

Abstract
Using a syngeneic p53 null mouse mammary gland tumor model that closely mimics human breast cancer, we have identified by limiting dilution transplantation as well as in vitro mammosphere assay a Lin−CD29HCD24H subpopulation of tumor-initiating cells. Upon subsequent transplantation, this subpopulation generated heterogeneous tumors that displayed properties similar to the primary tumor. Analysis of biomarkers suggests the Lin−CD29HCD24H subpopulation may have arisen from a bipotent mammary progenitor. Differentially expressed genes in the Lin−CD29HCD24H mouse mammary gland tumor-initiating cell population include those involved in DNA damage response and repair, as well as genes involved in epigenetic regulation previously shown to be critical for stem cell self-renewal. These studies provide in vitro and in vivo data that support the “cancer stem cell” hypothesis. Furthermore, this p53 null mouse mammary tumor model may allow us to identify new cancer stem cell markers and to test the functional importance of these markers.
Introduction
Cancer stem cells (CSCs), a limited subpopulation of tumor-initiating cells, are defined as cells that retain extensive self-renewal potential through a series of generations and have the ability to recreate the heterogeneity of the original tumor through asymmetric division. These cells have been posited to be responsible for resistance to conventional therapies, recurrence and metastasis (1, 2). CSCs were first identified and characterized in acute myeloid leukemia (AML) using antibodies which recognized specific cell surface markers and fluorescence-activated cell sorting (FACS) to isolate a small subpopulation of cells capable of self-renewal and tumor formation following transplantation into the bone marrow of immunodeficient mice (3–5). By applying a similar strategy of transplanting FACS sorted single cells from solid tumors into immunodeficient mice, a small subpopulation of tumor-initiating cells has been identified from a variety of solid tumors including breast (see review by Clarke and colleagues (6)).
Cells isolated from breast cancer pleural effusions with Lin−CD44+CD24−/low phenotype displayed increased tumorigenicity using serial limiting dilution transplantation assays into immunodeficient mice (7). A 186 gene “signature” was identified by comparing human breast cancer Lin−CD44+CD24−/low cells with normal breast epithelial and myoepithelial cells through gene expression profiling study (8). This signature, although derived from only a small number of patients, was able to predict the recurrence risk of breast, lung and prostate cancers and medulloblastoma, and also showed a strong correlation with overall and metastasis-free survival. The identification and characterization of tumor-initiating cells and the molecular pathways that account for their self-renewal and survival is critical to design therapies that preferentially target these cells and sensitize them to conventional chemo- and radiation therapies. From these studies, however, the relationship between the Lin−CD44+CD24−/low cells in breast cancer to normal breast stem cells is unclear. For example, are these markers expressed on normal stem cells and are they conserved in the mammary gland in mice and other species?
There are several caveats concerning the transplantation studies using xenografts of human breast cancer cells in immunocompromised mice, which need to be considered. Potential differences in the stroma and microenvironment in mice and human, as well as defects in the immune system, may have profound effects on tumor initiation and progression in these models. Difficulties in obtaining a renewable source of cells from primary patient biopsies also present serious obstacles to performing detailed mechanistic studies and for the development of preclinical models. With the exception of the hematopoietic system, detailed functional characterization of normal stem cells is usually lacking, making it difficult to perform a direct comparison of the “normal” and “cancer” stem cells.
In this regard, one notable exception is the mouse mammary gland. Stem cells have been identified in the mouse mammary gland using serial limiting dilution transplantation assays into the cleared mammary fat pad of syngeneic mice by two independent groups. A small proportion of cells isolated as β1 integrin (CD29)hiCD24+Lin− was able to reconstitute a complete and functional mammary gland (9). In parallel studies, α6 integrin (CD49f) and CD24 were also identified as mouse mammary gland stem cell markers facilitating the isolation of cells with mammary repopulating activity (10). Both of these studies indicated that mammary gland stem cells, as defined by these markers, are predominantly cycling and possess basal characteristics.
Genetically engineered mouse (GEM) models have been extremely important in helping to uncover the pathoetiology of human diseases. Numerous GEM models of breast cancer have been developed and characterized in detail (11). Many, but not all of these, generate diploid tumors, which have a uniform histology, and thus may not provide models in which to investigate the biology of tumor-initiating subpopulations (12). Tumorigenic cancer cells have recently been identified from the MMTV (mouse mammary tumor virus)-Wnt-1 mice in which Thy1+CD24+ cancer cells were more capable of regenerating tumors than non-Thy1+CD24+ cells (13). Tumor-initiating cells have also been reported in the homogeneous MMTV-Neu transgenic mouse model (14). However both of these models involve the targeted overexpression of oncogenes with specific MMTV that may target the more differentiated cells types, although in the case of the MMTV-Wnt model the secreted Wnt ligand may act on surrounding cells. As an alternative to these transgenic models, we have employed a p53 null mammary gland transplant model, in which p53 is deleted in the germline of Balb/c mice, and the mammary epithelium is transplanted into the cleared fat pads of syngeneic wild-type recipient mice (15, 16). Unlike many other mouse breast cancer models, a subset of the tumors arising in this model retain the estrogen receptor, and antiestrogens inhibit the formation of these tumors (17). Based upon extensive gene expression analyses of p53 null mammary epithelium as well as primary tumors, it has been suggested that tumors from this p53 null Balb/C breast cancer model mimic human tumors more closely than many other models (18–20). Finally, the p53 tumor suppressor is frequently (20–40%) mutated in human breast cancers and is a marker of poor prognosis as well as for chemo- and radiation resistance (21, 22).
Mammary epithelial cells (MECs) from p53 null tumors were FACS sorted based upon the mouse mammary gland stem cell markers, CD29 and CD24. The sorted populations were transplanted into cleared mammary fat pads of wild-type Balb/c mice. Using this approach, a small subpopulation (5–10%) of tumor cells with stem-like properties, including self-renewal and regeneration of heterogeneous tumors upon serial transplantation, was identified from ten independent primary tumors. In vitro mammosphere assays provided further evidence that this subpopulation possesses stem-cell properties. Finally, RNA microarray analyses on isolated p53 null tumor cell subpopulations as well as MECs from virgin Balb/c mice have permitted the identification of genes and regulatory pathways of potential importance in understanding tumor initiation and progression, as well as therapeutic resistance.
Materials and Methods
Antibodies
R-PE-conjugated rat anti-mouse CD44, FITC- and R-PE-conjugated rat anti-mouse CD24, biotin-conjugated mouse lineage panel (anti-CD3e, CD11b, CD45R/B220, Ly-6G/C, and TER-119), biotin-conjugated rat anti-mouse CD31, SAv-APC conjugate antibody, FITC-conjugated CD49f, and their corresponding isotype controls, FITC-conjugated rat IgG2a, R-PE-conjugated rat IgG2a, κ, and R-PE-conjugated rat IgG2b, κ, were all purchased from BD Biosciences. FITC anti-mouse/rat CD29 and FITC Armenian hamster IgG isotype control were from BioLegend. Anti-cytokeratins 5 and 14 (anti-K5 and –K14) rabbit polyclonal antibodies were from Covance. Anti-cytokeratin 8 (anti-K8) was from the Developmental Studies Hybridoma Bank, University of Iowa. Anti-estrogen receptor alpha (ERα) was obtained from Santa Cruz Biotechnologies. Biotin-conjugated CD140a was purchased from eBioscience. Biotinylated goat anti-rabbit or anti-rat secondary antibodies were obtained from Vector Laboratories, Inc. Texas Red- and Alexa 488-conjugated secondary antibodies for immunofluorescence were from Molecular Probes.
Preparation of single mammary tumor cells
All animal protocols were reviewed and approved by the Animal Protocol Review Committee at Baylor College of Medicine. Ten p53 null mammary tumors were generated as described (15), all without hormone treatment except tumor T3 with 2 weeks hormone stimulation, and tumors T6, T7, and T10 with pituitary isograft treatment. They were anesthetized with Avertin (Sigma) before tumors were removed. Tumors were minced using razor blades and digested in 10 ml digestion media per 1 g tissue (digestion buffer containing DMEM/F12, 100 μg/ml gentimycin, antibiotic-antimycotic from Invitrogen, and collagenase type III (225 U/ml, Worthington)) at 37°. Samples were pipetted every 30 min for 2 to 2.5 hrs while shaking on a rotary shaker at 125 rpm. Cells were filtered through 40 μm cell strainers and washed with washing buffer (F-12 medium (Invitrogen)/5% Fetal Bovine Serum (FBS, JRH Biosciences)/50 μg/ml gentimycin) until the supernatant was clear. Then, cells were resuspended in HBSS+ [HBSS (Invitrogen) containing 2% FBS and 10 mM HEPES Buffer (Invitrogen)] before labeling with antibodies.
Preparation of single normal mammary epithelial cells
This protocol is as described by Welm et al (23). Eight to ten-week old virgin female Balb/C mice were used to isolate single MECs. Briefly, the #4 inguinal mammary glands (lymph nodes removed) were minced into small pieces. The digestion buffer mentioned above was used to digest the gland for 1.5 hr at 37° on a rotary shaker shaking at 125 rpm. Samples were pipetted every 30 min, and centrifuged at 600 × g for 10 min to pellet the organoids. Samples were washed four times in washing buffer at 425 × g for 2 sec and, washed once with PBS before trypsinization in 0.5g/L trypsin/0.2g/L EDTA in saline 10 min. HBSS+ was added to dilute out trypsin. Cells were filtered through 40 μm filters and centrifuged at 671 × g for 5 min before labeling with antibodies.
Flow cytometry
Cells were labeled with antibodies at a concentration of 10 million cells/ml under optimized conditions (1:200 for CD29-FITC, and 1:100 for CD24-PE) and were subjected to FAC analysis and sorting on a triple laser MoFlo (Cytomation, Fort Collins, CO). Dead cells were excluded by using propidium iodide (2 μg/ml, Sigma). Data analysis was performed on FlowJo version 6.4.7, Tree Star, Inc.
Transplantation into the cleared fat pad
Clearance of MECs and transplantation procedures were performed as previously described (24). Following FACS, the designated number of cells were washed once with PBS and transplanted into the cleared fat pads of 21-day-old female Balb/C mice (Harlan).
Mammosphere assays
The protocol for mammosphere assays was as described by Dontu (25) with modification. Briefly, 10,000 sorted cells per well from distinct subpopulations were grown in 6-well Ultra Low Attachment plates (Corning, NY) with 2 ml serum-free mammosphere medium (DMEM/F12 with 20 ng/ml bFGF, 20 ng/ml EGF, B27, 100 μg/ml gentimycin, antibiotic-antimycotic, all from Invitrogen). The cells were fed every 3 days, and passaged using 0.05% trypsin/0.53 mM EDTA-4Na. 2,000 dissociated cells per well were re-plated in 2 ml mammosphere medium for passages thereafter. Mammospheres were counted using a Leica Dissecting scope.
Colony forming assay
Serial passaged mammospheres grown in the serum-free mammosphere medium, and collagenase-dissociated tumor cells after one week of growth on plastic with mammosphere medium supplemented with 5% FBS were dissociated with trypsin as described above. Viable cells from these two groups were sorted using Sytox Red (5 μM, Invitrogen) into 96 well plates with FACSAria (BD Biosciences). Eight hundred cells/well were placed in mammosphere medium supplemented with 5% FBS. The plates were irradiated, 0, 2, 4, or 6 Gy, immediately after being sorted. The cells were allowed to grow undisturbed for 2 weeks. The cells were then fixed with methanol: glacial acetic acid (2:1), stained with crystal violet, then washed with water. The colonies were then counted and data were graphed using SigmaPlot (Systat Software Inc.).
Immunohistochemistry
See http://www.bcm.edu/rosenlab/protocols.html for detailed procedures and the dilution concentration for each antibody.
Microarray analysis
Total RNA was isolated from the sorted subpopulations based upon Lin, CD29, CD24 expression using the PicoPure RNA isolation kit (Arcturus), following by mRNA amplification using a T7 global amplification method (Two-cycle Target Labeling kit; Affymetrix), DNA fragmentation, biotinylation, and hybridization onto Affymetrix 430 2.0 array chips. Microarray analysis was done with Affymetrix MG 430 2.0 chip with 45,037 probe sets. Statistical analysis was done with dChip (www.dchip.org) and BRB Array Tools (http://linus.nci.nih.gov/BRB-ArrayTools.html) software packages. Expression was estimated with dChip (26, 27) PM (Perfect-Match) model with quantile normalization. Differentially expressed genes were found with BRB Array Tools using paired (for tumors) or unpaired (for normal) t-test and ANOVA analysis. RVM (Random Variance Model) was used in all cases because of the small sample size (28). The method of Benjaminin and Hochberg (29) was used for the estimation of false discovery rate (FDR). The complete array data can be accessed at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=dpqnzeemqcqakdu&acc=GSE886 3.
Results
p53 null mammary gland mouse tumors are heterogeneous
Ten tumors from p53 null mouse model were studied as summarized in Supplemental Table S1 and depicted in Figure 1. Histological analysis showed that the resulting tumors are heterogeneous. Immunohistochemical staining using myoepithelial (K5, K14) and luminal markers (K8, ERα) was performed to characterize the various epithelial cell types in the p53 null mammary tumors (Figure 1A). Some tumors (T1–T5) express K5, K14, and K8. In contrast, other tumors displayed expression of K8, very low K14, but not K5 (T6–T10). Co-expression of K8 and K14 detected by immunofluorescence staining (Figure 1B), as shown in T1, T3, and T6 suggested the expansion of a putative bipotent progenitor, which has been suggested previously to be a potential target cell in Wnt-1 tumors (30, 31). In order to identify a tumor-initiating subpopulation, we generated a tumor bank and transplanted these primary tumors into the cleared fat pads of syngeneic hosts permitting the generation of sufficient numbers of tumors for further studies. Based upon marker expression, the phenotypes of these tumors remained stable during transplantation (Supplemental Figure S1).
Figure 1.
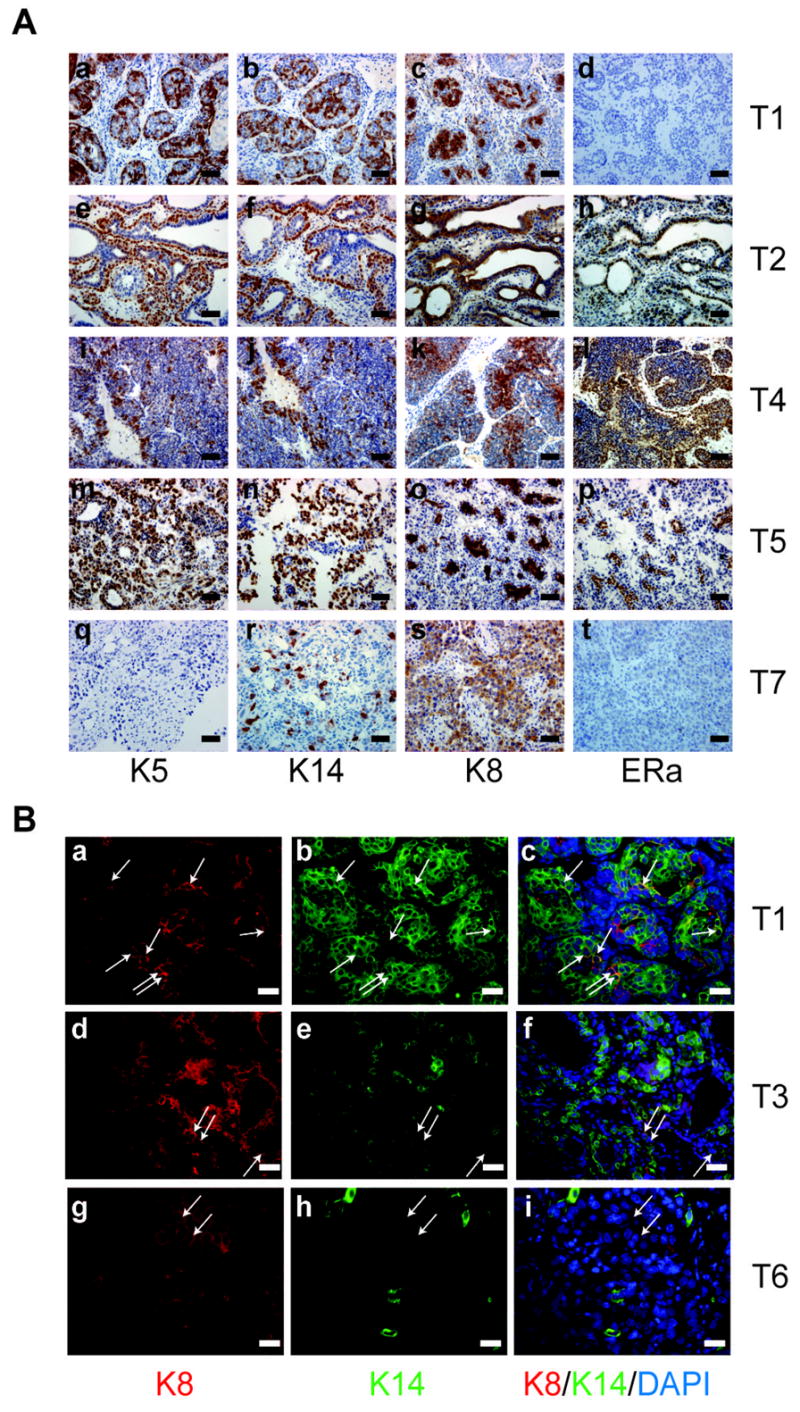
Immunostaining of paraffin-embedded sections illustrating the heterogeneity of p53 null mammary tumors. K5, K14, K8, ERα staining of five p53 null mammary tumors, A. (a)–(d) T1, (e)–(h) T2, (i)–(l) T4, (m)–(p) T5, (q)–(t) and T7. Scale bar, 50 μm. B. (a, d, g) K8 staining of T1, T3, T6. (b, e, h) K14 staining of T1, T3, T6. (c, f, i) K14, K8, DAPI co-staining of T1, T3, T6. Scale bar, 25 μm.
Identification of tumorigenic subpopulations with stem cell properties
In order to determine if Lin−CD44+CD24− were also the appropriate cell surface markers to identify the tumorigenic cells in the p53 null mouse model, subpopulations of cells obtained by FACS based upon expression of CD44 and CD24 (Supplemental Figure S2) were transplanted into the cleared mammary fat pad of syngeneic mice. No significant differences were found among the four Lin− subpopulations (CD44+CD24−, CD44+CD24+, CD44−CD24+, and CD44−CD24−) as shown in Supplemental Table S2. Histological analysis also showed no difference between these tumors (data not shown). Therefore, Lin−CD44+CD24−, which represents a highly tumorigenic human breast cancer population, was not a suitable marker set to identify tumor-initiating cells in the p53 null mouse model. Thus, cell surface markers, which have been used previously to identify mouse mammary stem cells, were employed to isolate subpopulations of p53 tumor cells.
Independent p53 null mammary tumors were dissociated with collagenase followed by FACS analysis based upon the cell surface markers, Lin, CD29 and CD24. Tumors displayed distinct FACS profiles (Figure 2A), again demonstrating the heterogeneity of tumors generated from this mouse model. Limiting dilution transplantation experiments into the cleared mammary fat pad of syngeneic Balb/c mice demonstrated that the Lin−CD29HCD24H (LineageNegativeCD29HighCD24High) subpopulation, representing overall approximately 5~10% of the total cell population, displayed significantly increased tumorigenic potential compared to the other subpopulations (Table 1; Supplemental TableS3). Thus, as few as 100 Lin−CD29HCD24H cells resulted in tumors (8 out of 14 transplants), while no tumors were observed from 100 cells of the corresponding Lin−CD29HCD24L (LineageNegativeCD29HighCD24Low), Lin−CD29LCD24L(LineageNegativeCD29LowCD24Low) or Lin−CD29LCD24H (LineageNegativeCD29LowCD24High) subpopulations. FACS analysis and immunostaining showed that the resulting tumors generated by Lin−CD29HCD24H subpopulation displayed similar FACS profiles (Figure 3A) and marker (K5, K14, K8, and ER α) expression (Figure 3B) when compared to the original tumors from which they were derived, suggesting that the tumor-initiating subpopulation was able to generate the heterogeneous characteristics of the original tumor. The Lin−CD29HCD24L subpopulation, which was usually <5% of the total, also displayed increased tumorigenicity when compared to the Lin−CD29LCD24L and Lin−CD29LCD24H subpopulations which represented the bulk (>90%) of the tumor cells. The purity, as indicated by FACS analysis, of the Lin−CD29HCD24H subpopulation was estimated to be approximately 70 to 88 % (Figure 2C). Therefore, the tumors observed when a large number (>2,500) of Lin−CD29LCD24L and Lin−CD29LCD24H cells were injected are most likely a result of the contamination with Lin−CD29HCD24H cells. This is supported by the observations that the Lin−CD29LCD24L- and Lin−CD29LCD24H-generated tumors had similar FACS profiles as well as expression of specific biomarkers when compared with the parental tumors (data not shown). Due to Lin−CD29HCD24L’s distinct FACS profile, contamination from Lin−CD29HCD24H is not likely the reason for the observed tumorigenicity of the Lin−CD29HCD24L subpopulation. FACS analysis of tumors arising from Lin−CD29HCD24L indicated that they did not recapitulate the phenotype of the parental tumor (Supplemental Figure S3). Notwithstanding, the Lin−CD29HCD24H subpopulation displayed approximately a 70-fold enrichment in tumorigenicity as compared to the total Lin− tumor cell population. The Lin−CD29HCD24H tumorigenic subpopulation from individual p53 null mammary tumors was enriched for K8+/K14+ cells as compared to other CD29CD24 subpopulations as shown by immunofluorescence co-staining (Supplemental Figure S4). CD49f, another putative mouse mammary gland stem cell marker, together with CD24, also defined a small percentage of tumorigenic cells by FACS and serial transplantation, as shown in Supplemental Figure S5 and Table S4.
Figure 2.
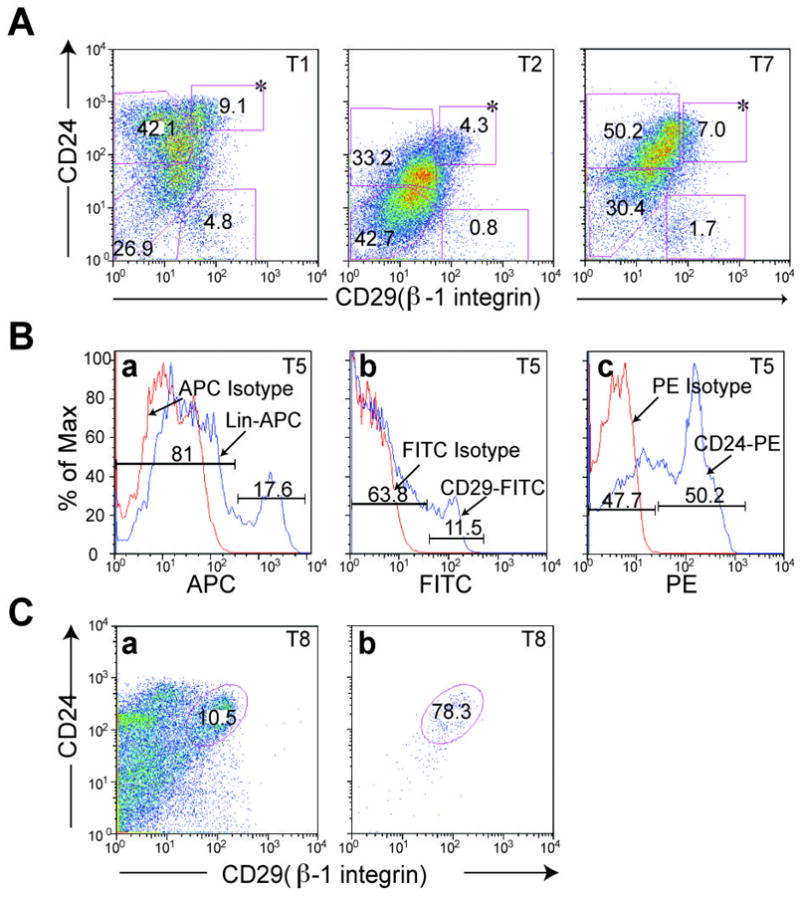
Flow cytometry analysis provides additional evidence of heterogeneity of p53 null mammary tumors. A. Tumors T1, T2, and T7 were FACS sorted based upon expression of cell surface markers, CD29 and CD24. Dead cells and lineage positive cells were gated out by PI and biotin-conjugated mouse lineage panel kit plus biotin-conjugated CD31 and CD140a. The percentage of individual subpopulations was determined according to isotype control from each assay. * Lin−CD29HCD24H subpopulation. B. Histograms of T5 were plotted after FACS sorting with red line indicating isotype control, (a) APC-conjugated rat IgG2a, (b) FITC-conjugated rat IgG2a, and (c) R-PE-conjugated rat IgG2a, and blue line representing corresponding antibodies, (a) Lin-APC, (b) CD29-FITC, and (c) CD24-PE. C. FACS sorted Lin−CD29HCD24H of T8 was collected (a) and re-applied to flow to check the purity of the collected fraction (b).
Table 1. Tumor-initiating cells were enriched in Lin−CD29HCD24H subpopulation.
Cells from T6, T7, T10 (primary tumor), T1–T5, T8, T9 (transplantation generation 1, TG1), were freshly digested and FACS sorted. All dead cells were gated out with PI. Mouse lineage panel plus anit-CD31 were used to exclude mouse lineage positive cells for tumors T1–T5, T8, and T9. Anti-CD140a was added as a lineage marker for tumors T6, T7, and T10. After FACS sorting, cells were washed with PBS and transplanted into the cleared fat pad of 3-week-old Balb/c female mice. Mice were monitored until tumors were observed or up to 18 months if no tumors were detected. Tumor-initiating cell frequency and the Poisson distribution analysis was generated using R software (The R Foundation for Statistical Computing). * T6 (primary), T4 (TG1) **T6, T7, T10 (primary), T1, T2 (TG1). Subpopulation-derived palpable tumors from 5,000 cells were detected between 4 to 6 weeks. 1,500 of Lin−CD29HCD24H and Lin−CD29HCD24L cells formed palpable tumors from 5 to 8 weeks, similar to tumors formed from 2,500 Lin−CD29LCD24L, Lin−CD29LCD24H and Lin− cells. Palpable tumors from 500 cells took 7 to 11 weeks, while those from 100 cells took 8 to 15 weeks. All primary tumors, TG1 tumors, and subpopulation-derived tumors were excised for FACS and immunostaining analysis between diameter(s) of 1 to 1.5 cm. Table 1
| 30,000 | 5,000 | 2,500* | 1,500** | 500 | 100** | Tumor-initiating cell frequency (95% CI) | |
|---|---|---|---|---|---|---|---|
| Lin− CD29HCD24H | 36/36 | 9/10 | 44/52 | 8/14 | 1/302 (1/413 - 1/221) | ||
| Lin− CD29HCD24L | 35/40 | 4/11 | 16/54 | 0/12 | 1/2156 (1/2934 - 1/1584) | ||
| Lin− CD29LCD24H | 7/44 | 1/4 | 0/10 | 1/14** | 0/6 | 1/25891 (1/49897 - 1/13434) | |
| Lin− CD29LCD24L | 2/44 | 1/4 | 0/10 | 0/13** | 0/6 | 1/81931 (1/254398 - 1/26387) | |
| Lin− | 8/10 | 3/18 | 1/4 | 0/10 | 1/21583 (1/38743 - 1/12023) |
Figure 3.

Lin−CD29HCD24H subpopulation-generated tumors mimic the parental tumors with respect to their CD29/CD24 profiling as well as histological staining for cytochemical markers. A. Flow plot for tumor T3, T6, and their respective resulting tumors from Lin−CD29HCD24H fraction. B. Immunostaining of parental tumors T3 (a–d), T6 (I-l) and resulting tumors from (e–h) 500 T3 Lin−CD29HCD24H cells generated tumor, and (m–p) 500 T6 Lin−CD29HCD24H cells generated tumor. Scale bar, 100 μm
Tumorigenic cells generate increased numbers of secondary mammospheres
Self-renewal is a unique property of stem cells which distinguishes them from more differentiated cells. A mammosphere (MS) assay has been developed to study self-renewal potential by plating cells in a serum-free medium with growth factor supplementation on a non-adherent substrata followed by subsequent quantitation of sphere formation (25). Secondary MSs from the Lin−CD29HCD24H subgroup as determined from 7 independent tumors were larger in size (up to 400 μm in diameter) and number (average 110 MSs per 2,000 Lin−CD29HCD24H cells) as compared with all other subpopulations (Figure 4A&B). Lin−CD29HCD24L cells can also form MSs, but with a reduced number, while barely any MSs were observed from Lin−CD29LCD24L. One potential limitation of the MS assay is the possible occurrence of cell aggregation. To address this concern, single Lin−CD29HCD24H cells were plated into individual wells. Under these conditions, the mammosphere forming efficiency was similar as that observed for the bulk Lin−CD29HCD24H cells (Supplemental Figure S6), suggesting that at the cell concentrations employed, the mammosphere assay is measuring the potential for self-renewal and not cell aggregation. FACS analysis on passaged mammospheres derived from tumor T7 revealed that p53 null mammospheres are enriched in the Lin−CD29HCD24H tumor-initiating cells, while cells cultured on plastic with serum are not (Supplemental Figure S7). In order to test whether the passaged mammospheres were resistant to radiation treatment, serial passaged mammospheres from a p53 null mammary tumor (T7) were treated with increasing doses of radiation up to 6 Gy, a lethal dose for the bulk of the p53 null mammary tumor cells when plated with serum on plastic as shown in Figure 4C. Our colony forming assay, results from passage 10 of tumor T7, suggested that mammospheres, which mainly consist of Lin−CD29HCD24H tumorigenic cells, displayed radiation resistance. The result has been repeated with an independent mammosphere passage.
Figure 4.
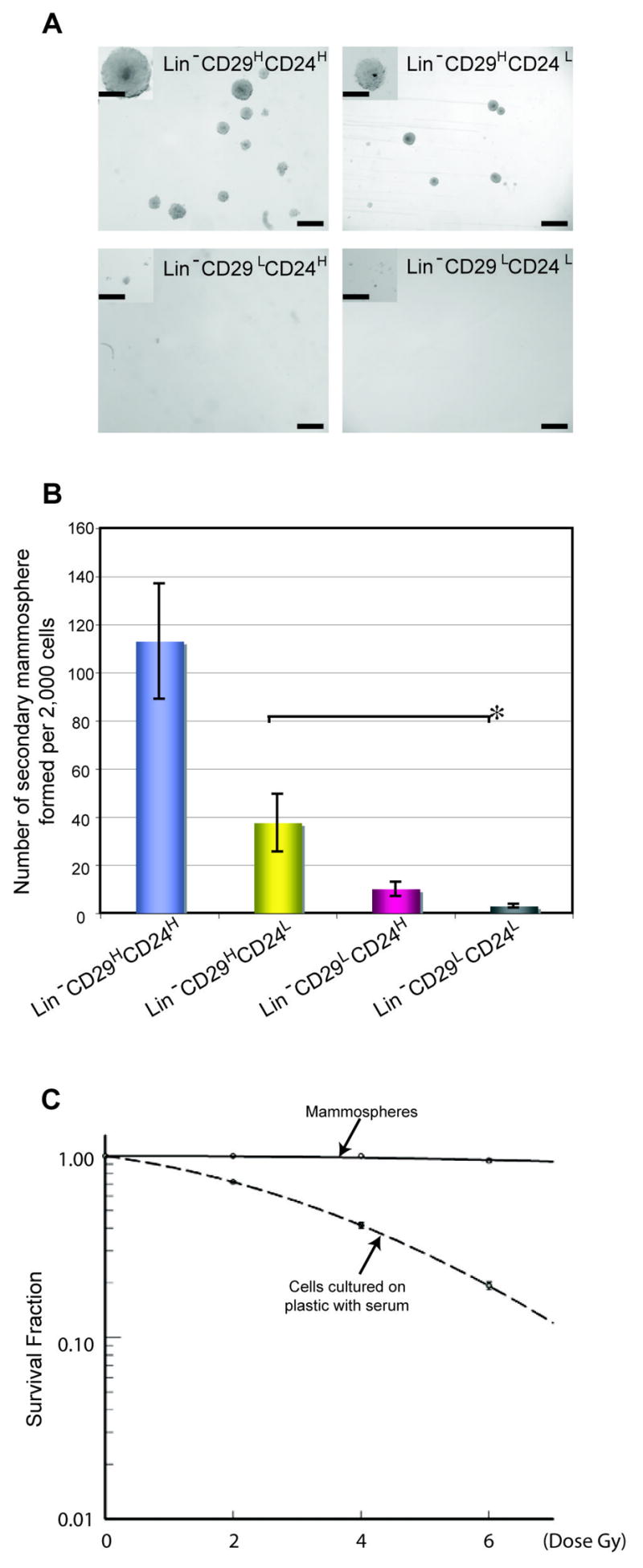
Mammosphere assay of sorted subpopulations of p53 null mammary tumors. A. Trypsin-digested primary mammospheres from T3 (a) Lin−CD29HCD24H, (b) Lin−CD29HCD24L, (c) Lin−CD29LCD24H, (d) Lin−CD29LCD24L were re-plated for secondary mammospheres. Pictures were taken on day 7 after plating secondary mammospheres. Scale bar, 250 μm. Scale bar in inserts, 100 μm. B. Plot was generated based on data from Tumors T1, T2, T3, T4, T6, T7, and T10. Two biological, three technical replicates from each tumor. P<0.05, *P<0.06. C. Serial passaged mammospheres (solid line, Tumor T7, passage 10) and collagenase-dissociated tumor T7 cells (spotted line) after grown on plastic for one week were dissociated with trypsin. Viable cells (Eight hundred cells/well in a 96-well plate) were irradiated, 0, 2, 4, or 6 Gy. After two weeks, the colonies were fixed, stained and counted. Relative survival rate were generated using SigmaPlot. Six technical replicates, two biological replicates were applied.
Microarray analysis
To begin to identify signaling pathways that distinguish the tumorigenic and non-tumorigenic subpopulations, microarray analysis was performed on the different subpopulations. For this analysis, subpopulations from three tumors (T1, T2, T7) were collected by FACS based upon expression of CD29 and CD24, and RNA was isolated from each of the subpopulations. Genes significant at p<0.01 using the univariate t-test with RVM (Random Variance Model) were identified in the comparisons of Lin−CD29HCD24H vs. each of the rest three groups (Lin−CD29HCD24L, Lin−CD29LCD24H, and Lin−CD29LCD24L). This p-value corresponded to false discovery rate (FDR) ~10%. There were 710 common probe sets (ps) in those three comparisons (representing 1.6 % of the total 45,037 ps) differentially expressed in the Lin−CD29HCD24H subpopulation as compared with the other three subpopulations, Lin−CD29HCD24L, Lin−CD29LCD24H, and Lin−CD29LCD24L (Figure 5A). Among 710 ps identified, 527 ps were up-regulated (from 1.4-fold to 4 -fold), and 183 ps were down-regulated (from 2-fold to 17-fold) (See Supplemental Table S5). A heat map was generated from microarray data of various subpopulations based on the Lin−CD29HCD24H cells differentially expressed 710 ps (Figure 5B) which were applied to Ingenuity Pathway Analysis for further analysis. Among 710 ps, 690 of them were mapped to known genes, 462 of them were found in the Ingenuity Pathway Analysis (IPA) knowledge database, and are labeled “focus” genes. Functional characterization of these differentially regulated genes assigns them to diverse biological processes including cell cycle, cellular assembly and organization, DNA replication, recombination and repair, gene expression, cell-to-cell signaling and interaction. Those top five IPA identified molecular and cellular functions in which either up- or down-regulated genes were involved are depicted in Figure 5B with the most significant genes among the networks highlighted below.
Figure 5.
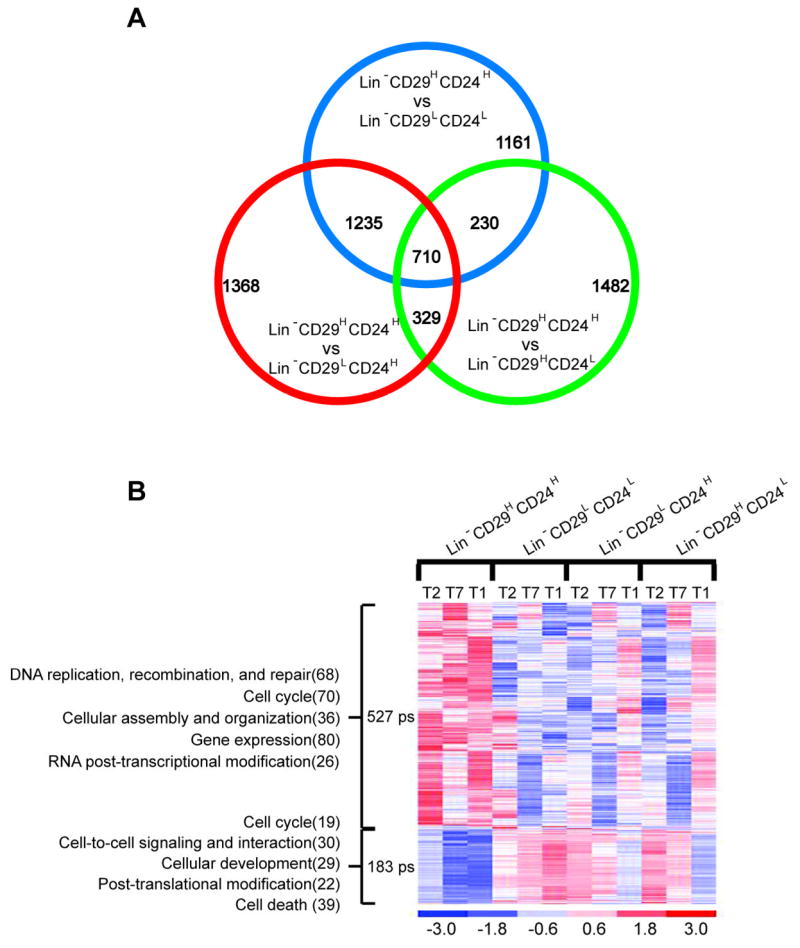
Differentially expressed transcripts in tumor-initiating cells of p53 transplantable mammary tumors. A. Venn diagram of transcripts differentially expressed in Lin−CD29HCD24H as compared with Lin−CD29HCD24L, Lin−CD29L:CD24H, and Lin−CD29LCD24Lsubpopulations of p53 null transplantable mammary gland tumors (p<0.01 for each comparison). B. The heat map of 710 differentially expressed transcripts in the tumorigenic cancer cell Lin−CD29HCD24H subpopulation. Each row represents a transcript; each column represents various subpopulation from three tumors. The red color indicates high level expression while blue indicates a low level of expression. The top five IPA picked molecular and cellular functions in which those down- and up-regulated genes involved are indicated on the left (number of molecules).
Lin−CD29HCD24H subpopulation showed increased expression of the polycomb genes Bmi1 and Ezh2, whose levels were up-regulated in a number of malignancies including breast cancer. Bmi1 plays an important role in regulating the self-renewal capacity of hematopoietic, as well as human mammary gland stem cell (32, 33). One of the more significant observations is the increased expression of DNA damage response and repair genes among Lin−CD29HCD24H cells. Nek1, Brca1, Chek1, Hus1, Ung, Xrcc5, Sfpq, Uhrf1 are among these genes. The altered expression of a subset of interesting genes was validated by quantitative PCR as shown in Supplemental Figure S8.
The ultimate goal of using mouse models is to investigate how the tumor-initiating cells identified in the tumors relate to normal stem cells, and which pathways might be deregulated, in order to provide more insight into the cell of origin, as well as to identify possible mechanisms involved in therapeutic resistance. The p53 null “normal” mammary epithelium has been shown to exhibit very similar global gene expression profiles as compared to p53 wild type Balb/c mammary epithelium through SAGE analysis (20). Thus, this allowed a comparison of the gene expression by microarray analysis of p53 null mammary tumor vs. that of p53 wild type epithelium.
Total RNAs from FACS-sorted subpopulations of wild type Balb/c mice MECs, Lin−CD29HCD24P(MEC) (Lin−CD29HighCD24Positive, putative mouse mammary gland repopulating unit, MRU, ~12%), Lin−CD29PCD24H(MEC) (Lin−CD29PostiveCD24High, 20%), Lin−CD24L(MEC) (Lin−CD24Low, ~46%) based upon expression of Lin, CD29, and CD24 were isolated (Supplemental Figure S9). The expression profiling of the MRU was compared with other subpopulations using a similar protocol to that used in the analysis of the p53 null tumor samples. Probe sets at p<0.01 were chosen and Ingenuity Pathway Analysis was used to identify target genes differentially expressed within the MRU. Consistent with the findings of Stingl et al. (9, 10), MRU cells expressed basal/myoepithelial markers with high expression of Krt14, Krt5, and smooth muscle genes, Actg2, Cnn1, Myh11, Myl9. On the other hand, expression of Krt8, Krt19, Muc1 was high in the Lin−CD29PCD24H(MEC) subpopulation.
Discussion
Evidence to support the CSC hypothesis has been derived primarily from studies of a variety of leukemias and solid human tumors in which FACS sorted cell populations were transplanted into immunocompromised mice. In a recent report, however, based upon the study of three syngeneic mouse lymphoma tumor models (34), the authors concluded that the majority of the cells isolated from these relatively homogeneous mouse tumors were able to initiate tumor formation. These results contrasted with those of John Dick and colleagues (5), who had identified a rare leukemia initiating population of “cancer stem cells” based primarily on xenografts of AML. Kelly et al. (34) questioned whether the overall microenvironment in xenograft models using immunocompromised mice might have accounted for the differences observed in these studies, thus casting doubt on the “cancer stem cell” hypothesis. However, an alternative interpretation is that these apparently disparate results most likely reflect the importance of selecting appropriate genetically engineered heterogeneous mouse models in which to perform these studies. The present study using the heterogeneous p53 null mouse mammary tumor model provides direct evidence using limited dilution transplantation experiments for the existence of a tumor-initiating subpopulation of CSCs in a syngeneic mouse model.
The anti-CD44 antibody successfully employed for the isolation of tumor-initiating cells from breast cancer cells present in pleural effusions (7) did not facilitate the isolation of a tumor-initiating subpopulation in the p53 null mouse model. This may reflect differences in the epitopes recognized by the specific anti-CD44 antibody expressed in human breast cancer as compared to mouse mammary tumor cells, possibly due to alternative splicing or post-translational modifications (35). CD44 has been identified as an important regulator of AML stem cell homing (36). However, the functional importance of CD44 in mouse mammary tumor-initiating cells has yet to be determined. While human breast cancer CSCs have been defined operationally as CD24−/LOW, the tumor-initiating population of p53 null tumors was predominantly CD24H, although some activity was observed in the Lin−CD29HCD24L subpopulation. Unlike in human, where CD24 was only expressed on luminal epithelial cells (37), in mice, the metastasis-associated CD24, a glycosyl phosphatidylinositol linked membrane protein, is expressed on major hematopoietic lineages, developing neural and epithelial cells, and has been suggested to be a marker for luminal MECs (38). The significance of this observation remains to be determined, but may be a consequence of the possible origin of these tumors from a bipotent progenitor as discussed below.
Breast cancer is not a disease that is driven through a simple mechanism, but through a complex set of changes (both genetic and epigenetic) in many pathways. BMI1 has been reported to play an important role in maintaining stem cell self-renewal. Up-regulation of Bmi-1 within the tumorigenic subpopulation may partially explain the increased mammosphere formation efficiency of such cells, but this remains to be demonstrated directly (32, 39). BMI1 can be recruited to histone H3 by EZH2, which was also increased in this tumorigenetic Lin−CD29HCD24H subpopulation. Overexpression of Ezh2 maintained long-term repopulating potential by preventing exhaustion of hematopoietic stem cells (40). Kamminga et al. identified 46 genes, categorized in three groups, that intereacted with Ezh2 in epigenetic chromatin modification of hematopoietic stem cells. Among these, 15 genes are among the up-regulated gene list in the present study, suggesting a similar mechanism may be active.
Bao et. al. (41) showed that following radiation, DNA damage checkpoint proteins were more activated in tumor cells bearing CD133+ (a marker for both neural and brain CSCs) than in CD133− tumor cells, suggesting that such tumor-initiating cells could be responsible for the recurrence of tumors following radiation therapy. Specific inhibitors of the checkpoint kinases, CHK1 and CHK2, were shown to sensitize CD133+ tumor cells to radiation treatment. Cell cycle checkpoint, DNA damage response and repair proteins are highly up-regulated in Lin−CD29HCD24H tumor-initiating cells. Interestingly, the proportion of Lin−CD29HCD24H tumor-initiating cells appears to increase upon successive mammosphere passaging, suggesting that the loss of p53 may promote symmetric division and expansion of this subpopulation. Taken together with the observation of radiation resistance of the mammospheres, a more efficient DNA damage repair mechanism may exist in Lin−CD29HCD24H cells as compared to the other subpopulations. Ongoing studies are directed at testing this hypothesis.
There are controversial reports as to ER status of mammary gland stem cells. Stem cells are believed to be slowly proliferating cells, and it has been reported that majority of long-lived MECs that retained the tritiated thymidine incorporation are ER+ luminal cells (42). “Side population” (SP) cells, a small percentage of cells with mammary stem cell properties, proportionally, contained six times as many ER+ cells as non-SP cells (43), though so far no in vivo transplantation experiments have supported the self-renewal property of the SP population. In contrast, two independent studies have reported that ER+ cells exhibit few stem cell properties, and that instead the basal population, which is enriched in mouse mammary stem cells, did not express ERα (44, 45). In agreement with these results, the mouse mammary gland MRU Lin−CD29HCD24P (MEC) cells exhibit basal features, with increased expression of K5, K14, and decreased expression of ERα. However, Lin−CD29HCD24H cells from p53 null mammary tumors, contain some ERα positive cells and cells with a mixed basal and luminal lineage (data not shown) supporting the hypothesis that they may have been derived from bipotent progenitors present in normal epithelium. The loss of p53 followed by other genetic changes appears to result in the deregulation of stem cell self-renewal and possibly an expansion of this progenitor population, which would then result in both basal and luminal, as well as ER+ and ER− cells in the tumorigenic Lin−CD29HCD24H subtypes. Therefore, the cell of origin for these tumors might be a bipotent progenitor, which possibly may be ER positive.
In summary, this study has identified a tumorigenic subpopulation within the p53 null mammary tumors as supported by both in vivo transplantation and in vitro mammosphere assay, and has identified genes that are preferentially expressed in the putative mouse mammary tumor-initiating cell population. The correlation of mammosphere with their tumorigenic outgrowth potential validates the use of the assay as in vitro surrogate, and suggests that it may be used for high throughput screens of small molecules and RNAi to identify pathways which are essential for the self-renewal of these cells. This may allow us, to identify new CSC markers, to test the functional importance of these markers in a syngeneic mouse model, and ultimately to improve the prognosis and treatment of breast cancer.
Supplementary Material
Microarray list of genes differentially expressed in CD29HCD24H vs. other subpopulations from p53 null mammary tumor c
Acknowledgments
We would like to thank the Texas Children’s Hospital Flow Laboratory for flow cytometry assistance, and the Baylor Microarray Core Facility for microarray analysis. We would also like to thank Drs. Margaret Goodell, Tracy Vargo-Gogola, Mike Lewis, Yi Li, and Stefan Siwko for constructive criticisms on this manuscript, and Drs. Edmund Chang, and Heidi Weiss for help with the array analysis, and statistical analysis. These studies were supported by grants CA16303 and U01-CA84243 from the National Institutes of Health (JMR), and grant PDF0504283 from the Komen foundation (MZ).
References
- 1.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 2.Huntly BJ, Gilliland DG. Cancer biology: summing up cancer stem cells. Nature. 2005;435:1169–70. doi: 10.1038/4351169a. [DOI] [PubMed] [Google Scholar]
- 3.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 5.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–43. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 6.Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–99. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 7.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu R, Wang X, Chen GY, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–26. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 9.Shackleton M, Vaillant F, Simpson KJ, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–8. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 10.Stingl J, Eirew P, Ricketson I, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–7. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 11.Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7:659–72. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- 12.Cardiff RD, Anver MR, Gusterson BA, et al. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000;19:968–88. doi: 10.1038/sj.onc.1203277. [DOI] [PubMed] [Google Scholar]
- 13.Cho RW, Wang X, Diehn M, et al. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells. 2008;26:364–71. doi: 10.1634/stemcells.2007-0440. [DOI] [PubMed] [Google Scholar]
- 14.Liu JC, Deng T, Lehal RS, Kim J, Zacksenhaus E. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res. 2007;67:8671–81. doi: 10.1158/0008-5472.CAN-07-1486. [DOI] [PubMed] [Google Scholar]
- 15.Jerry DJ, Kittrell FS, Kuperwasser C, et al. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19:1052–8. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- 16.Medina D, Kittrell FS, Shepard A, et al. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 2002;16:881–3. doi: 10.1096/fj.01-0885fje. [DOI] [PubMed] [Google Scholar]
- 17.Medina D, Kittrell FS, Hill J, et al. Tamoxifen inhibition of estrogen receptor-alpha-negative mouse mammary tumorigenesis. Cancer Res. 2005;65:3493–6. doi: 10.1158/0008.5472.CAN-04-3869. [DOI] [PubMed] [Google Scholar]
- 18.Aldaz CM, Hu Y, Daniel R, et al. Serial analysis of gene expression in normal p53 null mammary epithelium. Oncogene. 2002;21:6366–76. doi: 10.1038/sj.onc.1205816. [DOI] [PubMed] [Google Scholar]
- 19.Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abba MC, Fabris VT, Hu Y, et al. Identification of novel amplification gene targets in mouse and human breast cancer at a syntenic cluster mapping to mouse ch8A1 and human ch13q34. Cancer Res. 2007;67:4104–12. doi: 10.1158/0008-5472.CAN-06-4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langerod A, Zhao H, Borgan O, et al. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007;9:R30. doi: 10.1186/bcr1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardo FS, Su M, Borek C, et al. Transfection of rat embryo cells with mutant p53 increases the intrinsic radiation resistance. Radiat Res. 1994;140:180–5. [PubMed] [Google Scholar]
- 23.Welm AL, Kim S, Welm BE, Bishop JM. MET and MYC cooperate in mammary tumorigenesis. Proc Natl Acad Sci U S A. 2005;102:4324–9. doi: 10.1073/pnas.0500470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medina D. The mammary gland: a unique organ for the study of development and tumorigenesis. J Mammary Gland Biol Neoplasia. 1996;1:5–19. doi: 10.1007/BF02096299. [DOI] [PubMed] [Google Scholar]
- 25.Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–55. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing Journal of the Royal Statistical Society, Series B. Methodological. 1995;57:289–300. [Google Scholar]
- 30.Li Y, Welm B, Podsypanina K, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci U S A. 2003;100:15853–8. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu BY, McDermott SP, Khwaja SS, Alexander CM. The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Proc Natl Acad Sci U S A. 2004;101:4158–63. doi: 10.1073/pnas.0400699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–5. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 34.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 35.Choi CH, Roh CR, Kim TJ, et al. Expression of CD44 adhesion molecules on human placentae. Eur J Obstet Gynecol Reprod Biol. 2006;128:243–7. doi: 10.1016/j.ejogrb.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 36.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 37.Jones C, Mackay A, Grigoriadis A, et al. Expression profiling of purified normal human luminal and myoepithelial breast cells: identification of novel prognostic markers for breast cancer. Cancer Res. 2004;64:3037–45. doi: 10.1158/0008-5472.can-03-2028. [DOI] [PubMed] [Google Scholar]
- 38.Sleeman KE, Kendrick H, Ashworth A, Isacke CM, Smalley MJ. CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non-epithelial cells. Breast Cancer Res. 2006;8:R7. doi: 10.1186/bcr1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamminga LM, Bystrykh LV, de Boer A, et al. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107:2170–9. doi: 10.1182/blood-2005-09-3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 42.Zeps N, Bentel JM, Papadimitriou JM, D’Antuono MF, Dawkins HJ. Estrogen receptor-negative epithelial cells in mouse mammary gland development and growth. Differentiation. 1998;62:221–6. doi: 10.1046/j.1432-0436.1998.6250221.x. [DOI] [PubMed] [Google Scholar]
- 43.Clarke RB, Spence K, Anderson E, et al. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol. 2005;277:443–56. doi: 10.1016/j.ydbio.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 44.Sleeman KE, Kendrick H, Robertson D, et al. Dissociation of estrogen receptor expression and in vivo stem cell activity in the mammary gland. J Cell Biol. 2007;176:19–26. doi: 10.1083/jcb.200604065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asselin-Labat ML, Shackleton M, Stingl J, et al. Steroid hormone receptor status of mouse mammary stem cells. J Natl Cancer Inst. 2006;98:1011–4. doi: 10.1093/jnci/djj267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microarray list of genes differentially expressed in CD29HCD24H vs. other subpopulations from p53 null mammary tumor c