

Abstract
The proliferation of vascular smooth muscle cells is important in the pathogenesis of many vascular diseases. Reactive oxygen species (ROS) produced by NADPH oxidases in smooth muscle cells have been shown to participate in signaling cascades regulating proliferation induced by platelet-derived growth factor (PDGF), a powerful smooth muscle mitogen. We sought to determine the role of Nox5 in the regulation of PDGF-stimulated human aortic smooth muscle cell (HASMC) proliferation. Cultured HASMC were found to express four isoforms of Nox5. When HASMC stimulated with PDGF were pretreated with N-acetyl cysteine (NAC), proliferation was significantly reduced. Proliferation induced by PDGF was also heavily dependent on JAK/STAT activation, as the JAK inhibitor, AG490, was able to completely abolish PDGF-stimulated HASMC growth. Specific knockdown of Nox5 with a siRNA strategy reduced PDGF-induced HASMC ROS production and proliferation. Additionally, siRNA to Nox5 inhibited PDGF-stimulated JAK2 and STAT3 phosphorylation. ROS produced by Nox5 play an important role in PDGF-induced JAK/STAT activation and HASMC proliferation.
Keywords: NADPH oxidase, reactive oxygen species, Nox5, vascular smooth muscle cells, proliferation
In atherosclerosis and restenosis after percutaneous coronary intervention, cytokines elaborated by both vascular cells and cells invading the vessel wall induce vascular smooth muscle proliferation and contribute to lesion formation. In the normal vessel wall, smooth muscle cells (SMCs) maintain and regulate vascular tone. However, VSMCs change their phenotype to a synthetic, proliferative state when stimulated by a number of different growth factors.
One such growth factor, platelet-derived growth factor (PDGF), is expressed by all vascular cell types [1, 2] and by invading inflammatory cells, such as monocytes and lymphocytes, in atherosclerosis [1]. PDGF has long been recognized as a powerful VSMC mitogen [3] and the induction of PDGF receptors in VSMCs during atherogenesis has been demonstrated in several studies [4, 5]. Blockade of PDGF, whether by anti-PDGF antibody [6–8], PDGF receptor antisense therapy [9, 10], or chimeric knockout in mice [11], reduces lesion formation after vascular injury. Ligand binding to the PDGF receptor causes tyrosine autophosphorylation and the initiation of specific signaling pathways that have been linked to growth. Calcium appears to be one of the obligate mediators of PDGF-stimulated mitogenesis, as several studies have reported that blockade of the increase in intracellular calcium caused by PDGF inhibits VSMC proliferation [12–14]. Of particular interest, overexpression of the antioxidant enzyme catalase in VSMCs also reduces PDGF-stimulated growth, indicating that reactive oxygen species (ROS) may regulate proliferation as well [15, 16]. The mechanisms by which these two second messengers regulate growth are incompletely understood. One pathway that has been suggested to mediate the growth response is the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway. This pathway has been implicated in a host of cellular processes such as migration, apoptosis, and proliferation [17], and importantly, in fibroblasts [18] and human pulmonary artery SMCs [19], its activation by PDGF is modulated by ROS.
ROS have long been known as the agents responsible for the antimicrobial properties of phagocytic cells. However, the discovery that non-phagocytic cells also produce ROS, albeit at low concentrations, has expanded their role to include cell signaling. The source of ROS in phagocytes is the NADPH oxidase (Nox), an enzyme complex composed of membrane-bound and cytosolic subunits. Recently, homologues of the membrane-bound catalytic subunit of the NADPH oxidase have been discovered in several different cell types. The ROS produced by these homologues in VSMCs regulate signaling cascades that are activated by cytokines, hormones, and thrombin.[20] Currently, the Nox family is composed of Nox1, Nox2 (the phagocytic NADPH oxidase), Nox3, Nox4, and Nox5, as well as the Duox enzymes [21]. These enzymes appear to have different physiological roles. For example, Nox1 is responsible for angiotensin II- and PDGF-stimulated ROS-production in rat aortic VSMCs [22], while Nox4 has been shown to be important for the maintenance of VSMC differentiation [23]. Most of the work defining the function of the NADPH oxidases in the vasculature has been performed in rodents, but human VSMCs also express Nox5 [24], a homologue unique to humans. However, the exact role of Nox5 in human VSMCs has not been defined.
With regard to PDGF signaling, Nox5 is of particular interest because it is activated by calcium and produces ROS, both of which have been shown to be required for PDGF-induced proliferation. Nox5 has been detected in several cancer cell lines [25, 26] and, in a study with prostate cancer cells, appears to regulate cell growth [27]. Furthermore, the knockdown of Nox5 in human endothelial cells prevents proliferation [28]. Based on these observations, we hypothesized that Nox5 participates in PDGF-directed human VSMC proliferation via activation of the JAK/STAT pathway. Our results support this hypothesis and suggest that Nox5 may be an important regulator of a number of vascular pathologies in human disease.
Materials and Methods
Materials
PDGF-BB was purchased from R&D Systems, Inc. (Minneapolis, MN), and N-acetylcysteine (NAC) was from Sigma-Aldrich (St. Louis, MO). Non-silencing siRNA and siRNA to Nox5 (siNox5, which targets all Nox5 isoforms) were purchased from Ambion (Austin, TX). Phospho-JAK2 (Tyr1007/Tyr1008) antibody was from Santa Cruz Biotechnologies (Santa Cruz, CA), phospho-STAT3 (Tyr705) and STAT3 antibodies were from Cell Signaling Technology, Inc (Danvers, MA), and anti-JAK2 antibody was from Millipore (Billerica, MA). AG490 and LY29004 were purchased from Calbiochem Bio-Mol (La Jolla, CA). Diphenylene iodonium (DPI) was from Alexis Biochemicals (San Diego, CA), and dihydroethidium (DHE) and BAPTA-AM were from Invitrogen (Carlsbad, CA).
Cell culture
Human aortic SMCs (HASMCs) were purchased from Cambrex (East Rutherford, NJ) and maintained in smooth muscle growth medium (SmGM: smooth muscle basal medium (SmBM) with addition of 5% FBS, 5 μg/mL insulin, 0.5 ng/mL EGF, 2 ng/mL FGF-B, gentamicin, and amphotericin B) per the company’s instructions. Cells at passages 6 to 12 were used for the experiments.
Transfection
HASMCs were plated at 30% confluence, then transfected with 20 nmol/L siNox5 using oligofectamine (Invitrogen, Carlsbad, CA) as the transfection reagent in OPTI-MEM (Invitrogen). Four hours after transfection, the medium was changed to SmGM, and then to SmBM after 24 h. For the growth curve experiments, PDGF (25 ng/ml) was added to SmBM 4 h after transfection and the medium was changed daily thereafter. For the signaling experiments, the cells were left in SmBM for 2 days prior to use. For some experiments, the cells were transfected using nucleofector technology for siRNA delivery from Amaxa Biotechnologies (Gaithersburg, MD). The cells were transfected according to the manufacturer’s protocol for nucleofection of HASMCs.
Cloning
Total RNA was extracted from HASMCs with RNeasy kit (Qiagen, Valencia, CA) per the manufacturer’s instructions. Reverse transcription was performed with Superscript III (Invitrogen, Carlsbad, CA) oligo dT or a Nox5-specific primer (5′-GAGCTTGGAGAGGTGAGGCTAG -3′) at 55°C. PCR using forward primers for Nox5β and δ (5′-GAGCTTGGAGAGGTGAGGCTAG-3′) or Nox5α and γ (5′-GAGCTTGGAGAGGTGAGGCTAG-3′) was performed with 1x PCR Enhancer (Epicentre, Madison, WI) and Platinum Taq HiFi (Invitrogen, Carlsbad, CA). Products were verified by sequencing.
Quantitative real-time PCR
Total RNA was extracted as above. Superscipt II (Invitrogen) and random primers were used for reverse transcription. Quantitative real-time PCR was performed with a LightCycler (Roche) real time thermocycler with primers for Nox5 (detecting all isoforms), Nox4, Nox1, and gp91phox [29]. Primers for 18S (Classic II 18S alternate internal standards) were purchased from Ambion (Austin, TX). Results are expressed as copy number per 106 copies of 18S.
Cell counting
HASMCs were plated at 14,600 cells/well in a 24 well dish. After the appropriate treatment(s) and on the indicated day, cells were washed with Hank’s Balanced Salt Solution (Gibco), trypsinized with trypsin/EDTA (Gibco), diluted in Isoton II (Coulter) and counted with a Z1 Coulter counter.
Dichlorofluorescein-DA assay for ROS
Cells were plated at 30% confluence in 24 well plates and transfected with 20 nmol/L non-silencing siRNA (NSsiRNA) or 20 nmol/L siNox5. After overnight incubation in SmGM, the medium was changed to SmBM + 25 ng/mL PDGF-BB and then changed daily thereafter. At day four after transfection, cells were incubated with 5 μmol/L DCF-DA (Molecular Probes) for 1 hour. Cells were washed twice with colorless HBSS and then visualized with confocal microscopy.
2-Hydroxyethidium assay for superoxide
Cells were plated at 30% confluence in 60-mm dishes and transfected with 20 nmol/L NSsiRNA or siNox5. After overnight incubation in SmGM, the medium was changed to SmBM + 25 ng/mL PDGF-BB and changed daily thereafter. Three days after transfection, polyethylene glycolated-(PEG) superoxide dismutase (SOD) was added to half the samples to a final concentration of 50 U/mL. The following day, the cells were washed twice with chilled Krebs/HEPES buffer and incubated with 1 μmol/L DHE at 37°C for 20 min, and then scraped into amber microfuge tubes containing 300 μL methanol. Samples were stored at −80°C until assayed by high performance liquid chromatography as previously described [30] Results are expressed as the SOD-inhibitable signal.
Western Blotting
HASMCs at 70–90% confluence were made quiescent by incubation with SmBM containing 0.1% FBS for 24 to 48 hours. Cells were stimulated with PDGF (25 ng/mL) at 37°C in serum-free SmBM for specified durations. After treatment, cells were washed three times with phosphate buffered saline (PBS) and lysed with ice-cold lysis buffer containing 20 mmol/L Tris-HCl at pH 7.5, 1% Triton-X, 1% Glycerol, 0.1%SDS, 1% Na-deoxycholate and 2.5 mmol/L EDTA. The lysis buffer was supplemented with Na-orthovanadate, Na-pyrophosphate, NaF, aprotinin, leupeptin and PMSF. Western analysis was performed with SDS-PAGE and subsequent transfer to nitrocellulose membranes. Incubation with the primary antibody was then carried out and detection was achieved with ECL. ImageJ software was used to analyze band intensities.
Statistical Analysis
Results are expressed as mean ± standard error of mean (SEM). For two group analyses, the unpaired t-test was used. One-way ANOVA was used to analyze data comparing three or more groups with Bonferroni’s multiple comparison test for post hoc analysis (GraphPad Prism software). A value of P<0.05 was considered statistically significant.
Results
HASMCs predominantly express Nox4 and all isoforms of Nox5
Real time PCR with primers to Nox family members revealed that proliferating HASMCs express Nox4 and Nox5, with Nox4 mRNA more abundant than Nox5 mRNA (23,895 ± 7,150 vs. 395 ± 56 copies per 108 18S). Using primers to Nox1 and Nox2, we were able to measure copy numbers as low as 10 per reaction, but detected minimal Nox1 and no Nox2 mRNA in HASMCs. Five variants of Nox5 have been reported: α, β, γ, δ, and a short form that is missing exon 6 [24, 31, 32]. We used reverse transcriptase PCR using primers specific to the α/γ or β/δ isoforms to amplify products that were then cloned and sequenced. The sequencing of several clones revealed that HASMCs express all four isoforms (data not shown). The presence of the short form of Nox5 was not investigated. Because of the difference in the relative expression levels of Nox1, 2, 4, and 5, and the data demonstrating the importance of Nox4 in differentiation [23] and Nox5 in human cell growth [27, 28], we concentrated on examining the role of Nox5 in HASMC cell proliferation.
PDGF-directed HASMC proliferation is ROS-dependent
A previous study revealed that HASMC growth induced by 10% FBS is arrested by the addition of the antioxidant NAC [33]. We first determined if HASMC growth in response to PDGF is also ROS sensitive, as previous studies have only demonstrated this property in other cell types or in rat VSMCs [15, 16]. As demonstrated in Figure 1, PDGF strongly stimulates HASMC proliferation. NAC alone slightly decreases cell number in the absence of PDGF, but inhibits PDGF-induced HASMC proliferation by 41 ±3%. This finding indicates that ROS are important mediators of PDGF-induced HASMC proliferation.
Fig 1. PDGF-induced HASMC growth is ROS-sensitive.
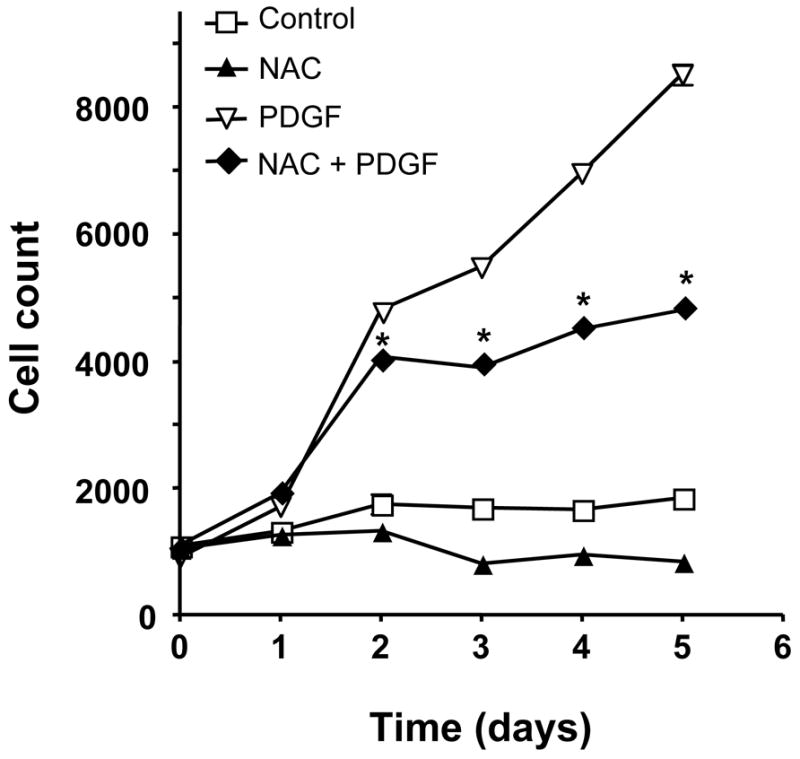
HASMCs were treated with 10 mmol/L NAC, 25 ng/mL PDGF-BB or both. Cells were counted at specified time points. A control group was maintained in SmBM + 0.1% FBS. Data are the mean ± SEM of 3 experiments. *P<0.001 compared to PDGF alone. Some error bars are smaller than the symbols.
Nox5 produces ROS in HASMCs in response to PDGF stimulation
The previous experiments indicate that ROS are required for proliferation, but do not provide insight into the source of ROS production. We hypothesized that the calcium-activated homologue Nox5 may mediate PDGF-induced ROS formation. To determine if Nox5 participates in PDGF-induced ROS production, we first tested its calcium sensitivity. As shown in Figure 2, pretreatment of HASMC with the intracellular calcium chelator BAPTA-AM (30 min, 10 μmol/L) essentially abolished superoxide production induced by PDGF (20 ng/mL). We next used siRNA to specifically downregulate all isoforms of Nox5 (Figure 3A), and used two assays to determine ROS production. The first was a qualitative measurement by DCF-DA staining. Figure 3B reveals that ROS production in cells treated with NSsiRNA or siNox5 alone is similar to that of control cells. However, cells treated with siNox5 failed to increase ROS production when treated with PDGF, while PDGF was able to increase ROS in cells treated with NSsiRNA. Figure 3C shows a quantitative decrease in superoxide production by 45 ±14% as measured by hydroxyethidium in cells treated with siNox5 and PDGF compared to cells treated with NSsiRNA and PDGF. In contrast, siNox4, under conditions that we have previously shown to be effective [23], had no effect on PDGF-induced superoxide production (Figure 3D). These findings demonstrate that Nox5 is a significant source of ROS in PDGF-stimulated HASMC.
Figure 2. PDGF-induced ROS formation is calcium-sensitive.
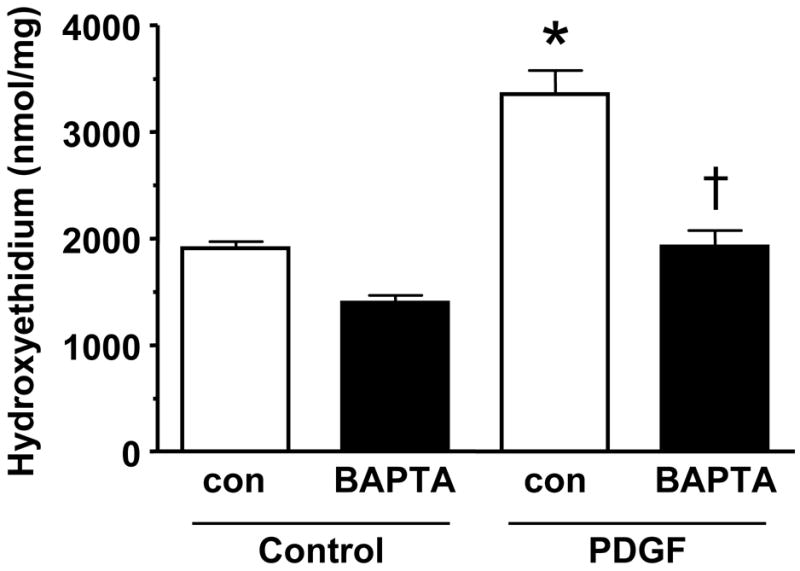
HASMCs were untreated or treated with 10 μmol/L BAPTA-AM for 30 min prior to addition of 20 ng/mL PDGF for 5 hours. Superoxide was measured using DH, followed by high performance liquid chromatography Data are the mean ± SEM of 4 experiments. *p<0.02 compared to PDGF alone, †p<0.05 PDGF vs. PDGF + BAPTA.
Figure 3. siNox5 downregulates Nox5 and inhibits PDGF-induced ROS production.
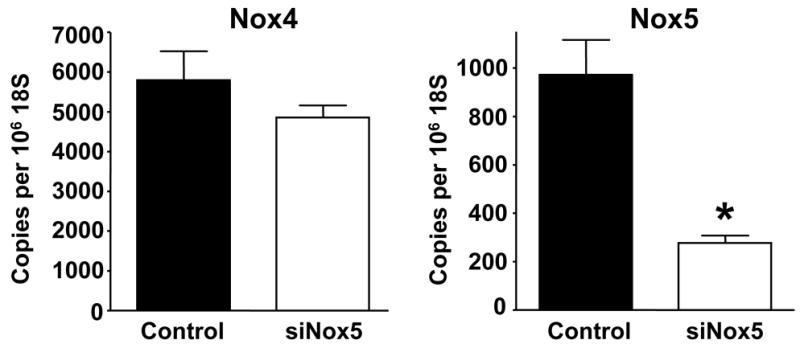
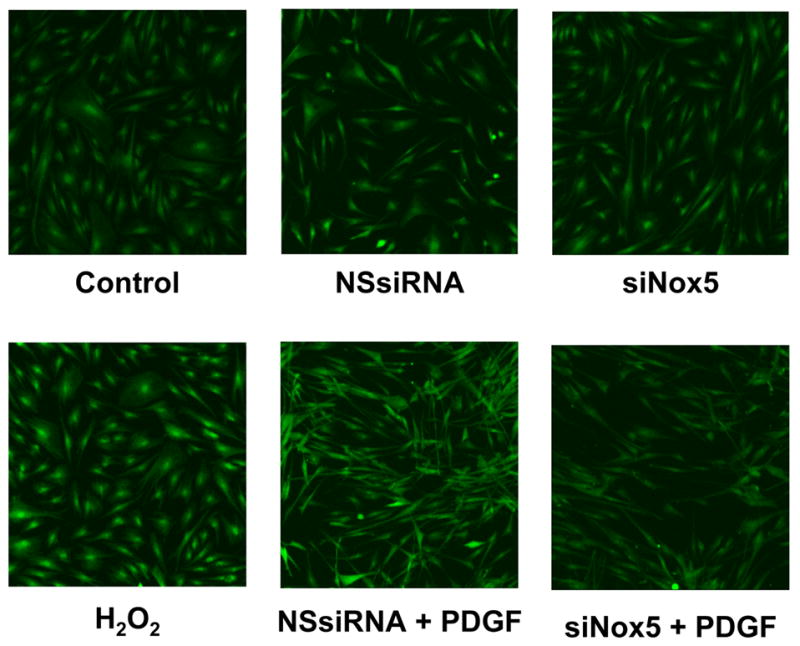
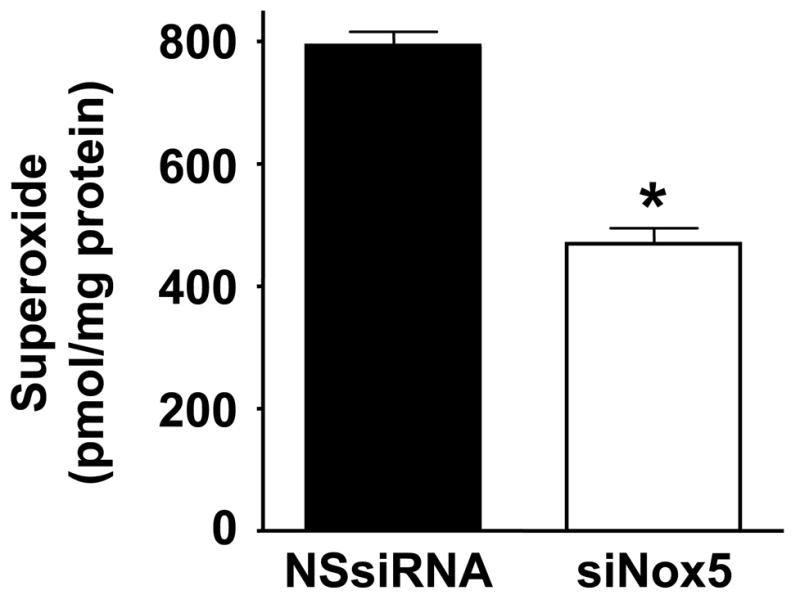
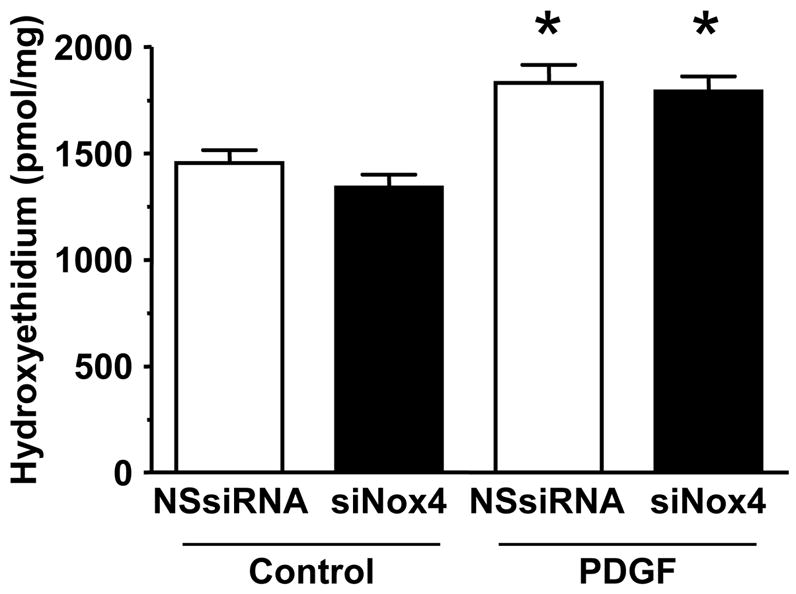
A. HASMCs were transfected with 20 nmol/L siNox5 and Nox4 and Nox5 mRNA were measured by quantitative real-time PCR. Data are expressed as copies per 106 copies of 18S cDNA and are the mean±SEM of 3 independent experiments. *p=0.008. B. HASMCs were not transfected, or transfected with non-silencing siRNA (NSsiRNA) or siNox5 (both at 20 nmol/L), and then treated with 25 ng/mL PDGF for 4 hours. DCF-DA staining for ROS was visualized with confocal microscopy. The response to H2O2 (100 μmol/L) is shown for comparison. Results are representative of 3 independent experiments. C. Superoxide measurement using DH, followed by high performance liquid chromatography was performed on HASMCs transfected with NSsiRNA or siNox5 (20 nmol/L) and stimulated with 25 ng/mL PDGF for 4 hours. Data are representative of 6 independent experiments and are expressed as mean±SEM. * indicates P=0.007. D. HASMCs were treated with NSsiRNA or siNox4 (25 nmol/L) and stimulated with 20 ng/mL PDGF for 5 hours prior to measurement of superoxide. Data are the mean of 6 independent experiments and are expressed as mean±SEM. *p=0.02 of PDGF vs. appropriate control.
Nox5 plays a role in PDGF-stimulated HASMC growth
To determine whether Nox5 participates in PDGF-induced HASMC proliferation, we used cells in which Nox5 was downregulated with siRNA, treated them with PDGF-BB, and counted them on a Coulter counter at predetermined time points. Viability of these cells was unaffected by PDGF treatment. As shown in Figure 4, non-transfected control cells and cells treated with the transfection reagent, oligofectamine, grow at similar rates. At day 5, cells treated with PDGF are increased in number by 134±5% when compared to untreated cells. The NSsiRNA does have a small effect on cell growth at day 5, because cell number increased by 91±4% under these conditions. However, on day 5 the cells treated with siNox5 were only able to increase their number by 31±6% when compared to the control cells. This result demonstrates that Nox5 is an important participant in PDGF-directed HASMC growth.
Fig 4. Nox5 regulates PDGF-induced HASM proliferation.

Cells were plated at 30% confluence, transfected with 20 nmol/L NSsiRNA or siNox5, and treated with or without 25 ng/mL PDGF-BB. At the indicated times, cells were trypsinized and counted with a Coulter counter. Data are mean±SEM of 3 separate experiments performed in triplicate. *P<0.001 compared to NSsiRNA. Some error bars are smaller than the symbols. Oligof= oligofectamine.
The JAK/STAT pathway mediates PDGF-induced HASMC proliferation
We next sought to identify effectors that depend on Nox5 for their activation and mediate PDGF-induced growth. One candidate is the JAK/STAT pathway, which has been shown to be ROS-sensitive in several different cell types including fibroblasts [18] and pulmonary artery SMCs [19]. As seen in Figure 5, pretreatment with the JAK inhibitor AG490 (10 μmol/L) completely inhibits HASMC proliferation in response to PDGF stimulation.
Fig 5. PDGF-induced HASMC proliferation is inhibited by the JAK/STAT inhibitor AG490.
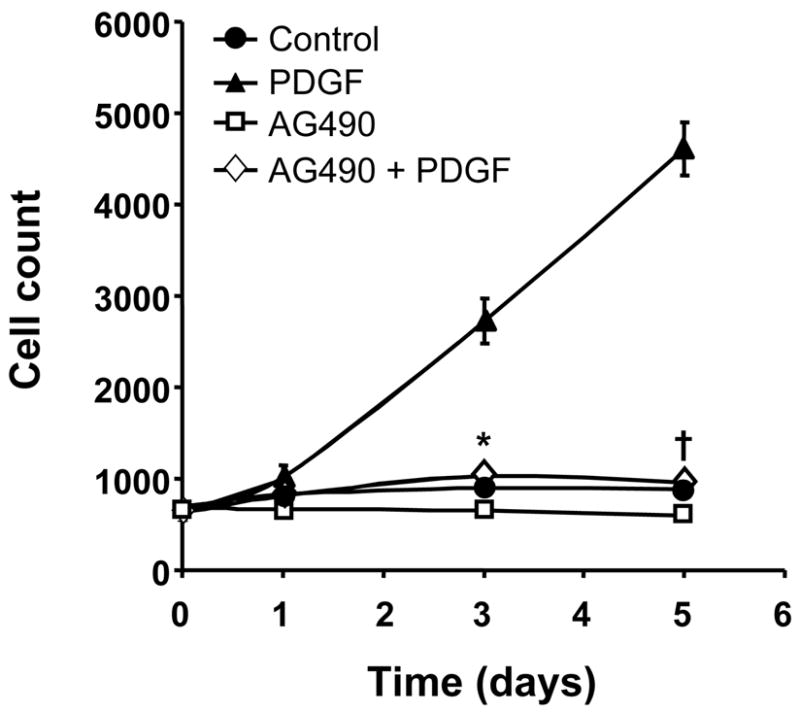
HASMCs were made quiescent, preincubated with AG490 (10 μmol/L) overnight and then stimulated with PDGF (25ng/ml). The cells were counted at specific time points. Values are mean ±SEM of 3 independent experiments performed in triplicate. Some error bars are smaller than the symbols. *P=0.002, †P<0.001 compared to cells treated with PDGF.
PDGF activation of JAK is flavin-containing enzyme-dependent in HASMC
To verify that activation of JAK/STAT in HASMC is ROS sensitive and that AG490 effectively inhibits JAK, we pretreated cells with either AG490 or the flavin-containing enzyme inhibitor DPI and then stimulated them with PDGF. Figure 6A demonstrates the prevention of PDGF-induced JAK2 phosphorylation with AG490 or DPI. The phosphorylation of STAT3 also appears to be ROS-sensitive (Figure 6B), but is not completely abolished with DPI treatment, suggesting that there may be another non-Nox-dependent, PDGF-activated JAK that can induce its phosphorylation.
Fig 6. PDGF-induced JAK2 and STAT3 phosphorylation is modulated by ROS and flavin-containing enzymes.
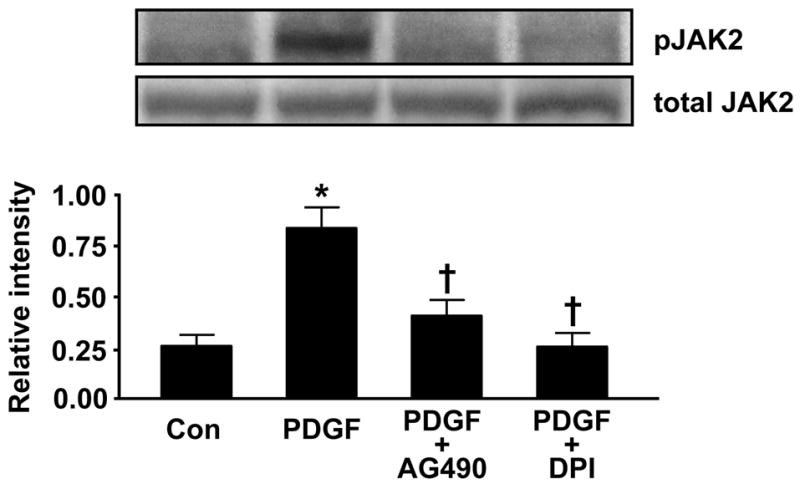
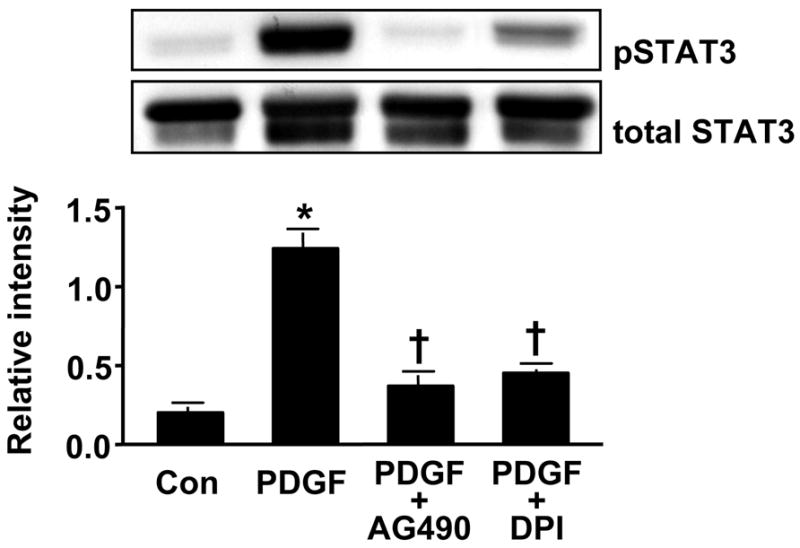
HASMCs were pretreated with the JAK inhibitor AG490 (100 μmol/L, 1h) or with the flavin-containing enzyme inhibitor DPI (10 μmol/L, 30min) before stimulation with PDGF (25 ng/mL) for 5 min. Phosphorylated JAK2 (A) or STAT3 (B) was analyzed by Western analysis. Total JAK2 or STAT3 was used as a loading control. Values are mean±SEM of 5 to 8 independent experiments. (A) * P<0.05 vs. unstimulated cells, † P<0.05 vs. cells treated with PDGF. (B) *P<0.001 vs. non-stimulated cells, † P<0.001 vs. cells stimulated with PDGF.
Nox5 is required for PDGF-induced JAK/STAT activation
To investigate the participation of Nox5 in PDGF-stimulated JAK/STAT phosphorylation, we transfected HASMCs with siNox5 and NSsiRNA then stimulated them with PDGF. As shown in Figure 7A, the transfection of HASMCs with siNox5, but not NSsiRNA, was able to significantly decrease Jak2 phosphorylation. Figure 7B demonstrates that the phosphorylation of STAT3 is also prevented by the knockdown of Nox5. These data demonstrate the importance of Nox5 for PDGF-induced JAK/STAT activation.
Fig 7. PDGF-induced JAK/STAT activation is mediated by Nox5.

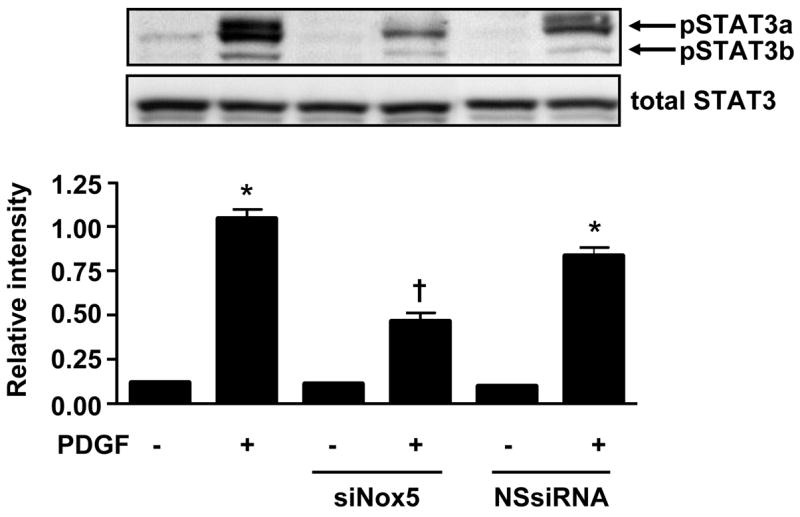
HASMCs were transfected with siNox5 (20 nmol/L) or NSsiRNA and 72h later stimulated with PDGF (25ng/mL) for 5min. Western blotting was performed using primary antibody to phosphoJAK2 (A) or phosphoSTAT3 (B). Total JAK or STAT3 was used as loading control. Values are mean ±SEM of 5 independent experiments performed in duplicate. (A) * P<0.001 vs. unstimulated cells with the indicated intervention, † P<0.001 vs. cells transfected with NSsiRNA and stimulated with PDGF. (B) * P<0.001 vs. unstimulated cells with the indicated intervention, † P<0.001 vs. cells transfected with NSsiRNA and stimulated with PDGF.
Discussion
The NADPH oxidases have emerged as important elements of signaling pathways in VSMCs. There is ample evidence that more than one Nox exists in this cell type and that the different homologues are located in different compartments [34] and serve different functions. In rat VSMCs, Nox1 appears to be required for angiotensin II- and PDGF-induced ROS production and growth [22], and Nox4 has been shown to be important for the maintenance of a contractile, non-proliferative phenotype [23]. However, human VSMCs appear to possess a different population of Noxes than rodent cells, with extremely low expression of Nox1 and much higher expression of Nox4 and Nox5. In this study, we show that Nox5 is a major modulator of PDGF-directed ROS production and HASMC growth. We also provide the first characterization of Nox5 in HASMCs, and identify one of its downstream targets.
Our data clearly demonstrate that Nox5 participates in PDGF-induced ROS production in HASMCs. Both the DCF-DA staining and the hydroxyethidium assay reveal that cells transfected with siNox5 produce significantly less ROS when treated with PDGF. Previous studies had shown that VSMCs produce ROS in response to PDGF stimulation. While in rat VSMC the source is likely Nox1 [22], we show that Nox5 plays an important role in ROS production in PDGF-treated HASMCs.
We found that HASMCs express more than one isoform of Nox5. Multiple isoforms of Nox5 have been found in other cell types such as Caco2 cells, endothelial cells, and SMCs isolated from pulmonary arteries [28]. We have also found Nox5 in human umbilical vein endothelial cells and human aortic endothelial cells (unpublished data). The cDNA sequences of Nox5α and γ consist of exons 3–19, while those of Nox β and δ consist of exons 1, 2, and 4–19. The majority of the differences in the isoforms are in the 5′ UTR. An alternative splice site at the end of exon 3 gives Nox5γ, a slightly longer transcript than Nox5α. The same is true for Nox5δ, which contains a longer exon 2 sequence than Nox5β. Our siNox5 targets all isoforms of Nox5, and thus we are not able to determine which isoform or isoforms are responsible for the response to PDGF. However, given the tight regulation of ROS needed in signaling pathways, it is plausible that the different isoforms in HASMC may regulate specific pathways and thus different biological functions. For example, PDGF has not only been shown to mediate VSMC proliferation, but also migration and apoptosis. It is also possible that one or more isoforms are important for the PDGF-mediated response at different steps along a single pathway, depending on their subcellular distribution. We show in this study that at least one isoform of Nox5 is responsible for PDGF-induced HASMC proliferation. This finding is in concert with studies of Nox5 in several cancer cell lines [25–27] and in human microvascular endothelial cells [28]. Further work will be necessary to investigate the participation of Nox5 in other cellular functions and to characterize the exact role of each of its isoforms.
One novel aspect of our findings is the identification of a signaling pathway that is dependent on Nox5 for its activation. A number of growth-related pathways have been shown to be regulated by ROS, including extracellular signal regulated kinases (Erk1/2), p38 mitogen-activated protein kinase and Akt [20]. However, none of these pathways is ROS-sensitive in PDGF-stimulated HASMCs (unpublished observations). The JAK/STAT pathway has also been shown to be activated by ROS [18, 19], and is interesting because it regulates PDGF-regulated cell growth in various cell types such as pancreatic stellate cells [35] and in human pulmonary SMCs [19]. As seen in Figures 6A and 6B, ROS are also important for PDGF-induced JAK/STAT activation in HASMCs. Furthermore, the source for these ROS appears to be Nox5, as cells transfected with siNox5 demonstrated reduced phosphorylation of both JAK2 and STAT3 (Figures 7A and 7B).
The studies published on Nox5 have thus far concentrated on its role in cell growth. However, its unique property of direct calcium activation that is not a feature of Nox1, 2, 3, or 4 provides it with potential to be involved in a number of different and important cellular processes. Moreover, the fact that it exists in different isoforms that may differ in location and mode of modulation (by phosphorylation [36] and/or calmodulin binding [37]) endows Nox5 with the complexity needed to participate in a variety of signaling pathways. If these postulations are true, Nox5 could prove to be a key regulator of calcium and ROS-dependent biological functions. The characterization of Nox5 activation has not yet been performed in HASMC. To date, the studies that have investigated the mechanism of Nox5 activation have been performed in cells transfected with Nox5 expression plasmids. The mRNA transcripts of the isoforms of Nox5 present in HASMC all contain calcium-binding EF hands, however, further studies would be needed to show that the native Nox5 isoforms in HASMC indeed bind calcium and produce superoxide as they do when heterologously expressed in other cells.
In summary, we have shown that HASMCs express several isoforms of Nox5, and that PDGF exerts its mitogenic properties on these cells by acting via Nox5 to stimulate the JAK/STAT pathway. This is the first evidence linking a specific Nox to JAK/STAT activation in this cell type. Furthermore, these findings provide a basis for understanding inter-species differences in Nox signaling and insight into a process important for several vascular diseases. Additional studies will be needed to determine which isoform(s) are responsible for mediating PDGF-induced HASMC proliferation and to discover if Nox5 plays a role in other PDGF-directed cellular processes.
Acknowledgments
This work was supported by NIH grant HL074604 to DBJ and HL058863 to KKG. We thank Dr. Peter Sayeski for his advice on the JAK/STAT experiments.
Abbreviations
- DCF-DA
dichlorofluorescein-DA
- DHE
dihydroethidium
- DPI
diphenylene iodonium
- HASMCs
human aortic smooth muscle cells
- NAC
N-acetylcysteine
- Nox
NADPH oxidase
- NSsiRNA
non-silencing siRNA
- PBS
phosphate buffered saline
- PEG-SOD
polyethylene glycolated superoxide dismutase
- PDGF
platelet-derived growth factor
- ROS
reactive oxygen species
- SEM
standard error of the mean
- SMC
smooth muscle cell
- SmBM
smooth muscle basal medium
- VSMC
vascular smooth muscle cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Funayama H, Ikeda U, Takahashi M, Sakata Y, Kitagawa S, Takahashi Y, Masuyama J, Furukawa Y, Miura Y, Kano S, Matsuda M, Shimada K. Human monocyte-endothelial cell interaction induces platelet-derived growth factor expression. Cardiovascular Research. 1998;37:216–224. doi: 10.1016/s0008-6363(97)00224-1. [DOI] [PubMed] [Google Scholar]
- 2.Wilcox JN, Nelken NA, Coughlin SR, Gordon D, Schall TJ. Local expression of inflammatory cytokines in human atherosclerotic plaques. Journal of Atherosclerosis & Thrombosis. 1994;1(Suppl 1):S10–13. doi: 10.5551/jat1994.1.supplemment1_s10. [DOI] [PubMed] [Google Scholar]
- 3.Ross R, Glomset JA, Kariya B, Harker L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc Natl Acad Sci USA. 1974;71:1207–1210. doi: 10.1073/pnas.71.4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohm M, Blasius S, Roessner A. Immunohistochemical localization of PDGF B, PDGF beta receptors and IGF I receptors during atherogenesis. Zentralblatt fur Pathologie. 1994;140:357–362. [PubMed] [Google Scholar]
- 5.Rubin K, Tingstrom A, Hansson GK, Larsson E, Ronnstrand L, Klareskog L, Claesson-Welsh L, Heldin CH, Fellstrom B, Terracio L. Induction of B-type receptors for platelet-derived growth factor in vascular inflammation: possible implications for development of vascular proliferative lesions. Lancet. 1988;1:1353–1356. doi: 10.1016/s0140-6736(88)92177-0. [DOI] [PubMed] [Google Scholar]
- 6.Lamb DJ, Avades TY, Ferns GA. Endogenous neutralizing antibodies against platelet-derived growth factor-aa inhibit atherogenesis in the cholesterol-fed rabbit. Arteriosclerosis, Thrombosis & Vascular Biology. 2001;21:997–1003. doi: 10.1161/01.atv.21.6.997. [DOI] [PubMed] [Google Scholar]
- 7.Rutherford C, Martin W, Carrier M, Anggard EE, Ferns GA. Endogenously elicited antibodies to platelet derived growth factor-BB and platelet cytosolic protein inhibit aortic lesion development in the cholesterol-fed rabbit. International Journal of Experimental Pathology. 1997;78:21–32. doi: 10.1046/j.1365-2613.1997.d01-237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 9.Noiseux N, Boucher CH, Cartier R, Sirois MG. Bolus endovascular PDGFR-beta antisense treatment suppressed intimal hyperplasia in a rat carotid injury model. Circulation. 2000;102:1330–1336. doi: 10.1161/01.cir.102.11.1330. [DOI] [PubMed] [Google Scholar]
- 10.Sirois MG, Simons M, Edelman ER. Antisense oligonucleotide inhibition of PDGFR-beta receptor subunit expression directs suppression of intimal thickening. Circulation. 1997;95:669–676. doi: 10.1161/01.cir.95.3.669. [DOI] [PubMed] [Google Scholar]
- 11.Raines E. PDGF and cardiovascular disease. Cytokine & Growth Factor Reviews. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Diliberto PA, Hubbert T, Herman B. Early PDGF-induced alterations in cytosolic free calcium are required for mitogenesis. Research Communications in Chemical Pathology & Pharmacology. 1991;72:3–12. [PubMed] [Google Scholar]
- 13.Nie L, Mogami H, Kanzaki M, Shibata H, Kojima I. Blockade of DNA synthesis induced by platelet-derived growth factor by tranilast, an inhibitor of calcium entry, in vascular smooth muscle cells. Molecular Pharmacology. 1996;50:763–769. [PubMed] [Google Scholar]
- 14.Seewald S, Sachinidis A, Seul C, Kettenhofen R, Ko Y, Vetter H. The role of platelet-derived growth factor-BB-induced increase in cytosolic free Ca2+ in activation of mitogen-activated protein kinase and DNA synthesis in vascular smooth muscle cells. Journal of Hypertension. 1997;15:1671–1675. doi: 10.1097/00004872-199715120-00071. [DOI] [PubMed] [Google Scholar]
- 15.Kappert K, Sparwel J, Sandin A, Seiler A, Siebolts U, Leppanen O, Rosenkranz S, Ostman A. Antioxidants relieve phosphatase inhibition and reduce PDGF signaling in cultured VSMCs and in restenosis. Arteriosclerosis, Thrombosis & Vascular Biology. 2006;26:2644–2651. doi: 10.1161/01.ATV.0000246777.30819.85. [DOI] [PubMed] [Google Scholar]
- 16.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 17.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. Journal of Cell Science. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 18.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol Cell Physiol. 1998;275:C1640–1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 19.Simon AR, Takahashi S, Severgnini M, Fanburg BL, Cochran BH. Role of the JAK-STAT pathway in PDGF-stimulated proliferation of human airway smooth muscle cells. American Journal of Physiology - Lung Cellular & Molecular Physiology. 2002;282:L1296–1304. doi: 10.1152/ajplung.00315.2001. [DOI] [PubMed] [Google Scholar]
- 20.Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovascular Research. 2006;71:216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambeth JD. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Current Opinion in Hematology. 2002;9:11–17. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways.[see comment] Circulation Research. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 23.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arteriosclerosis, Thrombosis & Vascular Biology. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. Journal of Biological Chemistry. 2001;276:37594–37601. doi: 10.1074/jbc.M103034200. [DOI] [PubMed] [Google Scholar]
- 25.Fu X, Beer DG, Behar J, Wands J, Lambeth D, Cao W. cAMP-response element-binding protein mediates acid-induced NAD PH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. Journal of Biological Chemistry. 2006;281:20368–20382. doi: 10.1074/jbc.M603353200. [DOI] [PubMed] [Google Scholar]
- 26.Kamiguti AS, Serrander L, Lin K, Harris RJ, Cawley JC, Allsup DJ, Slupsky JR, Krause KH, Zuzel M. Expression and activity of NOX5 in the circulating malignant B cells of hairy cell leukemia. Journal of Immunology. 2005;175:8424–8430. doi: 10.4049/jimmunol.175.12.8424. [DOI] [PubMed] [Google Scholar]
- 27.Brar SS, Corbin Z, Kennedy TP, Hemendinger R, Thornton L, Bommarius B, Arnold RS, Whorton AR, Sturrock AB, Huecksteadt TP, Quinn MT, Krenitsky K, Ardie KG, Lambeth JD, Hoidal JR. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. American Journal of Physiology - Cell Physiology. 2003;285:C353–369. doi: 10.1152/ajpcell.00525.2002. [DOI] [PubMed] [Google Scholar]
- 28.BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Gorlach A. NOX5 variants are functionally active in endothelial cells. Free Radical Biology & Medicine. 2007;42:446–459. doi: 10.1016/j.freeradbiomed.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 29.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts.[see comment] Circulation Research. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 30.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, Krause KH, Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5) Journal of Biological Chemistry. 2004;279:18583–18591. doi: 10.1074/jbc.M310268200. [DOI] [PubMed] [Google Scholar]
- 32.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- 33.Lee JS, Kypreos KE, Sonenshein GE. Synchronization of cultured vascular smooth muscle cells following reversal of quiescence induced by treatment with the antioxidant N-acetylcysteine. Experimental Cell Research. 1998;239:447–453. doi: 10.1006/excr.1997.3919. [DOI] [PubMed] [Google Scholar]
- 34.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arteriosclerosis, Thrombosis & Vascular Biology. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 35.Masamune A, Satoh M, Kikuta K, Suzuki N, Shimosegawa T. Activation of JAK-STAT pathway is required for platelet-derived growth factor-induced proliferation of pancreatic stellate cells. World Journal of Gastroenterology. 2005;11:3385–3391. doi: 10.3748/wjg.v11.i22.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. Journal of Biological Chemistry. 2007;282:6494–6507. doi: 10.1074/jbc.M608966200. [DOI] [PubMed] [Google Scholar]
- 37.Tirone F, Cox JA. NADPH oxidase 5 (NOX5) interacts with and is regulated by calmodulin. FEBS Letters. 2007;581:1202–1208. doi: 10.1016/j.febslet.2007.02.047. [DOI] [PubMed] [Google Scholar]