

Abstract
Unregulated activities of the matrix metalloproteinase (MMP) family have been implicated in primary and metastatic tumor growth, angiogenesis, and pathological degradation of extracellular matrix components, such as collagen and laminin. However, clinical trials with small molecule MMP inhibitors have been largely unsuccessful, with a lack of selectivity considered particularly problematic. Enhanced selectivity could be achieved by taking advantage of differences in substrate secondary binding sites (exosites) within the MMP family. In this study, triple-helical substrates and triple-helical transition state analog inhibitors have been utilized to dissect the roles of potential exosites in MMP-9 collagenolytic behavior. Substrate and inhibitor sequences were based on either the α1(V)436–450 collagen region, which is hydrolyzed at the Gly ↓ Val bond selectively by MMP-2 and MMP-9, or the Gly ↓ Leu cleavage site within the consensus interstitial collagen sequence α1(I–III)769–783, which is hydrolyzed by MMP-1, MMP-2, MMP-8, MMP-9, MMP-13, and MT1-MMP. Exosites within the MMP-9 fibronectin II inserts were found to be critical for interactions with type V collagen model substrates and inhibitors and to participate in interactions with an interstitial (types I–III) collagen model inhibitor. A triple-helical peptide incorporating a fibronectin II insert-binding sequence was constructed and found to selectively inhibit MMP-9 type V collagen-based activities compared with interstitial collagen-based activities. This represents the first example of differential inhibition of collagenolytic activities and was achieved via an exosite-binding triple-helical peptide.
Collagen catabolism (collagenolysis) is normally a well regulated physiological process critical to tissue and organ development, morphogenesis, and wound healing (1). Pathological conditions resulting from aberrant collagenolysis include primary and metastatic tumor growth, arthritis, arteriosclerosis, and periodontitis (1–4). A number of proteases have been described as exhibiting collagenolytic behavior, requiring that they catalyze the hydrolysis of the collagen triple helix (1). The most extensively studied are the matrix metalloproteinases (MMPs).2 MMPs that catalyze the hydrolysis of one or more of the interstitial collagens (types I–III) within their triple-helical domain include the secreted proteases MMP-1, MMP-2, MMP-8, MMP-9, and MMP-13 and the membrane-bound proteases MT1-MMP and MT2-MMP (5–8).
Considerable work has been performed to define the MMP domains and regions that participate in collagenolysis. In the cases of MMP-1, MMP-8, MMP-13, MT1-MMP, and MT2-MMP, efficient collagenolytic activity in the isolated enzyme requires both the catalytic (CAT) and hemopexin (HPX)-like domains (Fig. 1) (9–14). The linker region between these domains also participates in collagenolysis, either by direct binding of substrate (15) or by allowing for the proper orientation of the CAT and HPX domains (16). The “gelatinase” members of the MMP family (MMP-2 and MMP-9) incorporate three fibronectin type II (FN II) inserts within their CAT domains (Fig. 1). The FN II inserts possess binding sites for type I collagen, and thus are alternatively called the collagen-binding domains (17, 18). The FN II inserts, along with the CAT and HPX-like domains, contribute to MMP-2 collagenolytic activity (19).
FIGURE 1.

MMP family members and their structural domains. Pro indicates propeptide domain; CAT indicates catalytic domain; HPX indicates hemopexin-like domain; FN(II) indicates fibronectin II inserts.
Although the domains that participate in collagenolysis have been identified, little is known about the mechanism by which these domains collaborate to facilitate collagenolysis nor the specific sites that are necessary for the process. Collagenolysis may be divided into several steps as follows: (a) binding of the triple helix; (b) proper orientation of the MMP domains; (c) unwinding of the triple helix and/or trapping of relaxed triple-helical backbones; (d) binding of individual substrate strands to the active site; and (e) rapid sequential processing of all three strands (20). One approach to identify MMP residues and regions that participate in the individual collagenolytic steps is to use triple-helical substrates in combination with site-specific or domain mutagenesis (14, 20–25). We have also recently introduced triple-helical transition state analog inhibitors as probes of MMP function (26).
This study has utilized triple-helical substrates and inhibitors to dissect the role of the FN II inserts in triple-helical peptidase activity. Three forms of MMP-9 have been examined as follows: wild-type MMP-9, MMP-9 with the HPX-like domain deleted (MMP-9 CAT), and MMP-9 with the HPX-like domain and FN II inserts deleted (MMP-9 CAT-FN). MMP-2 and MMP-1 (with and without the HPX-like domain) have also been studied to provide a comparison with MMP-9. The triple-helical substrates are derived from sequences hydrolyzed by all collagenolytic MMPs (23, 25) or more selectively by MMP-2 and MMP-9 (27). The triple-helical inhibitors have sequences analogous to the substrates, except that a phosphinate moiety replaces the scissile bond and creates a transition state analog. We have recently described a selective MMP-2/MMP-9 triple-helical inhibitor that incorporates GlyΨ{PO2H-CH2}Val (26). A second, more general collagenolytic MMP triple-helical inhibitor, containing GlyΨ{PO2H-CH2}Leu, is described for the first time here. Activity has been examined as a function of triple-helical content to evaluate the role of triple helicity in MMP behaviors. Finally, the effects of a recently described inhibitor that binds to the MMP-2 FN II insert on different MMP-2 and MMP-9 triple-helical peptidase activities have been compared.
MATERIALS AND METHODS
All standard chemicals were peptide synthesis or molecular biology grade and purchased from Fisher. O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate, (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate, 1-hydroxybenzotriazole, Fmoc-amino acid derivatives, and MMP inhibitor III (a hydroxamic acid-Leu-homo-Phe dipeptide) were obtained from Novabiochem. Amino acids are of l-configuration (except for Gly). Knight SSP (Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2) and NFF-3 (Mca-Arg-Pro-Lys-Pro-Val-Glu-Nva-Trp-Arg-Lys(Dnp)-NH2; where Nva is norvaline) were synthesized by methods described previously (28, 29). The THP substrates fTHP-15 ((Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Gln-Gly ↓ Leu-Arg-Gly-Gln-Lys-(Dnp)-Gly-Val-Arg-(Gly-Pro-Hyp)5-NH2) and α1(V)436–447 fTHP ((Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Pro-Gly ↓ Val-Val-Gly-Glu-Lys(Dnp)-Gly-Glu-Gln-(Gly-Pro-Hyp)5-NH2) and the α1(V)-GlyΨ{PO2H-CH2}Val THPI (C6-(Gly-Pro-Hyp)4-Gly-Pro-Pro-GlyΨ{PO2H-CH2}(S)Val-Val-Gly-Glu-Gln-Gly-Glu-Gln-Gly-Pro-Pro-(Gly-Pro-Hyp)4-NH2) were synthesized and characterized as described previously (23, 25–27, 30). The synthesis of the protected Fmoc-GlyΨ{PO2H-CH2}Leu building block proceeded as described previously (26), with the exception of iodobutane being used instead of iodopropane to prepare the 2-isobutyl-3-oxobutyric acid allyl ester intermediate. The synthesis and purification of α1(I–III)GlyΨ{PO2H-CH2}Leu THPI (C6-Gly-Pro-Flp-(Gly-Pro-Hyp)4-Gly-Pro-Gln-GlyΨ{PO2H-CH2}-(RS)-Leu-Ala-Gly-Gln-Arg-Gly-Ile-Arg-(Gly-Pro-Hyp)4-Gly-Pro-Flp-NH2) proceeded as for α1(V)GlyΨ{PO2H-CH2}Val THPI (26). The synthesis and purification of α1(I)715–721 THP ((Gly-Pro-Hyp)4-Gly-Ala-Hyp-Gly-Ala-Hyp-Gly-Ser-Gln-Gly-Ala-Hyp-(Gly-Pro-Hyp)3-Gly-Pro-Tyr-NH2) proceeded as for other THPs (31–34).
Peptide Analyses—For α1(I–III)GlyΨ{PO2H-CH2}Leu THPI, analytical RP-HPLC was performed on a Waters 600E multisolvent delivery system with a model 486 tunable detector controlled by Empower software, equipped with a Vydac C18 RP column (15-mm particle size, 300-Å pore size, 100 × 8 mm). Eluants were 0.1% trifluoroacetic acid in water (A) and 0.1% trifluoroacetic acid in acetonitrile (B). The elution gradient was 10–70% B in 60 min with a flow rate of 1.0 ml/min. Detection was at λ = 220 nm. Fractions from analytical RP-HPLC were analyzed by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF-MS) on a Bruker Proflex III instrument with XMASS software. α1(I–III)GlyΨ{PO2H-CH2}Leu THPI gave [M + H]+ 3921.4 Da (theoretical 3920.2 Da). For α1(I)715–721 THP, fractions from preparative RP-HPLC were analyzed by MALDI-TOF-MS on an ABD Voyager DE-STR instrument. α1(I)715–721 THP gave [M + H]+ 3205.3 Da (theoretical 3201.4 Da).
Peptide Purification—For α1(I–III)GlyΨ{PO2H-CH2}Leu THPI, semi-preparative RP-HPLC was performed on a Waters Deltaprep System using a Vydac C18 RP column (15-μm particle size, 300-Å pore size, 200 × 25 mm) at a flow rate of 10.0 ml/min. Eluants were 0.1% trifluoroacetic acid in water (A) and 0.1% trifluoroacetic acid in acetonitrile (B). The elution gradient was 10–30% B in 100 min, with detection at λ = 220 nm. Isolated fractions were analyzed by PDA RP-HPLC. PDA RP-HPLC was performed on a Waters 625 pump with a model 996 diode array detector controlled by Millennium software, using a Vydac C18 RP column (5-mm particle size, 120-Å pore size, 250 × 4.6 mm) at a flow rate of 1.0 ml/min. Eluants were 0.1% trifluoroacetic acid in water (A) and 0.1% trifluoroacetic acid in acetonitrile (B). The elution gradient was 10–30% B in 25 min, with detection at λ = 200–400 nm.
CD Spectroscopy—CD spectra were recorded over the range λ = 190–250 nm on a JASCO J-810 spectropolarimeter using a 0.1-cm path length quartz cell. Thermal transition curves were obtained by recording the molar ellipticity ([Θ]) at λ = 225 nm, whereas the temperature was continuously increased in the range of 5–95 °C at a rate of 0.2 °C/min. Temperature was controlled using a JASCO PFD-425S temperature control unit. For samples exhibiting sigmoidal melting curves, the inflection point in the transition region (first derivative) is defined as the melting temperature (Tm). The highest [Θ]222 nm value was designated as 100% folded, and the lowest [Θ]222 nm value was designated as 0% folded (35–38).
Matrix Metalloproteinases—Pro-MMP-1 and pro-MMP-3 were expressed in Escherichia coli and folded from the inclusion bodies as described previously (39, 40). Pro-MMP-1 was activated by reacting with 1 mm 4-aminophenyl mercuric acetate (APMA) and 0.1 eq of MMP-3(Δ248–460) at 37 °C for 6 h. After activation, MMP-3(Δ248–460) was completely removed from MMP-1 by affinity chromatography using an anti-MMP-3 IgG Affi-Gel 10 column. Pro-MMP-3 was activated by reacting with 5 μg/ml chymotrypsin at 37 °C for 2 h. Chymotrypsin was inactivated with 2 mm diisopropyl fluorophosphate. Pro-MMP-2 was purified from the culture medium of human uterine cervical fibroblasts (41) and activated by incubating with 1 mm APMA for 2 h at 37 °C. Recombinant pro-MMP-9 was purchased from Chemicon International (Temecula, CA) and activated with 1 mm APMA for 1 h at 37 °C. Recombinant MMP-9 with the linker and the C-terminal HPX-like domain deleted (MMP-9 residues 107–443; designated MMP-9 CAT) was expressed in E. coli in the active form with Phe107 at the N terminus as described (42). Recombinant MMP-9 with the linker, C-terminal HPX-like domain, and FN II repeats deleted (MMP-9 residues 107–215 + 391–443; designated MMP-9 CAT-FN) was expressed in E. coli in the active form with Phe107 at the N terminus as described (42). The concentrations of active MMP-1, MMP-2, MMP-3, and MMP-9 were determined by titration with recombinant TIMP-1 or N-TIMP-1 over a concentration range of 0.1–3 μg/ml. Pro-MMP-1(Δ243–450) was expressed in E. coli using the expression vector pET3a (Novagen), folded from inclusion bodies and purified as described previously (43). The zymogen was activated as described above for the full-length pro-MMP-1. Active site titrations utilized either Knight SSP or NFF-3 as substrate (28, 29).
Fluorescence Resonance Energy Transfer Peptide Kinetic Assays—Substrate stock solutions were prepared at various concentrations in EAB buffer (50 mm Tricine, 50 mm NaCl, 10 mm CaCl2, 0.05% Brij-35, pH 7.5). MMP assays were conducted in EAB buffer by incubating a range of substrate concentrations (0.7–90 μm) with 2–5 nm enzyme at 25 °C. Fluorescence was measured on a Molecular Devices SPECTRAmax Gemini EM dual-scanning microplate spectrofluorometer using λexcitation = 324 nm and λemission = 393 nm. Rates of hydrolysis were obtained from plots of fluorescence versus time, using data points from only the linear portion of the hydrolysis curve. The slope from these plots was divided by the fluorescence change corresponding to complete hydrolysis and then multiplied by the substrate concentration to obtain rates of hydrolysis in units of micromolar/s. Kinetic parameters were evaluated by Lineweaver-Burk, Eadie-Hofstee, and Hanes-Woolf analyses, as well as by a nonlinear regression, one-site hyperbolic binding model with SigmaPlot software.
Gelatin Assay—DQ™ bovine skin type I gelatin (fluorescein-conjugated) was purchased from Molecular Probes (Eugene, OR). Fluorescein-labeled pig skin type A gelatin was prepared by dissolving 10 mg/ml gelatin in 0.2 m carbonate buffer (pH 9.0), incubating with a ∼30-fold excess of fluorescein isothiocyanate overnight at room temperature, and dialyzing. Stock solutions of labeled gelatins were prepared in EAB buffer. MMP assays were conducted in EAB buffer by incubating a range of gelatin concentrations (0.5, 1.0, and 2.0 μg/ml) with 1.0 nm enzyme. Fluorescence was measured using λexcitation = 485 nm and λemission = 535 nm. Rates of hydrolysis were obtained as described above.
Inhibition Kinetic Studies—Peptide substrates and inhibitors were dissolved in TSB buffer (50 mm Tris·HCl, pH 7.5, containing 100 mm NaCl, 10 mm CaCl2, 0.05% Brij-35, pH 7.5). 1–2 nm enzyme was incubated with varying concentrations of inhibitors for 2 h at room temperature. Residual enzyme activity was monitored by adding 0.1 volume of Knight SSP to produce a final concentration of <0.1 KM. Initial velocity rates were determined from the first 20 min of hydrolysis when product release is linear with time. Fluorescence was measured on a Molecular Devices SPECTRAmax Gemini EM dual-scanning microplate spectrofluorometer using λexcitation = 324 nm and λemission = 393 nm. Apparent Ki values were calculated using Equations 1 and 2,
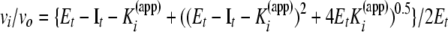 |
(Eq. 1) |
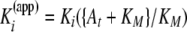 |
(Eq. 2) |
where It is the total inhibitor concentration; Et is the total enzyme concentration; At is the total substrate concentration; vo is the activity in the absence of inhibitor; and KM is the Michaelis constant. In our assays the value of Et/Kappi does not exceed 100 so that the inhibitor is distributed in both free and bound forms, and Kappi can be calculated by fitting inhibition data to Equation 1. Because the substrate concentration is less than KM/10, Kappi values are insignificantly different from true Ki values. In cases where weak inhibition occurred, Kappi values were calculated using vi = vo/(1 + It/Kappi).
It should be noted that the diastereomers present in α1(I–III)GlyΨ{PO2H-CH2}Leu THPI were not separable by RP-HPLC. Because the S stereoisomer is likely a better inhibitor than the R stereoisomer (26), the true Ki values for the S form are likely lower than illustrated here.
RESULTS
Triple-helical Substrates and Transition State Analogs—Two triple-helical substrates and two triple-helical transition state analog inhibitors were used as probes of MMP-9 function. One of the substrates, fTHP-15, is hydrolyzed by all collagenolytic MMPs (MMP-1, MMP-2, MMP-8, MMP-9, MMP-13, MT1-MMP, and MT2-MMP) (20, 25, 30, 44). The second substrate, α1(V)436–447 fTHP, is hydrolyzed more selectively by MMP-2 and MMP-9 among the collagenolytic MMPs (27). The α1(V)GlyΨ{PO2H-CH2}Val THPI, whose sequence is based on α1(V)436–450, is a low nanomolar inhibitor of MMP-2 and MMP-9 but not MMP-1, MMP-3, MMP-8, MMP-13, and MT1-MMP (26). The melting temperature (Tm) values for fTHP-15, α1(V)436–447 fTHP, and α1(V)GlyΨ{PO2H-CH2}Val THPI are 58.0, 49.5, and 25 °C, respectively (26, 27, 30).
The α1(I–III)GlyΨ{PO2H-CH2}Leu THPI, whose sequence is based on the interstitial collagen consensus sequence α1(I–III)769–783 (24, 25), has not been described previously. The consensus α1(I–III)769–783 is hydrolyzed by MMP-1, MMP-2, MMP-8, MMP-9, MMP-13, MT1-MMP, and MT2-MMP (20, 24, 25, 44). The P1-P1′ subsites of the triple-helical peptide, which are Gly ↓ Leu in the substrate, were substituted by a GlyΨ{PO2H-CH2}Leu transition state analog. Because the Tm value for α1(V)GlyΨ{PO2H-CH2}Val THPI was considerably lower than that for the analogous substrate (26), the α1(I–III)GlyΨ{PO2H-CH2}Leu THPI incorporated the non-natural amino acid (2S,4R)-4-fluoroproline (Flp) to enhance triple helicity (33, 45, 46). The CD spectrum of α1(I–III)GlyΨ{PO2H-CH2}Leu THPI was indicative of a collagen-like triple helix, with a strong negative molar ellipticity ([Θ]) at λ = 195 nm and a positive [Θ]at λ = 225 nm (data not shown). To examine the THPI thermal stability, [Θ]at λ = 225 nm was monitored as a function of increasing temperature. α1(I–III)GlyΨ{PO2H-CH2}Leu THPI exhibited a cooperative transition, indicative of the melting of a triple helix to a single-stranded structure, with Tm = 30 °C (Fig. 2).
FIGURE 2.

Thermal transition curve for α1(I–III)GlyΨ{PO2H-CH2}Leu THPI). The first derivative of the melting curve (shown in the inset) indicates a Tm of 30 °C. The sample was dissolved in 0.25% (v/v) TSB buffer at a substrate concentration of 25 μm. Molar ellipticity ([θ]) was recorded at λ = 225 nm while the temperature was increased from 5 to 85 °C.
Characterization of MMP-9 and MMP-9 Deletion Mutants at Different Temperatures—Three forms of MMP-9 were utilized in the present study as follows: full-length MMP-9, MMP-9 with the HPX-like domain deleted (MMP-9 CAT), and MMP-9 with the HPX-like domain and the FN II inserts deleted (MMP-9 CAT-FN). THPI activity against MMP-9, MMP-9 CAT, and MMP-9 CAT-FN was examined at 10 and 37 °C, based on the thermal stability of the inhibitor triple helix (see below). To evaluate the integrity of each MMP-9 at the two temperatures, inhibition by N-TIMP-1 and hydrolysis of type I gelatin was examined. N-TIMP-1 inhibited all MMP-9 species with sub-picomolar Ki values at both 10 and 37 °C (Table 1), indicative of the preservation of proper folding and exosite display for the enzymes. This result is consistent with a prior report of N-TIMP-1 inhibition of the same three MMP-9 species at 25 °C (42).
TABLE 1.
Inhibition of MMP-9 activity toward Knight SSP by N-TIMP-1
| Enzyme | Temperature | Kiapp |
|---|---|---|
| °C | pm | |
| MMP-9 | 10 | 165.6 ± 35.4 |
| MMP-9 | 37 | 83.3 ± 33.6 |
| MMP-9 CAT | 10 | 387.2 ± 0.3 |
| MMP-9 CAT | 37 | 245.3 ± 50.5 |
| MMP-9 CAT-FN | 10 | 479.7 ± 36.9 |
| MMP-9 CAT-FN | 37 | 200.9 ± 35.4 |
The initial velocity (relative fluorescence units/s) for MMP-9 and MMP-9 CAT hydrolysis of type I gelatin was quantified at 10 and 37 °C and compared. MMP-9 CD exhibited 76.1 and 69.0% of the activity of MMP-9 at 10 and 37 °C, respectively. As with the N-TIMP-1 results above, the gelatin hydrolysis indicated that MMP-9 and MMP-9 CAT were properly folded and retained comparable relative activities at 10 and 37 °C. MMP-9 CAT-FN could not be studied here, as the FN inserts are responsible for binding gelatin (18, 47).
Identification of MMP-9 Domains Participating in Triple-helical Peptidase Activity—We examined the role of the HPX-like domain and FN II inserts in MMP-9 catalysis using deletion mutants. Removal of the HPX-like domain (MMP-9 CAT) had no significant effect on MMP-9 activity toward linear (Knight SSP) or triple-helical (α1(V)436–447 fTHP, fTHP-15) substrates (Table 2). However, removal of the HPX-like domain and the FN II inserts (MMP-9 CAT-FN) dramatically altered triple-helical peptidase activity. MMP-9 CAT-FN did not hydrolyze the type V collagen-derived α1(V)436–447 fTHP and had only 19% of the activity of full-length MMP-9 toward the α1(I–III)769–783 consensus sequence fTHP-15 (Table 2). Thus, the FN II inserts appear to possess secondary binding sites (exosites) for optimum triple-helical peptidase activity. The decreased activity toward fTHP-15 was primarily because of a higher KM value (Table 2). Results with the α1(V)436–447 fTHP are consistent with a prior study, in which the FN II inserts were found to be necessary for MMP-9 activity toward type V collagen (48). It is interesting to note that removal of the FN II inserts did not have identical effects on the two triple-helical substrates, suggesting that for some triple-helical sequences (i.e. α1(V)436–447) the FN II inserts are critical for protease activity, although for others (i.e. α1(I–III)769–783) the FN II inserts participate in activity.
TABLE 2.
Kinetic parameters for MMP-9 and MMP-9 mutant hydrolysis of synthetic substrates at 25 °C
| Enzyme | Substrate | kcat/Km | Relative activity | kcat | Km |
|---|---|---|---|---|---|
| s-1m-1 | % | s-1 | μm | ||
| MMP-9 | Knight SSP | 224,513 ± 24,702 | 100 | 6.22 ± 0.68 | 27.7 ± 0.6 |
| MMP-9 CAT | Knight SSP | 201,499 ± 10,759 | 90 | 10.19 ± 0.80 | 51.6 ± 2.2 |
| MMP-9 CAT-FN | Knight SSP | 194,533 ± 15,248 | 87 | 10.40 ± 0.56 | 52.4 ± 0.5 |
| MMP-9 | α1(V)436-447 fTHP | 51,360 ± 1,676 | 100 | 3.99 ± 0.13 | 77.8 ± 5.8 |
| MMP-9 CAT | α1(V)436-447 fTHP | 52,249 ± 9,192 | 102 | 1.17 ± 0.21 | 22.5 ± 1.7 |
| MMP-9 CAT-FN | α1(V)436-447 fTHP | Not cleaved | 0 | Not cleaved | Not cleaved |
| MMP-9 | fTHP-15 | 64,851 ± 10,698 | 100 | 0.78 ± 0.13 | 12.1 ± 0.6 |
| MMP-9 CAT | fTHP-15 | 69,566 ± 9,138 | 107 | 1.05 ± 0.14 | 15.1 ± 1.4 |
| MMP-9 CAT-FN | fTHP-15 | 12,227 ± 1,077 | 19 | 1.75 ± 0.15 | 143.0 ± 13.9 |
The same three MMP-9 species as well as MMP-2 were studied for inhibition by α1(V)GlyΨ{PO2H-CH2}Val THPI. Because of the relative instability of this analog, experiments were performed at 10 °C to ensure triple-helical structure (26). Removal of the HPX-like domain (MMP-9 CAT) had no effect on α1(V)GlyΨ{PO2H-CH2}Val THPI inhibition of MMP-9 (Table 3). Removal of the both the HPX-like domain and the FN II inserts (MMP-9 CAT-FN) resulted in no measurable inhibition of MMP-9 (Table 3). Both results are consistent with the studies of MMP-9 activity toward the α1(V)436–447 fTHP (Table 2). Melting of the triple helix at 37 °C had no effect on the inhibitory potency of α1(V)GlyΨ{PO2H-CH2}Val THPI toward MMP-9 (Table 3). In contrast, inhibition of MMP-2 was sensitive to triple-helical conformation, as melting of the triple helix increased Ki 5-fold (Table 3) (26).
TABLE 3.
Inhibition of MMP-2 and MMP-9 activity toward Knight SSP by α1(V)Glyψ {PO2H-CH2}Val THPI
| Enzyme | Temperature | Kiapp |
|---|---|---|
| °C | nm | |
| MMP-9 | 10 | 1.76 ± 0.05 |
| MMP-9 | 37 | 1.29 ± 0.00 |
| MMP-9 CAT | 10 | 1.76 ± 0.11 |
| MMP-9 CAT | 37 | 1.80 ± 0.04 |
| MMP-9 CAT-FN | 10 | >>200 |
| MMP-9 CAT-FN | 37 | >>200 |
| MMP-2 | 10 | 4.14 ± 0.47 |
| MMP-2 | 37 | 19.23 ± 0.64 |
The activity of α1(I–III)GlyΨ{PO2H-CH2}Leu THPI was profiled with MMP-1, MMP-2, and MMP-9. This THPI exhibited low or sub-nanomolar Ki values for inhibition of MMP-1, MMP-2, and MMP-9 at 10 °C (Table 4). The α1(I–III)GlyΨ{PO2H-CH2}Leu THPI was more effective against a broader range of collagenolytic MMPs than α1(V)GlyΨ{PO2H-CH2}Val THPI (Table 3), which is not surprising based on the array of enzymes known to hydrolyze the parent sequence (see above). In similar fashion to the results with α1(V)GlyΨ{PO2H-CH2}Val THPI, removal of the MMP-9 HPX-like domain (MMP-9 CAT) had no effect on α1(I–III)GlyΨ{PO2H-CH2}Leu THPI inhibition of MMP-9, whereas removal of both the HPX-like domain and the FN II inserts (MMP-9 CAT-FN) resulted in a significant loss of inhibitory activity (Table 4). As seen with the THP substrates (Table 2), the MMP-9 FN II inserts were important for recognition of the both the α1(V)GlyΨ{PO2H-CH2}Val THPI (Table 3) and the α1(I–III)GlyΨ{PO2H-CH2}Leu THPI (Table 4). Melting of the α1(I–III)GlyΨ{PO2H-CH2}Leu THPI triple helix at 37 °C did not appear to effect MMP-9 inhibition substantially (Table 4). For MMP-2, melting of the α1(I–III)GlyΨ{PO2H-CH2}Leu THPI also had no effect on activity (Table 4), which was different from that observed for the α1(V)GlyΨ{PO2H-CH2}Val THPI (Table 3). This indicates that there is a sequence-dependent sensitivity to the triple-helical structure for MMP-2.
TABLE 4.
Inhibition of MMP activity toward Knight SSP by α1(I—III)Glyψ{PO2H-CH2}Leu THPI
Limitations in fluorescence detection are such that subnanomolar Kiapp values are likely overestimations due to an inability to test appropriately low [E].
| Enzyme | Inhibitor | Temperature | Kiapp |
|---|---|---|---|
| °C | nm | ||
| MMP-9 | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 10 | 0.02 ± 0.01 |
| MMP-9 | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 37 | 0.09 ± 0.00 |
| MMP-9 CAT | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 10 | 0.01 ± 0.00 |
| MMP-9 CAT | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 37 | 0.05 ± 0.01 |
| MMP-9 CAT-FN | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 10 | 3.51 ± 0.99 |
| MMP-9 CAT-FN | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 37 | 3.54 ± 0.62 |
| MMP-2 | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 10 | 0.18 ± 0.00 |
| MMP-2 | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 37 | 0.08 ± 0.01 |
| MMP-1 | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 10 | 7.83 ± 1.03 |
| MMP-1 | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 37 | 26.70 ± 5.21 |
| MMP-1 | MMP inhibitor III | 10 | 2.48 ± 0.35 |
| MMP-1 | MMP inhibitor III | 37 | 4.72 ± 0.38 |
| MMP-1 CATa | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 10 | 25.55 ± 8.00 |
| MMP-1 CATa | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 37 | 114.74 ± 7.61 |
| MMP-1 CATb | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 10 | 18.60 ± 2.33 |
| MMP-1 CATb | α1(I—III)Glyψ{PO2H-CH2}Leu THPI | 37 | 156.42 ± 0.69 |
| MMP-1 CATb | MMP inhibitor III | 10 | 1.70 ± 0.08 |
| MMP-1 CATb | MMP inhibitor III | 37 | 3.07 ± 0.14 |
This is self-processed (residues 259 - 450 or 270 - 450 removed (66)).
HPX-like domain is deleted (residues 243-450).
For comparison with MMP-9 and MMP-9 CAT, activity of the α1(I–III)GlyΨ{PO2H-CH2}Leu THPI was studied for MMP-1 and MMP-1 CAT at 10 °C. Removal of the HPX-like domain increased Ki ∼ 2–3 times (Table 4). MMP-1 was sensitive to the triple-helical structure of the inhibitor (Ki increased ∼4 times when the inhibitor was thermally unwound at 37 °C). This parallels MMP-1 behavior toward the analogous substrate (fTHP-15) (23, 30, 44).
To determine whether an increase in Ki as a function of temperature is a general trend for inhibition of MMP-1, inhibition of MMP-1 by a small molecule (MMP inhibitor III) was examined. The Ki values for both MMP-1 and MMP-1 CAT increased ∼2-fold between 10 and 37 °C (Table 4). Thus, for a small molecule inhibitor, an increase in temperature slightly decreased the affinity toward MMP-1. This suggests that the more substantial 4–8-fold change in Ki values for MMP-1 and MMP-1 CAT by α1(I–III)GlyΨ{PO2H-CH2}Leu THPI as a function of increasing temperature was because of unfolding of the inhibitor triple-helical structure.
Exosite Inhibition of MMP-2 and MMP-9 Triple-helical Peptidase Activities—Recently, a single-stranded peptide model of the α1(I)715–721 collagen sequence was identified as a ligand for the MMP-2 FN II insert and inhibited MMP-2 gelatinolysis (49). We assembled a triple-helical version of this ligand [α1(I)715–721 THP], which had a Tm value of 47 °C (data not shown). α1(I)715–721 THP was evaluated for its ability to inhibit MMP-2 and MMP-9 triple-helical peptidase and gelatinase activities. Dose-dependent inhibition of gelatinolysis was observed for both MMP-2 and MMP-9, with Ki values of 52 and 54 μm, respectively (Table 5). There was no inhibition of MMP-2 or MMP-9 hydrolysis of the Knight SSP or fTHP-15 by α1(I)715–721 THP (Table 5). However, MMP-2 and MMP-9 hydrolysis of α1(V)436–447 fTHP was inhibited by α1(I)715–721 THP, with Ki values of 144 and 123 μm, respectively (Table 5).
TABLE 5.
Inhibition of MMPs by α1(I)715-721 THP at 37 °C
| Enzyme | Substrate | Kiapp |
|---|---|---|
| μm | ||
| MMP-2 | DQ gelatin | 52.26 ± 5.110 |
| MMP-2 | Knight SSP | NIa |
| MMP-2 | fTHP-15 | NI |
| MMP-2 | α1(V)436-447 fTHP | 143.5 ± 11.40 |
| MMP-9 | DQ gelatin | 54.42 ± 7.616 |
| MMP-9 | Knight SSP | NI |
| MMP-9 | fTHP-15 | NI |
| MMP-9 | α1(V)436-447 fTHP | 122.7 ± 5.83 |
NI indicates no inhibition.
DISCUSSION
Roles of MMP-9 CAT and HPX-like Domains and FN II Inserts in Triple-helical Peptidase Activity—The “classic” collagenolytic MMPs (MMP-1, MMP-8, MMP-13, and MT1-MMP) cleave triple helices in a fashion that is distinct from that of the gelatinases of the MMP family (MMP-2 and MMP-9) (19–21, 23, 25, 50, 51). Triple-helical substrates and inhibitors have been used here to probe the functional subtleties of MMP-9 CAT domain, FN II insert, and HPX-like domain. The MMP-9 FN II insert participates in the binding of all triple-helical structures examined here. Deletion of the FN II insert from the MMP-9 CAT domain results in a complete loss of activity toward a type V collagen model THP substrate and complete loss of inhibition by a type V collagen model THPI (Tables 2 and 3). Activity toward a short single-stranded synthetic substrate (Knight SSP) had no significant decrease as a result of this deletion (Table 2). It was previously found that MMP-9 with the FN II insert deleted has similar activity as wild-type MMP-9 toward the Knight SSP and no activity toward soluble type V collagen (48). Also, the mutant enzyme did not bind to type V collagen films (48). Thus, the FN II insert was critical for binding of type V collagen, in similar fashion to our observations for type V collagen-derived triple helices.
Deletion of the FN II insert from the MMP-9 CAT domain results in an 80% loss of activity toward an interstitial (types I–III) collagen model THP substrate and a 150-fold decrease in inhibition by a types I–III collagen model THPI (Tables 2 and 4). Unfortunately, for comparative purposes, the role(s) of MMP-9 domains in interstitial collagenolysis have not been studied. However, the recombinant MMP-9 FN II insert binds type I–III and V collagens and inhibits MMP-2 and MMP-9 binding to type I collagen (18). Thus, we can make some comparisons to interstitial collagenolysis by MMP-2. The MMP-2 FN II insert competes for binding to type I collagen with the wild-type enzyme (17). Deletion of the MMP-2 FN II inserts results in a 60% reduction of type I collagenolytic activity (19). Thus, the FN II insert appears to have a similar role in interstitial collagenolysis and interstitial collagen model triple-helical peptidase activity.
In contrast to the FN II inserts, the HPX-like domain of MMP-9 does not appear to be important for binding or processing type V collagen- or interstitial collagen-derived triple-helical structure, as kcat/KM values for all substrates and apparent Ki values for all inhibitors do not change with its removal (Tables 2, 3, 4). In a similar fashion, the HPX-like domain does not appear to play a significant role in the type V collagenolytic activity of MMP-9 (52).
The role of the HPX-like domain in interstitial collagenolysis appears complex. Consistent with this study, the MMP-2 HPX-like domain and pro-MMP-2 with the FN II insert deleted do not bind type I collagen model triple helices (21). In apparent contrast to our results, deletion of the HPX-like domain of MMP-2 eliminates type I collagenolytic activity (19). In the latter case, fibrillar collagen was utilized, and thus the HPX-like domain may be critical for action on fibrillar collagen. Modeling of MMP-2 revealed a 70 Å distance between the enzyme active site and the most distal collagen interactions with HPX-like domain and FN II insert 3 (53). This distance corresponds to ∼24 residues of a triple helix, and thus our THP substrates and inhibitors are of sufficient length to engage in interactions with the HPX-like domain and FN II inserts.
It has been proposed that the HPX-like domain of MMP-2 is required to initiate collagenolysis, with the FN II inserts facilitating proteolysis in a two-phase process (19). The FN II inserts may promote gross unwinding of the triple helix prior to hydrolysis (51). Molecular modeling indicates that the collagen triple helix could contact MMP-2 FN II inserts 1 and 3, with a smaller portion contacting the HPX-like domain (53). The triple helix fits into the largest cavity of enzyme, with a portion of the molecule unwound and bent. The same modeling studies suggest that the HPX-like domain and FN II inserts are needed to induce specific motions within the CAT domain. The interplay between the HPX-like domain and FN II inserts unwinds the triple helix. The HPX-like domains in MMP-2 and MMP-9 may interact primarily with associated triple helices, as in fibrils, whereas the FN II inserts interact with individual triple helices. The recently described structural flexibility of MMP-9 (54) may enhance the proposed interactions between the CAT and HPX-like domains and FN II inserts in the gelatinases.
MMP-9 CAT-FN was able to process a THP substrate and was inhibited by a THPI modeled after the consensus MMP cleavage site in types I–III collagen (Tables 2 and 4). We observed previously that MMP-1 CAT and MT1-MMP CAT can process the same THP substrate (23, 24), and MMP-1 CAT is inhibited by this THPI (Table 4). Thus, the isolated CAT domain of the collagenolytic MMPs can hydrolyze a triple-helical sequence modeled after the interstitial collagens. However, the same is not true for type V collagen model sequences. MMP-9 activity toward triple-helical type V collagen sequences is completely lost upon removal of the FN II insert (Tables 2 and 3); thus, the CAT domain alone cannot hydrolyze this sequence. This result is consistent with previously reported observations for MMP-9 hydrolysis of type V collagen, in that deletion of the FN II insert resulted in a complete loss of activity (48). Interestingly, incorporation of the MMP-9 FN II insert into the CAT domain of MMP-1 resulted in activity toward type V collagen (52).
The Effects of Triple-helical Structure on Activity—Neither the CAT domain nor the full-length MMP-9 display any preference for binding a triple-helical inhibitor compared with a denatured one. Similar KD values are observed for MMP-9 FN II insert binding to types I and V collagen, whereas the KD value for binding to gelatin is better than for collagen (18). MMP-2 FN II insert binds triple-helical peptides of different stability with the same affinity (21). The MMP-2 FN II inserts bind to native and denatured type I collagen, and the two substrates compete for binding to the same site (17). Thus, the FN II repeats recognize denatured collagen as readily as triple-helical collagen and use the same regions within the FN II inserts to bind both substrate types. However, for MMP-9 hydrolysis of α1(V)436–450, a triple-helical construct is preferred over single-stranded (27). In consideration of both of these results, for initial binding a native triple helix is not preferred over a denatured structure, but a triple-helical construct is preferred for presentation of individual strands to the active site relative to a monomeric form. We have previously described different preferences for peptide strands presented from a triple-helical construct versus monomer for MMP-13 and MT1-MMP (20). It is interesting to note that the lack of discrimination in binding native relative to denatured triple helices may facilitate the action of MMP-9 at sites of inflammation (54), where temperatures above 37 °C generate a mixture of native and denatured collagens.
Differences between MMP-2 and MMP-9—An intriguing difference has been observed for MMP-9 and MMP-2. MMP-2 was found to display preferential binding for one inhibitor in triple-helical conformation (apparent Ki values increased 4–6-fold upon melting), although MMP-9 does not (Table 3). The current results are consistent with a prior study of substrate analogs containing the Gly ↓ Val cleavage site (27). MMP-9 retained the ability to cleave the substrate when it was presented in nontriple-helical form, whereas MMP-2 did not. MMP-2 and MMP-9 have been classified as the gelatinase members of the MMP family and are regarded as possessing similar substrate specificities (55). However, there are subtle differences in the orientation of the FN II inserts in MMP-2 and MMP-9. For example, of the three contiguous FN II inserts, insert 2 has numerous points of interaction with the MMP-2 CAT domain, but, due to a different orientation, insert 2 has no interaction with the MMP-9 CAT domain (54, 56, 57). The FN II inserts in MMP-2 and MMP-9 might have evolved differently to give rise to divergent binding specificities (48). Although MMP-2 and MMP-9 share many collagenolytic and gelatinolytic activities, MMP-2 cleaves type II collagen although MMP-9 does not (8). Also, MMP-9 cleaves soluble but not fibrillar type I collagen, although MMP-2 cleaves both (8, 19, 58). Finally, screening of a phage display library identified substrates that were differentially cleaved by MMP-2 and MMP-9 (59). This study points out a subtle difference between the two enzymes in recognition of a type V collagen-derived sequence. Further delineation of such differences may be crucial in developing inhibitors that can effectively select between MMP-2 and MMP-9 (42).
Exosite Inhibition of Select Triple-helical Peptidase Activities—For some substrates and inhibitors, removal of the FN II repeats from MMP-9 abrogates activity; for others, activity is retained at lower levels. Based on the differences seen for triple-helical substrates and inhibitors modeled after the interstitial collagens and type V collagen, it appears possible to inhibit one activity (type V collagenolysis) but not the other (interstitial collagenolysis) via an exosite (FN II insert) ligand. The MMP-2 FN II insert was found to bind to two cyanogen bromide fragments of type I collagen, α1(I)-CB7 (residues 552–822) and α1(I)-CB8 (residues 124–402) (17). Using a combinatorial peptide library approach, the α1(I)715–721 sequence within α1(I)-CB7 was identified as a ligand for MMP-2 FN II insert, and this peptide inhibited MMP-2 gelatinolysis (49). We assembled a triple-helical version of α1(I)715–721 and found it inhibited type V collagen model triple-helical peptidase activity but not interstitial collagen model triple-helical peptidase activity. To our knowledge, this demonstrates the first use of an exosite binder to inhibit one collagen-based MMP activity but not another.
Our triple-helical peptides and inhibitors interact with the FN II insert in MMP-9, and presumably in MMP-2. One can consider where these interactions are taking place. FN II inserts have been proposed to bind collagen based on hydrophobic interactions (60). The MMP-9 FN II insert 2 was found to have much greater affinity for gelatin than inserts 1 and 3 (47). Residues identified as important for gelatin binding to MMP-9 FN II insert 2 were Arg307, Asp309, Trp313, Cys314, Asn319, Tyr320, and Asp323 (47). These residues are at or around a hydrophobic pocket (57). The distance between Arg307–Asp323 and the MMP-9 active site is 40–45 Å, which is 13–15 residues of a triple helix. The FN II insert would interact with residues on the prime side of the scissile bond or zinc-binding group (61). Our triple-helical peptides and inhibitors have 23 residues on the C-terminal side of the scissile bond or zinc-binding group, and thus should easily interact with the FN II insert 2. Based on the distance, the residues from the THP substrate and inhibitor interacting with the FN II insert would be within the Gly-Pro-Hyp repeating sequences. It is possible that once the α1(I)715–721 THP binds to the FN II insert and displaces interactions with the Gly-Pro-Hyp repeats, substrate interactions with other regions within the CAT domain modulate activity. Such behavior is consistent with the results observed for MMP-9 CAT-FN, where discrimination of type V and interstitial collagen sequences was observed. Alternatively, the α1(I)715–721 THP may bind elsewhere in the FN II insert and discriminate between type V and interstitial collagen sequences. Additional structural information is needed to determine the exact mode of action of α1(I)715–721 THP, especially considering that the orientation of the FN II insert may differ upon substrate binding.
Comparison between Gelatinases and Collagenases—The HPX-like domain of MMP-1 was important for efficient binding to the α1(I–III)GlyΨ{PO2H-CH2}Leu THPI, whereas it did not play a significant role in mediating MMP-9 binding. Apparent Ki values for this inhibitor in its triple-helical form increased ∼2–3-fold when the MMP-1 HPX-like domain was removed. Although full-length MMP-1 appears to bind preferentially to the triple helix (based on an ∼3.5-fold difference in Ki with unfolding), the CAT domain alone shows a more marked preference given the ∼4.5–8-fold increase in Ki after thermal unfolding. The MMP-1 CAT and HPX-like domains clearly cooperate in binding triple-helical structures, with the isolated CAT domain showing less affinity for denatured triple helices than the intact enzyme. Although a prior study demonstrated that both MMP-1 and MMP-1 CAT hydrolyzed denatured type I collagen (gelatin), individual kinetic parameters were not determined (62). However, MMP-1 exhibited lesser affinity (higher KM values) for type I gelatin compared with collagen (63). This interesting phenomenon further underscores the functional differences between classic collagenases (such as MMP-1) and gelatinases (such as MMP-9) and illustrates the importance of developing and utilizing exosite inhibitors with defined topologies.
Future Prospects for Triple-helical Peptide Inhibitors—Most inhibitor design uses hydrophobicity to generate binding energy, which creates a favorable ΔG but poor selectivity (64), and this is generally a feature of low molecular weight inhibitors (<800 Da) (65). It may be possible to generate selective inhibitors that are more polar by incorporating interactions that are more specific for MMP subclasses. Protein-protein recognition sites usually have larger contact areas (>1500 Å2), and thus developing larger inhibitors might allow for greater specificity. One could design inhibitors that bind to regions that, in a family of enzymes, have similar structures but differing mobility (65) or where unique exosites exist within the family. The drawback of this approach has typically been the flexibility of larger inhibitors. However, THP inhibitor design optimizes conformational entropy because of imino acid restriction of peptide backbone mobility. Specificity can then be generated by inhibitor binding to multiple sites on the enzyme (active site and exosites). This can lead to inhibitors that are highly selective between members of an enzyme family (26) or inhibitors that are selective between activities of specific enzymes, as exemplified here for the hydrolysis of type V versus interstitial collagen model sequences by MMP-2 and MMP-9.
Acknowledgments
We gratefully acknowledge Dr. Hideaki Nagase for supplying MMP-1, MMP-2, and MMP-3 and Harinathachari Bahudhanapati for construction of the MMP-9 CAT and MMP-9 CAT-FN plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grant CA 98799 (to G. B. F. and R. P. H.). This work was also supported by a Glenn/American Federation for Aging Research Scholarship (to J. L. L.-F.), an ASBMB travel award (to J. L. L.-F.), and National Science Foundation IGERT Fellowship CHE-9987603 (to J. K. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: MMP, matrix metalloproteinase; RP-HPLC, reverse phase-high performance liquid chromatography; CAT, catalytic; HPX, hemopexin; MALDI-TOF-MS, matrix-assisted laser desorption-ionization time-of-flight mass spectrometry; FN, fibronectin; Fmoc, N-(9-fluorenyl)methoxycarbonyl; APMA, 4-aminophenyl mercuric acetate; Flp, (2S,4R)-4-fluoroproline; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; Dnp, 2,4-dinitrophenyl; Mca, (7-methoxycoumarin-4-yl)acetyl; Hyp, 4-hydroxy-l-proline; THP, triple-helical peptide; THPI, THP inhibitor; fTHP, fluorogenic THP.
References
- 1.Song, F., Wisithphrom, K., Zhou, J., and Windsor, L. J. (2006) Front. Biosci. 11 3100–3120 [DOI] [PubMed] [Google Scholar]
- 2.Overall, C. M., and Lopez-Otin, C. (2002) Nat. Rev. Cancer 2 657–672 [DOI] [PubMed] [Google Scholar]
- 3.Egeblad, M., and Werb, Z. (2002) Nat. Rev. Cancer 2 161–174 [DOI] [PubMed] [Google Scholar]
- 4.Fingleton, B. (2007) Curr. Pharm. Des. 13 333–346 [DOI] [PubMed] [Google Scholar]
- 5.McCawley, L. J., and Matrisian, L. M. (2001) Curr. Opin. Cell Biol. 13 534–540 [DOI] [PubMed] [Google Scholar]
- 6.Overall, C. M. (2002) Mol. Biotechnol. 22 51–86 [DOI] [PubMed] [Google Scholar]
- 7.Morrison, C. J., and Overall, C. M. (2006) J. Biol. Chem. 281 26528–26539 [DOI] [PubMed] [Google Scholar]
- 8.Bigg, H. F., Rowan, A. D., Barker, M. D., and Cawston, T. E. (2007) FEBS J. 274 1246–1255 [DOI] [PubMed] [Google Scholar]
- 9.Clark, I. N., and Cawston, T. E. (1989) Biochem. J. 263 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knäuper, V., Cowell, S., Smith, B., Lopez-Otin, C., O'Shea, M., Morris, H., Zardi, L., and Murphy, G. (1997) J. Biol. Chem. 272 7608–7616 [DOI] [PubMed] [Google Scholar]
- 11.Knäuper, V., Osthues, A., DeClerk, Y. A., Langley, K. E., Bläser, J., and Tschesche, H. (1993) Biochem. J. 291 847–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy, G., Allan, J. A., Willenbrock, F., Cockett, M. I., O'Connell, J. P., and Docherty, A. J. P. (1992) J. Biol. Chem. 267 9612–9618 [PubMed] [Google Scholar]
- 13.Ohuchi, E., Imai, K., Fujii, Y., Sato, H., Seiki, M., and Okada, Y. (1997) J. Biol. Chem. 272 2446–2451 [DOI] [PubMed] [Google Scholar]
- 14.Hurst, D. R., Schwartz, M. A., Ghaffari, M. A., Jin, Y., Tschesche, H., Fields, G. B., and Sang, Q.-X. A. (2004) Biochem. J. 377 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Souza, S. J., Pereira, H. M., Jacchieri, S., and Brentani, R. R. (1996) FASEB J. 10 927–930 [DOI] [PubMed] [Google Scholar]
- 16.Iyer, S., Visse, R., Nagase, H., and Acharya, K. R. (2006) J. Mol. Biol. 362 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steffensen, B., Wallon, U. M., and Overall, C. M. (1995) J. Biol. Chem. 270 11555–11566 [DOI] [PubMed] [Google Scholar]
- 18.Xu, X., Chen, Z., Wang, Y., Yamada, Y., and Steffensen, B. (2005) Biochem. J. 392 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patterson, M. L., Atkinson, S. J., Knäuper, V., and Murphy, G. (2001) FEBS Lett. 503 158–162 [DOI] [PubMed] [Google Scholar]
- 20.Minond, D., Lauer-Fields, J. L., Cudic, M., Overall, C. M., Pei, D., Brew, K., Moss, M. L., and Fields, G. B. (2007) Biochemistry 46 3724–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ottl, J., Gabriel, D., Murphy, G., Knäuper, V., Tominaga, Y., Nagase, H., Kröger, M., Tschesche, H., Bode, W., and Moroder, L. (2000) Chem. Biol. 7 119–132 [DOI] [PubMed] [Google Scholar]
- 22.Lauer-Fields, J. L., Tuzinski, K. A., Shimokawa, K., Nagase, H., and Fields, G. B. (2000) J. Biol. Chem. 275 13282–13290 [DOI] [PubMed] [Google Scholar]
- 23.Lauer-Fields, J. L., Broder, T., Sritharan, T., Nagase, H., and Fields, G. B. (2001) Biochemistry 40 5795–5803 [DOI] [PubMed] [Google Scholar]
- 24.Minond, D., Lauer-Fields, J. L., Nagase, H., and Fields, G. B. (2004) Biochemistry 43 11474–11481 [DOI] [PubMed] [Google Scholar]
- 25.Minond, D., Lauer-Fields, J. L., Cudic, M., Overall, C. M., Pei, D., Brew, K., Visse, R., Nagase, H., and Fields, G. B. (2006) J. Biol. Chem. 281 38302–38313 [DOI] [PubMed] [Google Scholar]
- 26.Lauer-Fields, J. L., Brew, K., Whitehead, J. K., Li, S., Hammer, R. P., and Fields, G. B. (2007) J. Am. Chem. Soc. 129 10408–10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauer-Fields, J. L., Sritharan, T., Stack, M. S., Nagase, H., and Fields, G. B. (2003) J. Biol. Chem. 278 18140–18145 [DOI] [PubMed] [Google Scholar]
- 28.Nagase, H., Fields, C. G., and Fields, G. B. (1994) J. Biol. Chem. 269 20952–20957 [PubMed] [Google Scholar]
- 29.Neumann, U., Kubota, H., Frei, K., Ganu, V., and Leppert, D. (2004) Anal. Biochem. 328 166–173 [DOI] [PubMed] [Google Scholar]
- 30.Lauer-Fields, J. L., Minond, D., Chase, P. S., Baillargeon, P. E., Saldanha, S. A., Stawikowska, R., Hodder, P., and Fields, G. B. (2008) Bioorg. Med. Chem., in press [DOI] [PMC free article] [PubMed]
- 31.Yu, Y.-C., Berndt, P., Tirrell, M., and Fields, G. B. (1996) J. Am. Chem. Soc. 118 12515–12520 [Google Scholar]
- 32.Yu, Y.-C., Tirrell, M., and Fields, G. B. (1998) J. Am. Chem. Soc. 120 9979–9987 [Google Scholar]
- 33.Malkar, N. B., Lauer-Fields, J. L., Borgia, J. A., and Fields, G. B. (2002) Biochemistry 41 6054–6064 [DOI] [PubMed] [Google Scholar]
- 34.Malkar, N. B., Lauer-Fields, J. L., Juska, D., and Fields, G. B. (2003) Biomacromolecules 4 518–528 [DOI] [PubMed] [Google Scholar]
- 35.Beck, K., Chan, V. C., Shenoy, N., Kirkpatrick, A., Ramshaw, J. A. M., and Brodsky, B. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 4273–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Persikov, A. V., Ramshaw, J. A. M., and Brodsky, B. (2000) Biopolymers 55 436–450 [DOI] [PubMed] [Google Scholar]
- 37.Persikov, A. V., Ramshaw, J. A. M., Kirkpatrick, A., and Brodsky, B. (2000) Biochemistry 39 14960–14967 [DOI] [PubMed] [Google Scholar]
- 38.Persikov, A. V., Xu, Y., and Brodsky, B. (2004) Protein Sci. 13 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung, L., Shimokawa, K., Dinakarpandian, D., Grams, F., Fields, G. B., and Nagase, H. (2000) J. Biol. Chem. 275 29610–29617 [DOI] [PubMed] [Google Scholar]
- 40.Lauer-Fields, J. L., Nagase, H., and Fields, G. B. (2004) J. Biomol. Techniques 15 305–316 [PMC free article] [PubMed] [Google Scholar]
- 41.Itoh, Y., Binner, S., and Nagase, H. (1995) Biochem. J. 308 645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamze, A. B., Wei, S., Bahudhanapati, H., Kota, S., Acharya, K. R., and Brew, K. (2007) Protein Sci. 16 1905–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki, K., Kan, C.-C., Huang, W., Gehring, M. R., Brew, K., and Nagase, H. (1998) Biol. Chem. 379 185–191 [DOI] [PubMed] [Google Scholar]
- 44.Lauer-Fields, J. L., and Fields, G. B. (2002) Biol. Chem. 383 1095–1105 [DOI] [PubMed] [Google Scholar]
- 45.Holmgren, S. K., Taylor, K. M., Bretscher, L. E., and Raines, R. T. (1998) Nature 392 666–667 [DOI] [PubMed] [Google Scholar]
- 46.Holmgren, S. K., Bretscher, L. E., Taylor, K. M., and Raines, R. T. (1999) Chem. Biol. 6 63–70 [DOI] [PubMed] [Google Scholar]
- 47.Collier, I. E., Krasnov, P. A., Strongin, A. Y., Birkedal-Hansen, H., and Goldberg, G. I. (1992) J. Biol. Chem. 267 6776–6781 [PubMed] [Google Scholar]
- 48.O'Farrell, T. J., and Pourmotabbed, T. (1998) Arch. Biochem. Biophys. 354 24–30 [DOI] [PubMed] [Google Scholar]
- 49.Xu, X., Chen, Z., Wang, Y., Bonewald, L., and Steffensen, B. (2007) Biochem. J. 406 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tam, E. M., Moore, T. R., Butler, G. S., and Overall, C. M. (2004) J. Biol. Chem. 279 43336–43344 [DOI] [PubMed] [Google Scholar]
- 51.Giola, M., Monaco, S., Fasciglione, G. F., Coletti, A., Modesti, A., Marini, S., and Coletta, M. (2007) J. Mol. Biol. 368 1101–1113 [DOI] [PubMed] [Google Scholar]
- 52.O'Farrell, T. J., and Pourmotabbed, T. (2000) J. Biol. Chem. 275 27964–27972 [DOI] [PubMed] [Google Scholar]
- 53.Falconi, M., Altobelli, G., Iovino, M. C., Politi, V., and Desideri, A. (2003) J. Comp. Aided Mol. Des. 17 837–848 [DOI] [PubMed] [Google Scholar]
- 54.Rosenblum, G., Van den Steen, P. E., Cohen, S. R., Grossmann, J. G., Frenkel, J., Sertchook, R., Slack, N., Strange, R. W., Opdenakker, G., and Sagi, I. (2007) Structure (Lond.) 15 1227–1236 [DOI] [PubMed] [Google Scholar]
- 55.Woessner, J. F., and Nagase, H. (2000) Matrix Metalloproteinases and TIMPs, Oxford University Press, Oxford
- 56.Morgunova, E., Tuuttila, A., Bergmann, U., Isupov, M., Lindqvist, Y., Schneider, G., and Tryggvason, K. (1999) Science 284 1667–1670 [DOI] [PubMed] [Google Scholar]
- 57.Elkins, P. A., Ho, S. H., Smith, W. W., Janson, C. A., D'Alessio, K. J., McQueney, M. S., Cummings, M. D., and Romanic, A. M. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 1182–1192 [DOI] [PubMed] [Google Scholar]
- 58.Aimes, R. T., and Quigley, J. P. (1995) J. Biol. Chem. 270 5872–5876 [DOI] [PubMed] [Google Scholar]
- 59.Chen, E. I., Kridel, S. J., Howard, E. W., Li, W., Godzik, A., and Smith, J. W. (2002) J. Biol. Chem. 277 4485–4491 [DOI] [PubMed] [Google Scholar]
- 60.Pickford, A. R., Potts, J. R., Bright, J. R., Phan, I., and Campbell, I. D. (1996) Structure (Lond.) 5 359–370 [DOI] [PubMed] [Google Scholar]
- 61.Overall, C. M., and Butler, G. S. (2007) Structure (Lond.) 15 1159–1161 [DOI] [PubMed] [Google Scholar]
- 62.Chung, L., Dinakarpandian, D., Yoshida, N., Lauer-Fields, J. L., Fields, G. B., Visse, R., and Nagase, H. (2004) EMBO J. 23 3020–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fields, G. B. (1991) J. Theor. Biol. 153 585–602 [DOI] [PubMed] [Google Scholar]
- 64.Rubin, A. J., Kiso, Y., and Freire, E. (2006) Chem. Biol. Drug Des. 67 2–4 [DOI] [PubMed] [Google Scholar]
- 65.Luque, I., and Freire, E. (2000) Proteins Struct. Funct. Genet. 4 63–7111013401 [Google Scholar]
- 66.O'Hare, M. C., Curry, V. A., Mitchell, R. E., and Cawston, T. E. (1995) Biochem. Biophys. Res. Commun. 216 329–337 [DOI] [PubMed] [Google Scholar]