in generating Fig. 1G–I. This work is supported by NIH grants Mark Ellisman (NS14718), John E. Johnson (GM34220), GM065937, GM072881 and NSF grant MCB-0131425 (GES). The Cell-Centered Database is supported by NIH grant Maryann Martone (DA016602). The work described here was conducted at the National Center for Microscopy and Imaging Research at San Diego, which is supported by National Institutes of Health Grant RR004050 (MHE).
Abbreviations
- CAF
conventional aldehyde fixation
- HPF
high pressure freezing or high pressure frozen
- CAF-HPF
conventional aldehyde fixation followed by high pressure freezing
- FS
freeze substituted or freeze substitution
- UA
uranyl acetate
- OsO4
osmium tetraoxide
- FHV
flock house virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al-Amoudi A, Dubochet J, Norlen L. Nanostructure of the epidermal extracellular space as observed by cryo-electron microscopy of vitreous sections of human skin. J Invest Dermatol. 2005;124:764–777. doi: 10.1111/j.0022-202X.2005.23630.x. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Dahlstrom A. Studies on the 3-dimensional architecture of dendritic spines and varicosities in human cortex by confocal laser scanning microscopy and Lucifer yellow microinjections. J Neurosci Methods. 1995;57:55–61. doi: 10.1016/0165-0270(94)00125-z. [DOI] [PubMed] [Google Scholar]
- Buhl EH. Intracellular injection in fixed slices in combination with neuroanatomical tracing techniques and electron microscopy to determine multisynaptic pathways in the brain. Microsc Res Tech. 1993;24:15–30. doi: 10.1002/jemt.1070240104. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle M, Corstens GJ, Wang L, Kozicz T, Denver RJ, Barendregt HP, Roubos EW. Evidence that urocortin I acts as a neurohormone to stimulate alpha MSH release in the toad Xenopus laevis. Brain Res. 2005;1040:14–28. doi: 10.1016/j.brainres.2004.12.056. [DOI] [PubMed] [Google Scholar]
- Capani F, Deerinck TJ, Ellisman MH, Bushong E, Bobik M, Martone ME. Phalloidin-eosin followed by photo-oxidation: a novel method for localizing F-actin at the light and electron microscopic levels. J Histochem Cytochem. 2001;49:1351–1361. doi: 10.1177/002215540104901103. [DOI] [PubMed] [Google Scholar]
- Chan FL, Inoue S, Leblond CP. Localization of heparan sulfate proteoglycan in basement membrane by side chain staining with cuprolinic blue as compared with core protein labeling with immunogold. J Histochem Cytochem. 1992;40:1559–1572. doi: 10.1177/40.10.1527375. [DOI] [PubMed] [Google Scholar]
- Chan FL, Inoue S, Leblond CP. The basement membranes of cryofixed or aldehyde-fixed, freeze-substituted tissues are composed of a lamina densa and do not contain a lamina lucida. Cell Tissue Res. 1993;273:41–52. doi: 10.1007/BF00304610. [DOI] [PubMed] [Google Scholar]
- Dahl R, Staehelin LA. High-pressure freezing for the preservation of biological structure: theory and practice. J Electron Microsc Tech. 1989;13:165–174. doi: 10.1002/jemt.1060130305. [DOI] [PubMed] [Google Scholar]
- Deerinck TJ, Martone ME, Lev-Ram V, Green DPL, Tsien RY, Spector DL, Huang S, Ellisman MH. Fluorescence photooxidation with eosin: a method for high resolution immunolocalization and in situ hybridization detect for light and electron microscopy. J. Cell Biol. 1994;126:910–910. doi: 10.1083/jcb.126.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe BS, Kang BH, Staehelin LA. Identification and characterization of COPIa- and COPIb-type vesicle classes associated with plant and algal Golgi. Proc Natl Acad Sci U S A. 2007;104:163–168. doi: 10.1073/pnas.0609818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epperlein HH, Radomski N, Wonka F, Walther P, Wilsch M, Muller M, Schwarz H. Immunohistochemical demonstration of hyaluronan and its possible involvement in axolotl neural crest cell migration. J Struct Biol. 2000;132:19–32. doi: 10.1006/jsbi.2000.4298. [DOI] [PubMed] [Google Scholar]
- Epperlein HH, Schwarz H, Piendl T, Lofberg J, Studer D, Spring H, Muller M. Improved preservation of the subepidermal extracellular matrix in axolotl embryos using electron microscopical techniques based on cryoimmobilization. J Struct Biol. 1997;118:43–61. doi: 10.1006/jsbi.1996.3838. [DOI] [PubMed] [Google Scholar]
- Friesen PD, Rueckert RR. Synthesis of Black Beetle Virus Proteins in Cultured Drosophila Cells: Differential Expression of RNAs 1 and 2. J Virol. 1981;37:876–886. doi: 10.1128/jvi.37.3.876-886.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Zhao S, Graber W, Drakew A, Studer D. New ways of looking at synapses. Histochem Cell Biol. 2007;128:91–96. doi: 10.1007/s00418-007-0305-7. [DOI] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Gaietta GM, Giepmans BN, Deerinck TJ, Smith WB, Ngan L, Llopis J, Adams SR, Tsien RY, Ellisman MH. Golgi twins in late mitosis revealed by genetically encoded tags for live cell imaging and correlated electron microscopy. Proc. Natl. Acad. Sci. U S A. 2006;103:17777–17782. doi: 10.1073/pnas.0608509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A, Charara A, Pare JF, Levey AI, Smith Y. Differential subcellular and subsynaptic distribution of GABA(A) and GABA(B) receptors in the monkey subthalamic nucleus. Neuroscience. 2004;127:709–721. doi: 10.1016/j.neuroscience.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233–245. doi: 10.1016/j.cardiores.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH. Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots. Nat. Methods. 2005;2:743–749. doi: 10.1038/nmeth791. [DOI] [PubMed] [Google Scholar]
- Hawes P, Netherton CL, Mueller M, Wileman T, Monaghan P. Rapid freeze-substitution preserves membranes in high-pressure frozen tissue culture cells. J Microsc. 2007;226:182–189. doi: 10.1111/j.1365-2818.2007.01767.x. [DOI] [PubMed] [Google Scholar]
- Hayat M. Fixation for Electron MIcroscopy. New York: Academic Press; 1982. [Google Scholar]
- Hohenberg H, Tobler M, Muller M. High-pressure freezing of tissue obtained by fine-needle biopsy. J Microsc. 1996;183:133–139. doi: 10.1046/j.1365-2818.1996.820642.x. [DOI] [PubMed] [Google Scholar]
- Hsieh CE, Leith A, Mannella CA, Frank J, Marko M. Towards high-resolution three-dimensional imaging of native mammalian tissue: electron tomography of frozen-hydrated rat liver sections. J Struct Biol. 2006;153:1–13. doi: 10.1016/j.jsb.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Kieval JZ, Hubert GW, Charara A, Pare JF, Smith Y. Subcellular and subsynaptic localization of presynaptic and postsynaptic kainate receptor subunits in the monkey striatum. J Neurosci. 2001;21:8746–8757. doi: 10.1523/JNEUROSCI.21-22-08746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschning E, Rutter G, Hohenberg H. High-pressure freezing and freeze-substitution of native rat brain: suitability for preservation and immunoelectron microscopic localization of myelin glycolipids. J Neurosci Res. 1998;53:465–474. doi: 10.1002/(SICI)1097-4547(19980815)53:4<465::AID-JNR8>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Konig P, Braunfeld M, Agard DA. Use of surface affinity enrichment and cryo-embedding to prepare in vitro reconstituted mitotic chromosomes for EM tomography. Ultramicroscopy. 2005;103:261–274. doi: 10.1016/j.ultramic.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Lawrence A, Bouwer JC, Perkins G, Ellisman MH. Transform-based backprojection for volume reconstruction of large format electron microscope tilt series. J. Struct. Biol. 2006;154:144–167. doi: 10.1016/j.jsb.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Marsh BJ, Volkmann N, McIntosh JR, Howell KE. Direct continuities between cisternae at different levels of the Golgi complex in glucose-stimulated mouse islet beta cells. Proc Natl Acad Sci U S A. 2004;101:5565–5570. doi: 10.1073/pnas.0401242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone M, Gupta A, Wong M, Qian X, Sosinsky G, Ludascher B, Ellisman M. A cell-centered database for electron tomographic data. J. Struct. Biol. 2002;138:145. doi: 10.1016/s1047-8477(02)00006-0. [DOI] [PubMed] [Google Scholar]
- Martone ME, Armstrong DM, Young SJ, Groves PM. Ultrastructural examination of enkephalin and substance P input to cholinergic neurons within the rat neostriatum. Brain Res. 1992;594:253–262. doi: 10.1016/0006-8993(92)91132-x. [DOI] [PubMed] [Google Scholar]
- Martone ME, Zhang S, Gupta A, Qian X, He H, Price DL, Wong M, Santini S, Ellisman MH. The cell-centered database: a database for multiscale structural and protein localization data from light and electron microscopy. Neuroinformatics. 2003;1:379–395. doi: 10.1385/NI:1:4:379. [DOI] [PubMed] [Google Scholar]
- Masich S, Ostberg T, Norlen L, Shupliakov O, Daneholt B. A procedure to deposit fiducial markers on vitreous cryo-sections for cellular tomography. J Struct Biol. 2006;156:461–468. doi: 10.1016/j.jsb.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Dual-axis tomography: an approach with alignment methods that preserve resolution. J. Struct. Biol. 1997;120:343–352. doi: 10.1006/jsbi.1997.3919. [DOI] [PubMed] [Google Scholar]
- McDonald K. High-pressure freezing for preservation of high resolution fine structure and antigenicity for immunolabeling. Methods Mol Biol. 1999;117:77–97. doi: 10.1385/1-59259-201-5:77. [DOI] [PubMed] [Google Scholar]
- McDonald KL, Auer M. High-pressure freezing, cellular tomography, and structural cell biology. Biotechniques. 2006;41:137, 139, 141. doi: 10.2144/000112226. passim. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Schwartz MD, Ahlquist P. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J Virol. 2001;75:11664–11676. doi: 10.1128/JVI.75.23.11664-11676.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobius W, Ohno-Iwashita Y, van Donselaar EG, Oorschot VM, Shimada Y, Fujimoto T, Heijnen HF, Geuze HJ, Slot JW. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J Histochem Cytochem. 2002;50:43–55. doi: 10.1177/002215540205000105. [DOI] [PubMed] [Google Scholar]
- Monaghan P, Perusinghe N, Muller M. High-pressure freezing for immunocytochemistry. J Microsc. 1998;192:248–258. doi: 10.1046/j.1365-2818.1998.00387.x. [DOI] [PubMed] [Google Scholar]
- Moor H. Theory and practice of high pressure freezing. In: RAZ Steinbrecht K, editor. Cryotechniques in Biological Electron Microscopy. Berlin: Springer-Verlag; 1987. pp. 175–191. [Google Scholar]
- Moor H, Bellin G, Sandri C, Akert K. The influence of high pressure freezing on mammalian nerve tissue. Cell Tissue Res. 1980;209:201–216. doi: 10.1007/BF00237626. [DOI] [PubMed] [Google Scholar]
- Murk JL, Posthuma G, Koster AJ, Geuze HJ, Verkleij AJ, Kleijmeer MJ, Humbel BM. Influence of aldehyde fixation on the morphology of endosomes and lysosomes: quantitative analysis and electron tomography. J Microsc. 2003;212:81–90. doi: 10.1046/j.1365-2818.2003.01238.x. [DOI] [PubMed] [Google Scholar]
- Nicolas G, Gaill F, Zylberberg L. In situ localization of two fibrillar collagens in two compact connective tissues by immunoelectron microscopy after cryotechnical processing. J Histochem Cytochem. 1997;45:119–128. doi: 10.1177/002215549704500115. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster Hd. Fine Structure of the Nervous System: Neurons and Their Supporting Cells. New York: Oxford University Press, New York; 1991. 494 pp. [Google Scholar]
- Potrebic S, Ahn AH, Skinner K, Fields HL, Basbaum AI. Peptidergic nociceptors of both trigeminal and dorsal root ganglia express serotonin 1D receptors: implications for the selective antimigraine action of triptans. J Neurosci. 2003;23:10988–10997. doi: 10.1523/JNEUROSCI.23-34-10988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reipert S, Fischer I, Wiche G. High-pressure cryoimmobilization of murine skin reveals novel structural features and prevents extraction artifacts. Exp Dermatol. 2004;13:419–425. doi: 10.1111/j.0906-6705.2004.00180.x. [DOI] [PubMed] [Google Scholar]
- Rostaing P, Real E, Siksou L, Lechaire JP, Boudier T, Boeckers TM, Gertler F, Gundelfinger ED, Triller A, Marty S. Analysis of synaptic ultrastructure without fixative using high-pressure freezing and tomography. Eur J Neurosci. 2006;24:3463–3474. doi: 10.1111/j.1460-9568.2006.05234.x. [DOI] [PubMed] [Google Scholar]
- Schwab M, Bauer R, Zwiener U. The distribution of normal brain water content in Wistar rats and its increase due to ischemia. Brain Res. 1997;749:82–87. doi: 10.1016/s0006-8993(96)01165-1. [DOI] [PubMed] [Google Scholar]
- Senda T, Iizuka-Kogo A, Shimomura A. Visualization of the nuclear lamina in mouse anterior pituitary cells and immunocytochemical detection of lamin A/C by quick-freeze freeze-substitution electron microscopy. J Histochem Cytochem. 2005;53:497–507. doi: 10.1369/jhc.4A6478.2005. [DOI] [PubMed] [Google Scholar]
- Shimoni E, Muller M. On optimizing high-pressure freezing: from heat transfer theory to a new microbiopsy device. J Microsc. 1998;192:236–247. doi: 10.1046/j.1365-2818.1998.00389.x. [DOI] [PubMed] [Google Scholar]
- Siksou L, Rostaing P, Lechaire JP, Boudier T, Ohtsuka T, Fejtova A, Kao HT, Greengard P, Gundelfinger ED, Triller A, Marty S. Three-dimensional architecture of presynaptic terminal cytomatrix. J Neurosci. 2007;27:6868–6877. doi: 10.1523/JNEUROSCI.1773-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JV. Organization of actin in the leading edge of cultured cells: influence of osmium tetroxide and dehydration on the ultrastructure of actin meshworks. J Cell Biol. 1981;91:695–705. doi: 10.1083/jcb.91.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosinsky GE, Deerinck TJ, Greco R, Buitenhuys CH, Bartol TM, Ellisman MH. Development of a model for microphysiological simulations: small nodes of Ranvier from peripheral nerves of mice reconstructed by electron tomography. Neuroinformatics. 2005;3:133–162. doi: 10.1385/NI:3:2:133. [DOI] [PubMed] [Google Scholar]
- Sosinsky GE, Giepmans BNG, Deerinck TJ, Gaietta GM, Ellisman MH. Markers for correlated light and electron microscopy. In: McIntosh JR, editor. Cellular Electron Microscopy. Vol. 79. San Diego, CA: Elsevier; 2007. pp. 573–589. [DOI] [PubMed] [Google Scholar]
- Studer D, Michel M, Wohlwend M, Hunziker EB, Buschmann MD. Vitrification of articular cartilage by high-pressure freezing. J Microsc. 1995;179:321–332. doi: 10.1111/j.1365-2818.1995.tb03648.x. [DOI] [PubMed] [Google Scholar]
- Vanharreveld A, Crowell J. Electron Microscopy after Rapid Freezing on a Metal Surface and Substitution Fixation. Anat Rec. 1964;149:381–385. doi: 10.1002/ar.1091490307. [DOI] [PubMed] [Google Scholar]
- Vanharreveld A, Crowell J, Malhotra SK. A Study of Extracellular Space in Central Nervous Tissue by Freeze-Substitution. J Cell Biol. 1965;25:117–137. doi: 10.1083/jcb.25.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhecke D, Eggli P, Graber W, Studer D. A new microbiopsy system enables rapid preparation of tissue for high-pressure freezing. Methods Mol Biol. 2006;319:463–477. doi: 10.1007/978-1-59259-993-6_21. [DOI] [PubMed] [Google Scholar]
- Vanhecke D, Graber W, Herrmann G, Al-Amoudi A, Eggli P, Studer D. A rapid microbiopsy system to improve the preservation of biological samples prior to high-pressure freezing. J Microsc. 2003;212:3–12. doi: 10.1046/j.1365-2818.2003.01226.x. [DOI] [PubMed] [Google Scholar]
- Wang LC, Meijer HK, Humbel BM, Jenks BG, Roubos EW. Activity-dependent dynamics of coexisting brain-derived neurotrophic factor, pro-opiomelanocortin and alpha-melanophore-stimulating hormone in melanotrope cells of Xenopus laevis. J Neuroendocrinol. 2004;16:19–25. doi: 10.1111/j.1365-2826.2004.01110.x. [DOI] [PubMed] [Google Scholar]
- Zechmann B, Muller M, Zellnig G. Membrane associated qualitative differences in cell ultrastructure of chemically and high pressure cryofixed plant cells. J Struct Biol. 2007;158:370–377. doi: 10.1016/j.jsb.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Zuber B, Nikonenko I, Klauser P, Muller D, Dubochet J. The mammalian central nervous synaptic cleft contains a high density of periodically organized complexes. Proc Natl Acad Sci U S A. 2005;102:19192–19197. doi: 10.1073/pnas.0509527102. [DOI] [PMC free article] [PubMed] [Google Scholar]