

Summary
In Drosophila, a phospholipase C-mediated signaling cascade links photo-excitation of rhodopsin to the opening of the TRP/TRPL channels. A lipid product of the cascade, DAG (diacylglycerol) and its metabolite(s), polyunsaturated fatty acids (PUFAs), have both been proposed as potential excitatory messengers. A crucial enzyme in the understanding of this process is likely to be DAG lipase (DAGL). However, DAGLs which might fulfill this role have not been previously identified in any organism. In this work, the Drosophila DAGL gene, inaE, has been identified from mutants that are defective in photoreceptor responses to light. The inaE-encoded protein isoforms show high sequence similarity to known mammalian DAG lipases, exhibit DAG lipase activity in vitro, and are highly expressed in photoreceptors. Analyses of norpA inaE double mutants and severe inaE mutants show that normal DAGL activity is required for the generation of physiologically meaningful photoreceptor responses.
Introduction
Visual transduction in Drosophila utilizes a G-protein-coupled, phospholipase C-mediated signaling cascade. Phospholipase C, upon activation via rhodopsin and G-protein, Gq, catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into two potential second messengers, diacylglycerol (DAG) and inositol trisphosphate (IP3). A body of evidence suggests that IP3 is not involved in Drosophila phototransduction (Acharya et al., 1997; Raghu et al., 2000), leaving the DAG branch as a likely source of messenger(s) of activation for the phototransduction channels, TRP (transient receptor potential) and TRPL (TRP-like). The mechanism by which the diacylglycerol (DAG) branch might activate the TRP/TRPL channels is still unresolved. The first indication that a lipid messenger might be involved was provided by Chyb et al. (1999), who showed that polyunsaturated fatty acids (PUFAs) could activate both TRP and TRPL channels either in intact photoreceptors or heterologous expression systems. Later, the same group presented evidence that DAG is required for photoreceptor excitation using DAG kinase mutants, rdgA (Masai et al., 1993). Because the conversion of DAG to phosphatidic acid is blocked in these mutants, they should have an elevated DAG basal level. These investigators showed that TRP/TRPL channels are constitutively active in rdgA (Raghu et al., 2000) and that diminished responses of hypomorphic PLC (norpA) mutants could be greatly enhanced by rdgA mutations (Hardie et al., 2002), in support of the contention that DAG might be excitatory to the channels. However, rdgA mutations are expected to raise the basal levels of not only DAG but also its metabolites. In addition to these two molecules, phosphatidylinositol 4,5-bisphosphate (PIP2) has also been suggested to play a role in channel excitation (review: Hardie, 2002). Currently, no consensus exists as to which, if any, of these might be the excitatory agent for TRP/TRPL channels.
Drosophila TRP is the founding member of a superfamily of TRP channel proteins. There are now nearly 30 mammalian members of this superfamily comprising seven subfamilies (reviews: Montell, 2005; Minke, 2006; Hardie, 2007). Although these channels are heterogeneous in their modes of activation, at least four mammalian TRP channels have been reported to be activated by DAG: TRPC2, 3, 6, and 7. While there may be variations in the mechanisms of activation of these channels, elucidation of Drosophila TRP/TRPL channel activation could provide insight into activation of these channels as well.
Since both DAG and its potential metabolite, PUFA, have been implicated in the activation of TRP/TRPL channels, a key enzyme in this process is likely to be DAG lipase, which catalyzes the hydrolysis of DAG. Little is known about DAG lipases. Two mammalian DAG lipase genes, DAGLα and β, have been identified by a bioinformatics approach and characterized both biochemically and molecularly (Bisogno et al., 2003), and many proteins homologous to DAGα and β have been identified across species. In the case of Drosophila, Huang et al. (2004) have described a mutant, rolling blackout (rbo), which they suggested might be in a DAG lipase gene. The protein encoded by the rbo gene, however, shows little homology to the known mammalian DAG lipases. Moreover, conditional loss of the RBO protein leads to rapid depletion of DAG, the opposite of what one would expect if RBO catalyzes the hydrolysis of DAG. Furthermore, in the absence of previous activity, the receptor potential is normal in rbo mutants, making it unlikely that RBO has any direct involvement in the activation of TRP/TRPL channels (Huang et al., 2004). Other than rbo, no candidate DAG lipase that might function in phototransduction has been reported in any species.
In this work, we report on a Drosophila DAG lipase (DAGL) gene, inaE, identified from mutants that are defective in photoreceptor responses to light. The protein isoforms encoded by this gene show high sequence similarity to the two known mammalian DAGLs, exhibit DAGL activity in vitro, are highly expressed in photoreceptors, and have access to rhabdomeres. Genetic evidence suggests that the inaE-encoded DAGLs interact in vivo with the DAG generated in the phototransduction cascade. Analysis of mutants generated by imprecise excision of P-element insertion in inaE, show that no physiologically meaningful photoreceptor responses can be generated if inaE gene is severely impaired.
Results
Mutant ERG phenotypes
The inaE gene was identified by two EMS (ethylmethane sulfonate)-induced allelic mutants, N125 and P19. These mutants are characterized by their “ina” (inactivation, no afterpotential) ERG (electroretinogram) phenotype. Wild-type flies, when placed on a white-eye (w) background, respond to a bright blue stimulus with a large response during light stimulus followed by a prolonged depolarizing afterpotential (PDA) after the light is turned off (Fig. 1A). A second blue stimulus elicits only a small response, originating from R7/8 photoreceptors, superposed on the PDA. By contrast, in ina mutants, the response begins to decay during stimulus (inactivation; arrowhead, Fig. 1A), and the decay continues after the stimulus (arrow, Fig. 1A). As a result, the PDA is greatly diminished in amplitude (no afterpotential). This phenotype can also be viewed as a mild form of the “trp” phenotype displayed by strong mutants of the TRP channel gene. As illustrated in Fig. 1A, in trp mutants, the response to the first blue stimulus decays nearly to baseline during stimulus, and there is no PDA.
Fig. 1. ERG analyses of inaEN125.

A) Comparison of ERG phenotypes of inaEN125 (N125), trpP343 (P343), and wild type (wt). ERGs recorded from wild type, inaEN125, and trpP343 are shown superimposed. The inaEN125 phenotype fell between those of wild type and trp in severity. B, Or: blue and orange stimuli generated by interposing, respectively, Corning 5433 and CS2–73 filters in the light path. B) Comparison of refractory periods among wild type, inaEN125, and trpP343. For each genotype, the fly was given a 5 s orange stimulus and allowed to dark adapt for 3 min before being exposed to the first stimulus. Immediately after the first stimulus, a 5 s orange stimulus was given and the second stimulus was delivered after a dark duration of 10, 30, 70, 110, 150, or 180 s. C-a) Semidominance of TrpP365 (P365). P365 heterozygotes (P365/+) generated responses that were about half-way between those of wild type and P365 homozygotes in size. C-b) Enhancement of P365/+ by N125. C-c) Non-complementation between KG08585 (P-line) and N125. KG08585 enhanced the P365/+ phenotype. Moreover, a heteroallelic combination of KG08585 and N125 also enhanced P365/+. C-d,e) The interaction between inaEN125 and TrpP365 was nullified by the expression of the D form (d) but not the A form of INAE. (e). D) Rescue of the inaEN125 ERG phenotype by the rescue construct harboring the inaE-RD isoform but not by that containing the inaE-RA isoform. Quantification of the above data based on multiple recordings is presented in Table 1.
Moreover, responses of inaE mutants resemble those of trp in that they both display refractory properties. Following a response to the first stimulus, only very small responses can be elicited from trp until they recover over a period of minutes (Fig. 1B, bottom traces), while wild-type responses recover almost immediately (Fig. 1B, top traces). Likewise, inaE mutants exhibit a similar refractory period, although the degree and duration of response suppression are not as pronounced or prolonged as in trp (Fig. 1B, middle traces).
In addition to the above similarities, inaEN125 acts as a genetic enhancer of TrpP365. TrpP365 is a semidominant allele of trp, which causes constitutive activation of TRP channels and, as a result, massive photoreceptor degeneration from excessive Ca2+ influx (Yoon et al., 2000). In TrpP365 homozygotes, degeneration is already so advanced in 1–2 d-old flies that essentially no ERGs can be elicited (Fig. 1C-a, top trace). TrpP365 heterozygotes exhibit a much milder phenotype and elicit ERGs of substantial amplitude at the same age (Fig. 1C-a, middle trace). However, if inaEN125 is introduced into the TrpP365/+ background, the resulting phenotype is as severe as that of TrpP365 homozygotes (Fig. 1C-b). The genetic enhancement of TrpP365/+ by inaEN125 and the basic similarity of ERG phenotypes between inaE and trp mutants (Fig. 1A–B) led to the hypothesis that the protein products of these two genes may interact and/or subserve closely related functions.
inaE cloning
A. Identification of an inaE candidate gene
Although we initially attempted traditional positional cloning approaches, the approach that ultimately proved successful was one based on DNA microarrays (review: Leung et al., 2007). Mutations that affect the ERG almost always also affect the level of mRNA produced by the mutated genes. Accordingly, DNA microarrays were carried out on inaEN125 and its corresponding wild type to look for genes that are altered in mRNA levels within the mapped limit of the mutant.
Deficiency mapping placed the inaE gene within the cytogenetic interval 12C5-6 to 12C8-D1 on the X chromosome. The deficiencies that were critical for this mapping were Df(1)benCO2 (12C5-6;12E) and Df(1)AR10 (12B1-2;12C8-D1), which both uncovered inaE (Fig. 2A). To be conservative, we examined microarray data from a much larger region than that determined by mapping, 12B4 to 12D4, a ca. 350 kb region with 41 genes. Within this interval, the following three genes exhibited the greatest and/or statistically most significant changes in mRNA level in inaEN125 compared to wild type: Yp3 at 12C1, CG33174 at 12C4-5, and CG32626 at 12C6-7 (Fig. 2B). The only RNA changes to which statistical analysis assigned a p-value of 0 in this interval were those corresponding to CG33174. Complementation tests between inaEN125 and the existing Yp3 mutants ruled out Yp3 as a candidate gene. Southern blots of deficiency heterozygotes probed with DNA fragments from CG32626 showed that this gene is outside the mapped limits of inaE, leaving CG33174 as the only viable candidate for inaE.
Fig. 2. Maps of the inaE region, gene, transcript, and protein.

A) The inaE genomic region. inaE mutations are uncovered by the deficiencies Df(1)benCO2 (first line) and Df(1)AR10(second line). The third line shows cytogenetic regions, and the fourth line indicates nucleotide coordinates. B) The CG33174 (inaE) gene and its transcripts. Yp3 and CG32626, shown in black, were identified along with CG33174 in microarrays as potential candidate genes. The inaE gene generates two transcripts, RA and RD, by alternative splicing. In the A form, the 13th intron is not spliced out and transcription ends in the 13th intron, thereby adding one amino acid residue to the A form of the protein (indicated by “643 + 1” in (C)). Untranslated regions are shown in grey. The sites of the inaEN125 mutation and of KG08585 insertion are indicated. C) The INAE protein. The regions of the four transmembrane segments and the lipase domain, as well as the 20-mer peptide used in antibody generation, are indicated. The C-termini of the A and D forms of the INAE protein are indicated by arrows and the C-terminal amino acid residue number. The vertical lines correspond to coding exon boundaries.
B. Validation of candidate gene identification
To verify the identification, we carried out the following series of validation experiments. 1) Sequencing: Sequencing the CG33174 gene in inaEN125 showed that inaEN125 carries a G->A transition at the 5′ splice site of the 11th intron (Fig. 2B). 2) P-insertion lines: We obtained four P element-insertion lines in this gene listed in Flybase. None of these showed any obvious ERG phenotype. However, one of them, KG08585, which carries a P insertion just outside the 13th exon (Fig. 2B), mimicked inaEN125 in enhancing the TrpP365/+ phenotype, when introduced into the TrpP365/+ background in a homoallelic combination (KG08585/KG08585; TrpP365/+) (Fig. 1C-c). More importantly, KG08585 in a heteroallelic combination with inaEN125 also enhanced the TrpP365/+ phenotype (KG08585/inaEN125; TrpP365/+) (Fig. 1C-c). Thus, KG08585 failed to complement inaEN125. 3) In vivo rescue: The CG33174 gene is predicted to encode two protein isoforms, PA and PD, through alternative splicing (Fig. 2B). We made cDNA rescue constructs of both the D and A forms, driven by the trp promoter. The D construct, but not the A construct, when introduced into the inaEN125 background by germline transformation, rescued the mutant ERG phenotype (Fig. 1D). Moreover, in the presence of the construct, inaEN125 no longer enhanced the TrpP365/+ phenotype (Fig. 1C-d). The A construct, on the other hand, did not have this effect (Fig. 1C-e). The above three lines of evidence unequivocally established CG33174 as the inaE gene.
Identification of the INAE proteins as diacylglycerol lipases
The CG33174 gene had not been characterized previously, and its function was electronically inferred as “triacylglycerol lipase (TAGL) activity” (Flybase). However, we considered the possibility that the above annotation could simply reflect dearth of information on DAGLs. The first two human DAGLs, DAGLα and β, were cloned and characterized by Bisogno, et al. (2003) by a bioinformatic approach and shown to be sn-1 type DAGLs. Multiple alignment of INAE-A and INAE-D with DAGLα and β revealed extensive sequence and domain conservations (Fig. 1, Supplemental Material). All four proteins are predicted to have four transmembrane segments near the N-terminal region, and they all have a lipase_3 domain with a highly conserved serine active site (Fig. 1, Supplemental Material). Overall sequence homology between INAE-D and the two human DAGLs is 39% identity and 56% similarity for DAGLα and 30% identity and 50% similarity for DAGLβ, respectively. In the lipase_3 domain, the sequence homology between INAE-A/D and the mammalian proteins rises to 60% and 45% identity and 73% and 63% similarity for DAGLα and DAGLβ, respectively.
To demonstrate that the INAE proteins have DAG lipase activity, the INAE-A and -D protein isoforms were expressed in E coli and purified to >95% purity to carry out DAG lipase assays using 1-stearoyl-2-arachidonoyl-sn-glycerol as substrate, and the lipase assay products were analyzed by LC-MS (liquid chromatography-mass spectrometry) and GC-MS (gas chromatography-mass spectrometry) for identification of hydrolysis products and kinetic studies (Experimental Procedures).
LC-MS detected four products from the analysis of both INAE isoforms: two primary products, stearic acid (18:0) and 2-arachidonoyl glycerol (2-AG), and two minor products, arachidonic acid (20:4) and 1-stearoyl glycerol (1-SG), eluting at 6.3, 9.3, 5.8, and 8.7 min, respectively (Fig. 3B). Two primary products corresponded to hydrolysis at the sn-1 position of DAG substrate and the two minor products corresponded to hydrolysis at the sn-2 position (Fig. 3A). Thus, in vitro, the two recombinant INAE isoforms are both DAG lipases highly preferential for hydrolysis at the sn-1 position, with the D form having much higher activity than the A form.
Fig. 3. DAG lipase assay.
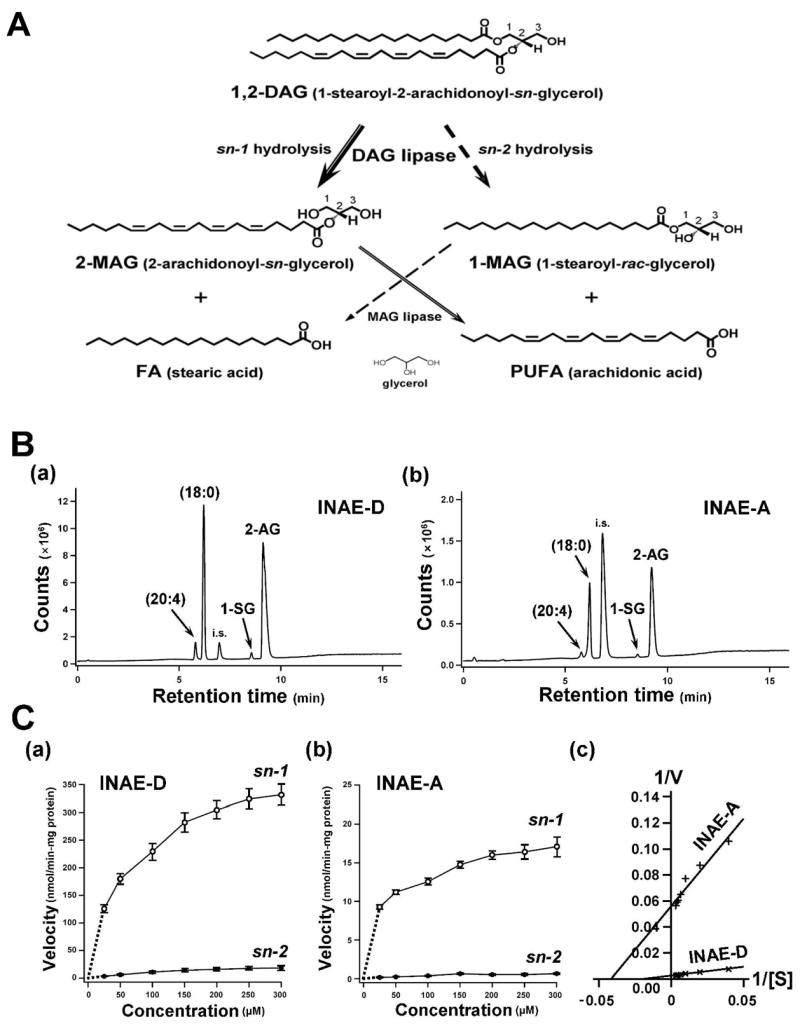
A) Potential metabolic pathways of the DAG substrate. B) Enzymatic characterization of INAE isoforms. Representative LC-MS total ion chromatograms (m/z 65–500) of INAE-D (B-a) and INAE-A (B-b) lipase assay products, using 1-stearoyl-2-arachidonoyl-sn-glycerol as substrate. Arachidonic acid (20:4), stearic acid (18:0), 1-stearoyl glycerol (1-SG), and 2-arachidonoyl glycerol (2-AG) were eluted at 5.8, 6.3, 8.7, and 9.3 min, respectively. Samples contained palmitic acid as an internal standard (i.s.). C) Velocity dependence plots as a function of substrate concentration obtained by GC-MS. Assays were performed on recombinant INAE-D (C-a) and INAE-A (C-b) using the same substrate as in (B). Hydrolysis rates at sn-1 and sn-2 positions were estimated by the amount of released stearic acid and arachidonic acid, respectively (Fig. 3A). Open and closed circles represent sn-1 and sn-2 reactions, respectively. Values represent the mean ± SD (n = 4). Note that the ordinate scales are different between (B-a) and (B-b) and between (C-a) and (C-b). C-c) Determination of kinetic parameters by Lineweaver-Burk plots. Calculations were performed by pooling sn-1 and sn-2 data for each isoform. V: velocity; [S]: substrate concentration.
Enzyme kinetics were studied by GC-MS quantification of the lipase assay products. Amounts of liberated stearic acid and arachidonic acid were used to estimate the hydrolysis rates at the sn-1 and sn-2 positions, respectively (Fig. 3A). Enzyme kinetic characteristics were extracted from velocity dependence plots, plotting hydrolysis rates against substrate concentrations (Fig. 3C-a and b). In both INAE-D (Fig. 3C-a) and INAE-A (Fig. 3C-b), sn-1 selectivity was about tenfold higher than that of sn-2. The kinetic values were calculated by pooling sn-1 and sn-2 data for each isoform (Fig. 3C-c). They were: Vmax = 400.00 nmol/min-mg protein and Km = 53.08 nmol (Y = 0.1327X + 0.0025, R2 = 0.9906) for INAE-D, and Vmax = 17.79 nmol/min-mg and Km = 23.62 nmol (Y = 1.3278X + 0.0562, R2 = 0.9377) for INAE-A (Fig. 3C-c). Thus, the D form represented about 20 fold higher lipase activity than the A form.
Molecular characterization of mutants and protein expression
The coding region and exon-intron boundaries of inaE were sequenced in both N125 and P19. N125 was found to harbor a G->A mutation at the 5′ splice site of the 13th intron (Fig. 2B). If splicing does not occur, translation would proceed into the 13th intron until a stop codon is encountered 39 bp downstream of the splice site. However, as described below, the mutation appears to be leaky. As for P19, no mutations were detected in either its inaE coding region or exon-intron boundaries. The P element insertion in KG08585 is 20 bp downstream of the 5′ junction of the 13th intron (Flybase; Fig. 2C).
Polyclonal antibodies were raised against a 20-mer peptide near the C-terminal end of the INAE-A protein (Fig. 2C; Experimental Procedures) and are expected to recognize both isoforms of the INAE protein. In Western blots of wild-type heads, the antibody recognized two bands of approx. 70 and 110 kD (Fig. 4B). The heavier band migrated somewhat larger than the predicted molecular weight of ~90 kD. Neutralizing the antibody with excess amounts of the 20-mer peptide completely eliminated any recognizable bands in Western blots (results not shown), demonstrating the specificity of the antibody for the 20-mer peptide. Fig. 4B shows a representative set of Western blot data comparing the amounts of INAE proteins detected in N125, P19, and KG08585 mutant heads with those in wild-type and eya (eyes absent) heads. Fig. 4C presents quantification of data based on three sets of measurements. The mutant eya, which has no eyes (Sved, 1986; Bonini, et al., 1997), had approximately 60 and 40% of the wild-type amounts of INAE-D and –A proteins, respectively, suggesting that INAE is expressed throughout the head, with substantial expression in the eye. All three inaE mutants, N125, P19 and KG08585, had protein bands of the correct sizes but at reduced levels (~70–90% for INAE-D and 60–70% for INAE-A). The results suggested that all three mutants are relatively mild, partial loss-of-function mutants, inaEN125 being the most severely affected of the three. The splice junction mutation in inaEN125 appeared to be leaky, since apparently correctly spliced INAE proteins were produced at a reduced level.
Fig. 4. Western blot analysis of inaE mutants with eya and wt controls.

A) A representative blot obtained in Western blot analyses carried out on the isolated heads of the inaE mutants, N125, P19, and KG08585. Beta actin was used as loading control; eya: eyes absent. B) Quantification of the data. Each data point was normalized with respect to the corresponding beta actin loading control, and the normalized data points in a given blot were in turn normalized to the wild-type PD value. Based on three repetitions.
The ERG phenotype reflected the mild nature of these mutations. N125, when placed on a white-eye background, consistently showed the slowly decaying ERG phenotype in response to a long white stimulus, shown in Fig. 1D. P19 also generated decaying ERGs under similar conditions, but the amount of decay was about one half of that seen in N125 and the phenotype was best seen after dark adaptation. However, N125 and P19 did not complement each other in complementation tests. None of the four P insertion mutants showed any obvious ERG phenotype, when tested as obtained on a w+ background.
Subcellular distribution of the INAE protein
The polyclonal anti-INAE antibody was used in fluorescence confocal microscopy to examine the distribution of the INAE proteins in the eye. There was very little difference in the pattern of antibody labeling between N125 and wild type except for the lower density of labeling in N125. Within the photoreceptors, the staining appeared as amorphous punctate material distributed unevenly throughout the photoreceptor cell body (Fig. 5A & B). Staining tended to be denser near the rhabdomeres (Fig. 5A), but it was not confined to any specific regions of the photoreceptor cell and seemed to be present throughout the length of the cell bodies (Fig. 5A & B). Although longitudinal images showed many labeled puncta that seemingly colocalized with rhabdomeres (Fig. 5B), most of these probably were located outside the rhabdomeres. Cross-sectional images also showed that most of the labeled puncta resided in photoreceptor cell bodies (Fig. 5A). Occasionally, however, some of the puncta were found localized within the rhabdomeres (arrowheads, Fig. 5A). Considering the geometry of rhabdomeres in cross-sectional images, the colocalizaiton was most likely real, suggesting that some of the INAE protein had access to the rhabdomeres.
Fig. 5. Fluorescence confocal microscopy of the photoreceptor layer.

A) A cross-sectional image taken ~35 μm from the distal tip of the ommatidia. Insets show the two highlighted ommatidia in higher magnification. Occasionally, some puncta are seen well inside the rhabdomres (arrow). B) A longitudinal section showing nearly the entire length of the ommatidia. Green: anti-INAE; red: F-actin in rhabodomeres. Rh: rhabdomere.
Analysis of inaE mutants
Because the available inaE mutants were all hypomorphic, they were relatively uninformative in revealing the role of DAGL in phototransduction. We attempted to resolve this problem by (1) generating strong inaE mutant alleles, and (2) using a norpA inaE double mutant to limit the production of DAG and hence its metabolites during phototransduction.
A. Generation of strong inaE alleles
We generated strong inaE mutants by remobilizing the P element in the P insertion line, KG08585 (Fig. 2B), to induce imprecise excision events. The offspring of 460 plus remobilization lines were examined by ERG (Experimental Procedures). None of the viable, excision-carrying progeny displayed severe ERG phenotypes. However, about 30 additional lines produced no viable offspring that were hemi- or homozygous for excision events. These results suggested that severe inaE mutations were homozygous lethal and that these mutations would have to be studied as somatic mosaics. Accordingly, we selected 25 of the lethal lines and generated, for each line, mosaic flies that were homozygous for imprecise excisions only in the eye using the EGUF/hid method of Stowers and Schwarz (1999) (Experimental Procedures).
Mosaic flies had slightly smaller than normal, mildly rough eyes (Stowers and Schwarz, 1999). Robust ERGs could be readily obtained from them, and the ERG phenotypes of the mosaic lines varied in severity. Three of these lines with ERG phenotypes varying from the mildest to severest, named xl29, xl15, and xl18, were chosen for analyses by electron microscopy, sequencing, real-time RT-PCR, and intracellular recordings.
A-1. Electron microscopy of xls (excision lines)
The structural integrity of photoreceptors in xl mutants was examined by electron microscopy. Fig. 6 compares cross-sectional images of groups of xl and wild-type ommatidia taken at about 35 μm depth from the distal tips of rhabdomeres at 2–3 days post-eclosion. The size of xl eyes and cross-sectional areas of rhabdomeres were significantly smaller than those of wild type. Moreover, the xl ommatidia were less orderly in their arrangement than wild type, and many contained less than the normal complement of seven rhabdomeres. However, all these differences appeared to be common to all three xl mosaics. To quantify the variations in the number of rhabdomeres per ommatidium, the entire eye sections were viewed in low magnification EM to determine the fractions of ommatidia containing 7, 6, 5, etc. rhabdomeres. This was done on 3–4 different eyes for each genotype at the same depth of section and at the same age. The results are presented in Fig. 6B. There were no statistically significant differences in the percentage of ommatidia containing various numbers of rhabdomeres among the three xl mosaics. Thus, while distinct structural differences existed between xl mosaics and wild type, they were common to all three mosaics.
Fig. 6. Electron microscopy of xl ommatidia.
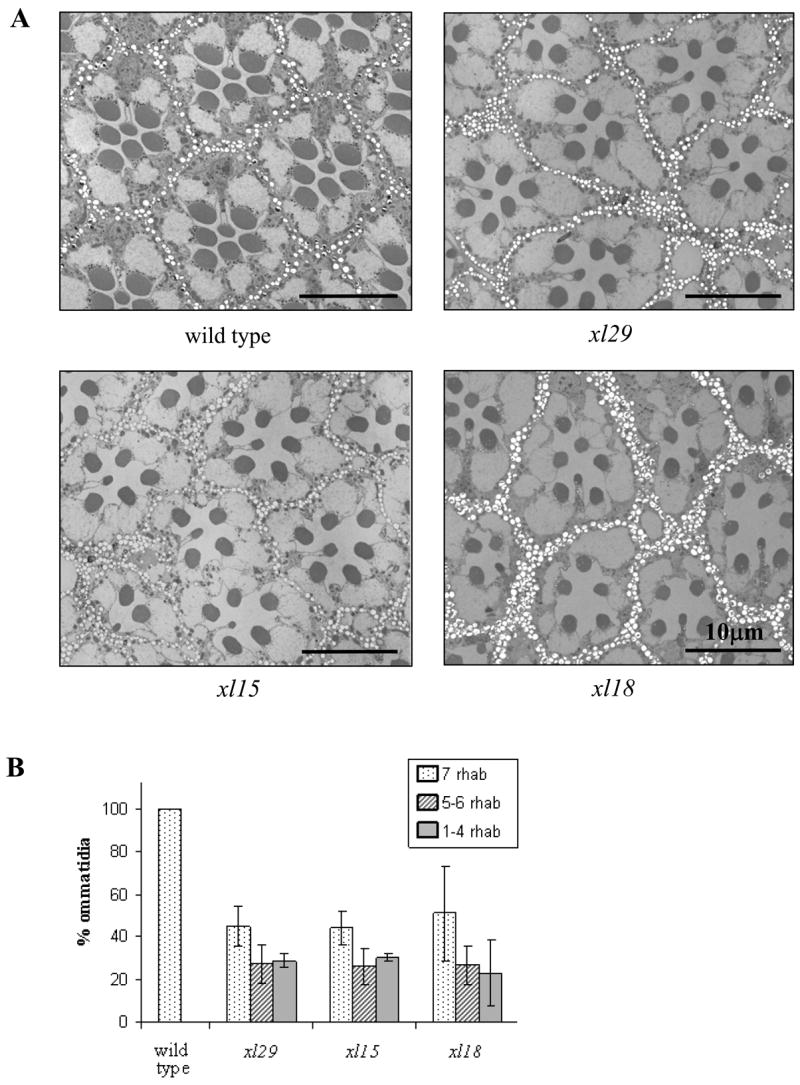
A) Transverse sections of groups of ommatidia obtained at ~35 μm depth from the distal tips of rhabdomeres in 2–3 d post-eclosion wild type, xl129, xl15 and xl18. B) Fractions of ommatidia containing 7, 5–6, and 1–4 rhabdomeres in wild type, xl29, xl15 and xl18. The number of rhabdomeres in each ommatidium was counted in low magnification views of the entire section in 4, 4, 3, and 4 different eyes of wild type, xl29, xl15, and xl18, respectively.
A-2. Molecular identification of alterations in xl
Because xl mutations are homozygous lethal in the adult, PCR amplifications for inaE gene sequencing or transcript quantification could not be performed in the adult. These were carried out in xl male embryos, which were viable and which could be identified through the use of a GFP-tagged balancer chromosome (Hagedorn et al., 2006; Experimental Procedures).
As in the case of N125 (Fig. 1D), the rescue construct containing the D form of inaE cDNA rescued the mutant phenotype of xl15 and xl18. The A form, while improving the phenotype slightly, was ineffective in rescuing it. By contrast, the lethality phenotype of xl15 and xl18 could be rescued by rescue constructs containing either the A or D form. However, the lethality of xl29 could not be rescued by either form. The results suggested that the lethality-causing alterations generated by imprecise excision resided within the inaE coding sequences in xl15 and xl18, but outside them in xl29. Consistent with this interpretation, no mutation was detected in the inaE coding sequences of xl29. The most severely affected of the xl lines, xl18, carried a ~1.5 kb deletion extending from the middle of the 12th intron to near the beginning of the 14th intron, eliminating both the 13th and 14th exons (Fig. 7A). The deleted sequence was replaced by a ~690 bp translocation from the right arm of the third chromosome. xl15 carried a >1.5 kb insertion that was too large to be amplified by PCR. The insertion site, determined within ~50 bp, was located just upstream of exon 13 (Fig. 7A).
Fig. 7. A) Molecular alterations in xl mutants.
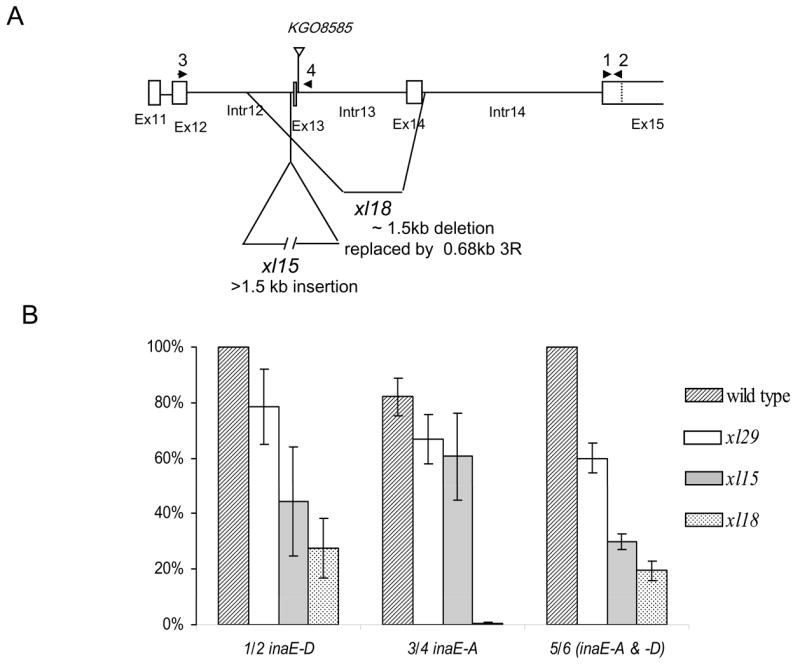
xl29 had no mutation in the inaE gene, xl15 carried a >1.5 kb insertion just upstream of Exon 13 (determined within 50 bp), and xl18 carried a ~1.5 kb deletion, which was replaced by a 0.68 kb sequence translocated from the right arm of the third chromosome. B) Quantitative RT PCR measurements, based on three repetitions. All values obtained with primer pairs 1/2 and 3/4 were normalized with respect to those of inaE-D specific transcript of wild type. Those values obtained with the primer pair 5/6 were normalized with respect to wild type values within that series. No inaE-A transcript was detected in xl18 because the A-form specific sequence was deleted in the mutant. Because the primer pair 5/6 amplified a much larger product than 1/2 or 3/4 (~400 bp vs. <150 bp), the amplification was less efficient. Locations of primer pairs 1/2 and 3/4 are shown in (A). The primer pairs 5/6 were located in the 4th and 7th exons, respectively.
A-3. RNA expression in xl mutants
The amounts of transcripts in xl mutants were determined by real-time RT-PCR of xl embryos (Experimental Procedures) and are shown normalized with respect to wild-type values of inaE-D transcript in Fig. 7B. The transcripts detected by primers common to both forms are shown normalized to the wild-type values within that series. inaE-D RNA expression in xl29 was about 80% that of wild type and that of xl15 was about 40%, while xl18 had about 25% expression. xl18 clearly is not a null mutant. Expression detected by primers common to both the A and D forms (5/6 in Fig. 7B) also showed a similar progressive decrease in relative amounts of RNA with severity of mutations.
B. Analysis of norpA inaE double mutants
The norpA gene encodes phospholipase Cβ (Bloomquist et al., 1988), which catalyzes the hydrolysis of PIP2 to DAG and IP3 during phototransduction (Fig. 8A). In a norpA inaE double mutant, the norpA mutation limits the production of DAG, hence restricting the amount of DAG metabolites generated even if inaE is hypomorphic. In order to be able to monitor the effects of mutations by electrophysiology, we used a hypomorphic norpA allele, norpAH43, which encoded a mutant PLC with ~7% of wild-type PLC activity that still allowed a receptor potential of sizable amplitude to be generated (Yoon et al., 2004). The amount of DAG generated in both the single and double mutants was expected to be very small compared to wild type because of the norpAH43 mutation (Fig. 8A). If inaE, indeed, encoded DAGL that acted on DAG generated by norpA-encoded PLC, then, in the double mutant, the small amount of DAG generated would be inefficiently metabolized because of the inaEN125 mutation. Thus, the amount of DAG metabolites generated was expected to be reduced and the amount of DAG was correspondingly increased in the double mutant compared to the single mutant. On the other hand, if inaE-encoded DAGL did not act on the DAG generated in the phototransduction cascade, introduction of inaE into the norpA background would have little or no effect on the NORPA-generated DAG.
Fig. 8. Analyses of inaE mutants and double mutants.
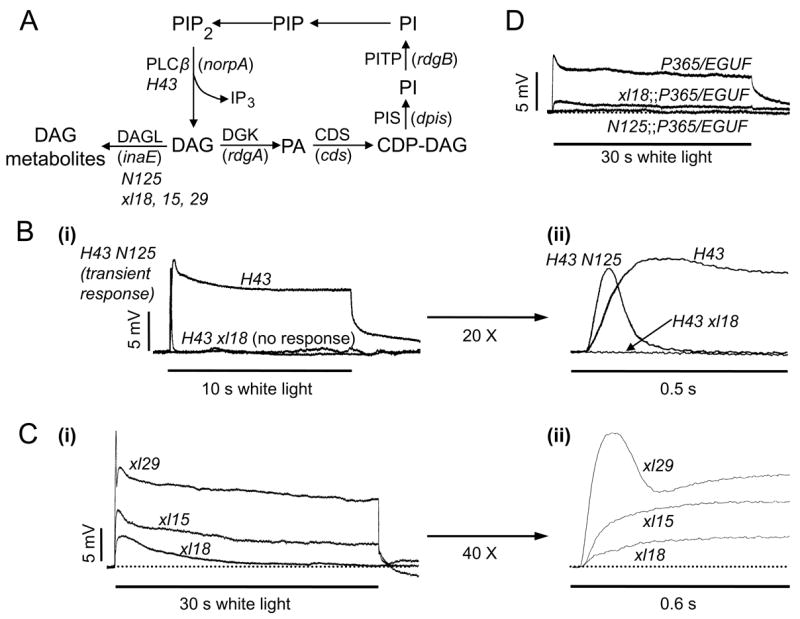
A) A diagram depicting a part of the phototransduction cascade and the phosphoinositide recycling pathway. Names of the genes encoding the proteins are shown in parentheses, and the mutants used in the present study are shown in italics under the protein names. H43: norpAH43; N125: inaEN125; xl18: inaExl18. B) norpA inaE double mutant study. B-i) Intracellular recordings of the receptor potentials elicited from the H43 mutant are compared with those obtained from the H43 N125 or H43 xl18 double mutant. Receptor potentials of 12.5 ± 2.9 mV peak amplitude and 6.4 ± 2.3 mV steady-state amplitude were obtained from H43 by a bright white stimulus of 10 s duration. The same stimulus elicited a fast transient response of 11.5 ± 2.4 mV peak amplitude with no steady-state component from the H43 N125 double mutant and no response at all from the H43 xl18 double mutant. B-ii) Initial portions of the responses shown in B-i are displayed in an expanded time scale. C) inaE mutations, xl15, xl18 and xl29. C-i) Intracellular recordings of receptor potentials elicited by a bright, white, 30 s stimulus from xl18, xl15, and xl29 are shown superimposed. B-ii) The initial portions of the responses in C-i are shown expanded in time scale by 40x. Quantification of the data, including those from N125, is presented in Table 2. D) Enhancement of the P365/+ phenotype. Introducing N125 to the P365/+ background abolishes the small, ~7 mV receptor potential present in P365/+. Replacing N125 with xl18 makes the phenotype of the double mutant less severe. EGUF is wild type for P365. Because it is needed in xl18;P365/EGUF to make the fly mosaic for xl18, it was also introduced in other genotypes to maintain the same genetic background.
The double mutants were generated using both the mild N125 and the severe xl18 alleles, and all recordings were performed intracellularly. As seen in Fig. 8B-i, a receptor potential of ~12.5 mV peak amplitude (~40% of wild-type amplitude) and ~6.5 mV steady-state amplitude could be elicited from norpAH43 by a bright 10 s white stimulus. In the norpAH43 inaEN125 double mutant, on the other hand, the same stimulus elicited a receptor potential of comparable peak amplitude (11.5 mV) that rose to peak within 0.1 sec and rapidly and completely decayed to baseline within ~0.25 sec (Fig. 8B-i & ii). If N125 is replaced with the severe inaE allele, xl18, in the double mutant, no response at all could be elicited (Fig. 8B-i & ii). Thus, under conditions in which the amount of DAG produced was restricted (hypomorphic norpA background), DAG metabolite(s) appeared to be required for the generation of the receptor potential.
C. Analysis of xl mutants
Shown in Fig. 8C are representative intracellular recordings of photoreceptor potentials elicited from the mosaic lines xl29, xl15, and xl18, by bright white stimuli of 30 sec duration. Quantification of various response parameters based on multiple recordings is presented in Table 2. The response parameters obtained from the xl29 flies were indistinguishable from those of wild-type. Thus, the xl29 data could be considered an internal wild-type control. By contrast, flies of the most severely affected of the three lines, xl18, gave only a small response of about 5 mV peak amplitude that rose slowly and decayed completely to baseline in about 15 s (Fig. 8C-i & ii). It lacked both the maintained steady-state and the fast initial phasic components compared to xl29 responses. Flies of the third mosaic line, xl15, responded with receptor potentials of intermediate phenotype. They had a small but distinct steady-state component of ~4 mV amplitude but lacked the rapidly rising, initial phasic component, present in xl29. Nevertheless, the initial rate of rise (activation rate) of the receptor potentials was substantially faster in xl15 than xl18 (Fig. 8C-ii; Table 2). These differences in receptor potential phenotype were observed reliably and consistently in multiple intracellular (n = 7–9, Table 2) and ERG recordings (n ≥ 15 each). Since no significant differences in morphological parameters were observed among these mutants, the phenotypic differences could not be attributed to structural differences.
Table 2.
Quantification of receptor potential parameter measurements.
Measurements were made on the following parameters of the receptor potential by intracellular recordings: peak amplitude, activation rate, latency, and amplitude at 30 s. The activation rate was defined as the slope of the straight line running from the initiation of the response to one-half peak amplitude. A) inaE mutants. The parameters obtained from xl29 were indistinguishable from those of wild type. B) Enhancement of P365/+. The milder inaE mutation, N125, displayed conspicuously stronger enhancement of P365/+ than xl18.
| Peak amplitude (mV) | Activation rate (mV/s) | Latency (ms) | Amplitude at 30s (mV) | |
|---|---|---|---|---|
| A. inaE mutants | ||||
| xl29 (n = 9) | 20.4 ± 1.4 | 407 ± 58 | 12.9 ± 1.4 | 9.8 ± 1.3 |
| N125 (n = 10) | 19.6 ± 2.7 | 219 ± 19 | 16.5 ± 1.3 | 3.0 ± 0.5 |
| xl15 (n = 7) | 8.6 ± 1.4 | 110 ± 17 | 16.3 ± 1.6 | 4.3 ± 0.8 |
| xl18 (n = 8) | 4.9 ± 0.5 | 53 ± 11 | 16.0 ± 1.0 | 0.5 ± 0.5 |
| B. Enhancement of P365/+ (n = 7 for all) | ||||
| P365/EGUF | 7.4 ± 2.4 | 77 ± 11 | 13.2 ± 1.2 | 4.0 ± 1.5 |
| xl18;;P365/EGUF | 1.2 ± 0.3§ | * | 16.2 ± 2.3 | 0.3 ± 0.2 |
| N125;;P365/EGUF | 0.2 ± 0.3§ | * | * | * |
Too small to be determined accurately.
The peak amplitude of xl18;P365/EGUF is significantly larger than that of N125;P365/EGUF (P < 0.0001).
We considered the possibility that blocking DAGL might have caused the DAG level to rise in xl15 and xl18 (Fig. 6A) leading to constitutive activity of TRP/TRPL channels (Raghu et al., 2000), collapse of the resting potential, and hence the degraded receptor potentials. Accordingly, resting potentials were determined for all three xl lines, inaEN125 and rdgAP35 single mutants, norpA inaE double mutants, as well as wild type. Resting potentials were difficult to determine in rdgA mutants, because strong depolarizations of cells made it difficult to judge penetration of cells reliably. The cells for which the resting potentials could be determined reliably tended to be less depolarized ones, biasing the sample toward larger resting potentials. Nevertheless, the average resting potential determined for rdgAP35 was −43 mV, significantly smaller than the wild type value of −56 mV (Table 3). Likewise, resting potentials were smaller than normal in all lines heterozygous for TrpP365, in which a substantial fraction of the TRP channels is expected to be constitutively active (Table 3). By contrast, resting potentials were normal in flies of all three mosaic lines (Table 3), ruling out collapse of the membrane potential as the basis of the small degraded responses of xl15 and xl18.
Table 3.
Quantification of resting potential measurements.
A) Resting potentials were determined from inaE mutants (N125, xl15, xl18), norpA inaE double mutants (H43 N125, H43 xl18), rdgA mutant (P35), and controls (wt, xl29). Resting potentials were normal in all genotypes except in rdgA. B) Interaction with P365/+. Introduction of N125, but not xl18, into the P365/EGUF background significantly depolarized the resting potential.
| Resting potential (mV) | |||
|---|---|---|---|
| A. inaE mutants, double mutants, rdgB mutant, and controls | |||
| wt | −56 ± 6 (n = 7) | N125 | −58 ± 3 (n = 7) |
| xl29 | −55 ± 5 (n = 7) | H43 xl18 | −51 ± 3 (n = 7) |
| xl15 | −53 ± 5 (n = 7) | H43 N125 | −55 ± 5 (n = 6) |
| xl18 | −52 ± 4 (n = 8) | P35 | −43 ± 5* (n = 8) |
| B. Interaction with P365/+ | |||
| P365/EGUF | −41 ± 3* (n = 7) | ||
| xl18;;P365/EGUF | −41 ± 4* (n =7) | ||
| N125;;P365/EGUF | −28 ± 3 § (n = 7) | ||
Significantly smaller than the wild-type value (P < 0.0002);
Significantly smaller than those marked with * (P < 0.0001).
Taken together, the above results suggested that the small degraded response obtained from xl18 most likely had its origin in the residual DAGL activity in xl18, since it is not a null mutant. Furthermore, responses of the mosaic lines xl15 and xl29 were larger and faster because of the increasingly active presence of the DAGL, INAE.
D. Enhancement of TrpP365/+ by inaEN125
However, the above line of evidence seemingly contradicted the observation that the TrpP365/+ phenotype was enhanced by inaEN125 (Fig. 1C-b & 8D). As discussed earlier, placing inaEN125 on a TrpP365/+ background abolished the small receptor potential that remained in TrpP365/+ (Fig. 1C-b; Fig. 8D). Since the reduction in size of the receptor potential in TrpP365+ was due to constitutive activity of a substantial fraction of TRP channels (Yoon et al., 2000) and adding inaEN125 was expected to increase the basal DAG level, a reasonable explanation was that DAG activated the remaining TRP channels. Consistent with this explanation, the resting potential was even more depolarized in inaEN125; TrpP365/+ than in TrpP365/+ (Table 3). To understand this phenomenon further, we placed inaExl18, instead of inaEN125, on a TrpP365/+ background (Fig. 8D). Now a small receptor potential could be recovered from the double mutant (Fig. 8D; Table 2), and the resting potential also returned to the same level as in TrpP365/+ (Table 3). Adding inaEN125 to the TrpP365/+ background would have increased DAG and decreased DAGL, and replacing it with inaExl18 would have sharply enhanced these conditions. Thus, the above results were opposite of what one would expect if DAG were excitatory to the channels.
Discussion
A growing body of evidence suggests that Drosophila phototransduction utilizes the DAG branch of the G-protein-coupled, PLCβ-mediated signaling pathway (reviews: e.g., Hardie, 2003). Although DAG lipase is expected to play a critical role in this pathway, no DAG lipase that could play such a role had been identified previously in Drosophila. Here we report on a DAG lipase identified from the Drosophila mutants, inaE. The inaE gene was found to encode two protein isoforms, INAE-A and INAE-D, by alternative splicing. Both these proteins are highly homologous to the two previously identified mammalian sn-1 type DAG lipases, and in vitro DAG lipase assays of recombinant INAE-A and INAE-D showed that both are DAG lipases highly preferential for hydrolysis at the sn-1 position (Fig. 3). Expression of the INAE protein is not restricted to the eye but occurs throughout the head (Fig. 4A & B), consistent with the finding that strong mutations in this gene are homozygous lethal. In photoreceptors, anti-INAE antibody labeling occurs as punctate staining scattered throughout the photoreceptor cytoplasm (Fig. 5A). Occasionally, some of the puncta are found within the rhabdomeres, indicating that some DAGL enters the rhabdomeres. Results of the norpA inaE double mutant study (Fig. 8B) provide strong functional support for the above observation. In this study, the receptor potential disappears in an inaE allele-dependent manner – the stronger the inaE allele in the double mutant, the more severe the double mutant phenotype. The allele dependence strongly suggests that the action of inaE-encoded DAGL is responsible for the observed double mutant phenotype. Furthermore, to affect the receptor potential phenotype, DAGL must act on the DAG generated by norpA-encoded PLCβ, and, for that to occur, DAGL must enter the rhabdomeres.
Because inaE mutations already available were all relatively mild, severe mutations were generated by imprecise excisions of a P element insertion in the inaE gene. These imprecise excision alleles were homozygous lethal and had to be studied as eye mosaics. Quantitative RT-PCR results showed that even the severest of these imprecise excision mutants, inaExl18, is not a null mutant and expresses RNA at ~25% of the normal level. This mutation profoundly affects the photoreceptor responses to light. If xl18 is placed on a norpAH43 background to reduce the amount of DAG generated, the light stimulus generates no response at all (Figs. 8B). In xl18 flies themselves, a bright prolonged stimulus generates only a small response of slow kinetics that decays to baseline completely during the stimulus (Fig. 8C). This response most likely represents the residual DAGL activity in this severely affected mutant. As the severity of mutation progressively decreases in the xl series of mutants, the receptor potential phenotype returns to normal in an allele-dependent manner (Fig. 8C). Again, the inaE allele-dependence strongly argues that the action of inaE-encoded DAGL is responsible for the observed change in the receptor potential phenotype. These results, taken together, thus, suggest (1) that the production of DAG metabolite(s) through the action of the inaE-encoded DAGL is required for the generation of photoreceptor responses to light and (2) that, in the absence of the metabolite, DAG plays little direct role in the activation of channels. However, the identity of the excitatory molecule cannot be specified from this work. It could be one or more of the products generated by INAE, such as monoacylglycerol (2-AG) or stearic acid (Fig. 3A), or even DAGL (INAE) itself.
While DAG may not have a direct role in channel activation, we found evidence suggesting that it may be important in regulating the action of the DAG metabolite that acts as an excitatory agent, although the evidence is still largely indirect. As discussed earlier, the ability of inaEN125 to act as an enhancer of TrpP365/+ (Fig. 1D-b; 8D) seems to present a quandary when considered in relation to the results summarized above. If DAG has little or no direct role in channel activation, how does one explain the disappearance of the small response present in P365/+ when N125 is added to this background (Figs. 1C-a & b; Fig. 8D, bottom trace)? A simple explanation for the phenomenon would be that DAG is excitatory to the channels and that adding N125 to the P365/+ background raised the level of DAG to make more channels to become constitutively active in N125;P365/+ than in P365/+. However, results of the experiment replacing N125 with a stronger inaE allele, xl18, in the N125;P365/+ double mutant (Fig. 8D) run counter to this simple explanation. Replacing N125 with xl18 should have sharply raised the basal DAG level further in the double mutant. If DAG were excitatory, the resting potential should have depolarized even more than before the N125 replacement and no receptor potential at all should have been obtained. Just the opposite results were obtained. A small but distinct receptor potential could be recorded from xl18;P365/+ (Fig. 8D, middle trace), and the resting potential has returned to the level in P365/+ (Table 3). These results are incompatible with the hypothesis that DAG is excitatory to the channel and instead provides another line of evidence for the conclusions summarized earlier.
However, the fact that a much more severe phenotype is obtained in N125;P365/+ than in P365/+ or xl18;P365/+ suggests that DAG may have a role in facilitating, enhancing and orchestrating the action of the DAG metabolite that serves as the excitatory agent. This action of DAG would be more noticeable under conditions in which a sufficient amount of the excitatory product is produced, as in N125;P365/+ than in xl18;P365/+. A similar action of DAG can also be inferred from the norpA inaE double mutant studies (Fig. 8B). In this series of experiments, hypomorphic norpA mutation, H43, was used to restrict the amount of DAG generated both in the single and double mutants. The response obtained from H43 N125 is short in duration but has nearly the same maximum amplitude as the H43 response and much faster time course of rise than the H43 response (Fig. 8B). The shortness of response duration may be due to the fact that under the conditions of this experiment (restricted DAG generation), the response cannot be sustained during a bright prolonged stimulus. This response arises as a result of DAGL activity because further reducing the DAGL activity (xl18 mutation) abolishes the response (Fig. 8B). However, the fact that adding N125 to the H43 background resulted in a response of faster time course may also be a manifestation of the enhancing and facilitatory effects of DAG on the excitatory agent. Speculating further, the facilitatory action of DAG might be in place to ensure that the excitation of channels is light-regulated since DAGL activity is not light-regulated while the generation of DAG is.
Experimental Procedures
Experimental animals
The wild-type strains were Berlin and Oregon R marked with white (w). Both inaEP19 and inaEN125 were recovered in ethylmethane sulfonate mutagenesis in the 70’s, P19 in the Pak laboratory and N125 by Martin Heisenberg, in Tübingen, Germany. The P-insertion mutant KG08585 was obtained from the Bloomington Stock Center. The xl mutants were obtained by inducing imprecise excision through remobilization of the P element insertion in the KG08585 mutant.
Microarray analysis, statistical analysis and candidate gene identification
Three independent replicate RNA samples (>5 μg) each were prepared from inaEN125 and a wild-type control. Total RNA was extracted from isolated heads using the RNeasy mini kit (Qiagen), and each sample was checked for quality and concentration before being handed over to the Center for Medical Genomics, Indiana University School of Medicine for processing. The chips used were the Affymetrix “GeneChip Drosophila Genome 2.0 Arrays,” based on Drosophila Genome Annotation release 3.1 and each comprising 18,952 probe sets. The processing of samples was all carried out by the Center according to the Affymetrix protocol (http://www.affymetrix.com/Auth/support/downloads/manuals/expression_s2_manual.pdf).
Statistical analysis of data, carried out by LA and RWD, consisted of using an analysis of variance (ANOVA) to statistically test for differences in mRNA steady-state levels between the mutant and the control using normalized data (Craig et al., 2003). It was necessary to adjust for the type I error rate to accommodate multiple testing issues, using both the false discovery rate (FDR) approach (Benjamin and Hochberg, 1995) and Holm’s sequential Bonferroni correction procedure (Holm, 1979). The significance level was chosen as 0.05.
Candidate genes for inaE were identified by looking for those genes within the mapped region that showed the largest and/or most statistically significant changes in the mRNA level in inaEN125compared to wild type. The Affymetrix Drosophila 2.0 annotation file was used to determine the identity of genes corresponding to probe sets detecting significant alterations in the mRNA level (http://www.affymetrix.com/support/technical/byproduct.affx?product=fly-20).
Validation of gene identification: rescue constructs
Total RNA was extracted from Berlin (wild type) fly heads, and cDNA was synthesized using the Superscript II reverse transcriptase and a primer mix of oligo (dT)15 and d(N)6 (Invitrogen).The cDNA for inaE-RA and inaE-RD were amplified from Berlin head cDNA using high fidelity pfuTurbo DNA polymerase (Stratagene). The inaE-RA and inaE-RD clones were completely sequenced for accuracy and extracted by endo-free plasmid maxi kit (Qiagen). They were cloned into the pCaSpeR[trp, w+] vector at the XbaI and BglII sites, allowing expression of the inserts to be driven by the trp promoter. The constructs were injected into inaEN125 mutant embryos.
Electrophysiology
Light stimuli originated from a 300 W Halogen lamp (OSRAM) and were delivered to the specimen with a light guide, using neutral density filters to achieve desired intensity, when needed. The unattenuated intensity at the level of the fruitfly was 810 μW/cm2. Both the recording and ground electrodes were filled with Hoyle’s solution. Orange and blue stimuli were generated by interposing, respectively, Corning CS2-73 and 5433 filters in the light path without attenuation. For other purposes, unattenuated white stimuli were used. All ERG recordings were performed at 25°C.
Intracellular recordings were performed as described in Johnson and Pak (1986). Both the ground and recording electrodes were inserted through a small cut made in the corneal-head junction. The recording electrode had 25–50MΩ resistance when filled with 2M KCl. The light source and the filters were as in ERG recordings. Signals were sampled at 2 kHz with an analog-to-digital converter (Digidata 1200A), and the data were acquired and analyzed with Axoscope software (Axon Instruments).
Expression of recombinant INAE-PD and INAE-PA
The inaE-RA and inaE-RD sequences were ligated onto the pET24a expression vector (Novagen) containing a C-terminal 6×His tag. Recombinant INAE-PA and INAE-PD were expressed in E. coli Rosetta (DE3) (Novagen) according to the standard protocol. The recombinant INAE protein isoforms were purified in a Ni-NTA agarose column (Qiagen) from the soluble fraction of the E. coli cell culture according to the manufacturer’s instructions. The recombinant proteins were concentrated by Amicon Ultrapure-15 centrifugal filter (Sigma) with exchange of buffer to 20 mM MOPS, pH 7.0. Corresponding fractions from the products of E. coli containing empty vectors served as negative controls. SDS-PAGE analysis indicated that the purity of the recombinant proteins obtained was >95%.
Diacylglycerol Lipase Assay
Using 1-stearoyl-2-arachidonoyl-sn-glycerol (Sigma) as substrate, assays were performed at 37 °C for 15 min in 20 mM MOPS buffer (pH 7.0) containing 0.25% (w/v) fatty acid-free bovine serum albumin (BSA) (Sigma) and 0.5% (w/v) gum arabic (Sigma). 20 μg recombinant proteins (INAE-PD or INAE-PA) were used per assay in 0.5 ml of reaction mixture. The substrate (10% w/v) was emulsified in 10 % (w/v) gum arabic and diluted into the reaction mixture to the final amounts of substrate. Assay products were extracted by chloroform/methanol (1:2, v/v) for molecular species analysis by LC-MS and kinetic analysis by GC-MS.
High performance liquid chromatography–mass spectrometry (LC-MS)
The HPLC-MS system consisted of an Agilent series 1100 HPLC coupled to an Agilent 6210 LC-TOF mass spectrometer via ESI interface. Reversed phase HPLC separation was conducted on a Zorbax SB-C8 column (50 mm × 4.6 mm I.D., 3.5 μm) (Agilent) at a flow rate of 1.0 ml/min using a gradient mobile phase. The mobile phase initially consisted of acetonitrile, water, and ammonium acetate in the following proportions: 45:55:0.1 (v/v/w). The acetonitrile fraction was then increased to 90% in a linear gradient over 10 min, and then held at 90 % for 10 min. The column was maintained at 35 °C. The MS system was operated in the negative ion mode with capillary temperature of 350 °C and capillary voltage of 4 kV. The spectra were recorded in the m/z 50–1699 range.
Gas liquid chromatography–mass spectrometry (GC-MS)
Samples were TBDMS-derivatized by adding anhydrous pyridine (Sigma) and MTBSTFA (Regis Technologies) and heating at 60 °C for 1 hr. GC-MS was performed using an Agilent 6890N model gas chromatograph interfaced to a LECO Pegasus III TOF-MS (LECO Corp.). A 30 m × 0.25 mm I.D., 0.25 μm HP-5MS capillary column (Agilent) was used under the following conditions: injector temperature, 280 °C; GC column temperature, 225 °C for 2 min, then 8 °C/min to a final temperature of 300 °C and held for 15 min; transfer-line temperature, 280 °C; ion-source temperature, 200 °C; and carrier gas, helium. Analyses were performed in the electron ionization (EI) mode at 70 eV. Mass spectra were acquired in the m/z 50–900 range.
Polyclonal antibody
Polyclonal anti-inaE antibodies were raised in rabbit against the 20-mer peptide (TARSTSAHPTDSSIALTLHQ), located within the peptide encoded by exon 12 (Fig. 2C). The antibody was purified by affinity chromatography using INAE peptide-conjugated agarose beads (Bethyl).
Confocal microscopy
Heads of 1 d-old flies were immersed in the fixative (2% paraformaldehyde, 0.21% sodium m-periodate, 1.37% DL-lysine, 0.001g saponin in 1 ml PBS buffer) overnight at 4°C. Eyes were dissected out and allowed to continue fixation for 1 hr at 4°C in the same fixative. Dissected eyes were washed 3x in PBST (phosphate buffered saline, 0.05% Tween20) and blocked for 2 hour in PBST/4% BSA/10% normal donkey serum. They were incubated in an anti-INAE antiserum diluted 1: 150 with PBS containing 4% BSA and 10% normal donkey serum overnight at 4°C. For actin staining, Texas Red-conjugated phalloidin (Molecular Probes) was added to the diluted primary antiserum in a ratio of 1:100. After 3x PBST washes, eyes were incubated for 4 hours at room temperature in donkey anti-rabbit IgG labeled with Alexa Fluor 488 (Molecular Probes) diluted 1:200 with PBS containing 4% BSA and 10% normal donkey serum. Specimens were washed 3x in PBST, mounted with ProLong Gold Antifade Reagents (Molecular Probes), and viewed on a Bio-Rad MRC1024 confocal microscope ( Nikon 60x, 1.4NA lens).
Electron microscopy
Heads of adult flies were bisected and immersed in the primary fixative (2.5% glutaraldehydde and 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4) and post-fixed in reduced OsO4 (1% OsO4 + 1.5% K3Fe(CN)6). The tissue was dehydrated in a graded ethanol/water series (10, 30, 50, 70, 90, and 2× 100% ethanol, 40 s each), followed by an exchange of ethanol with propylene oxide, infiltrated with Epon/Spurr (1:1) in graded mixtures with propylene oxide (25, 50, 75%, 2 hr each, 100% Epon/Spurr, 6 hr), imbedded in Epon/Spurr in a pyramid-tip mold, and polymerized for 48 hr at 60°C. Fixation and infiltration were accelerated by microwaving. Blocks were sectioned on Leica EM UC6 with a low-compression Diatome diamond knife, sections were stained with uranyl acetate followed by lead citrate, and images were viewed on the FEI/Philips CM-10 Biotwin transmission electron microscope (FEI/Philips, Hillsboro, OR).
Generation of imprecise excision lines (xl)
The P element in the P insertion line KG08585 (Fig. 2B) was mobilized by crossing the KG08585 stock with the (Δ2–3) stock. To track each excision event independently, balanced virgin female offspring carrying a P element excision were mated individually to males of the balancer stock to establish ~500 single female P-excision lines. All lines that produced viable male progeny hemizygous for excision were tested by screening for ERG phenotypes, and they all gave normal or nearly normal ERGs. However, there were about 30 lines that produced no viable males that are hemizygous for P excision events. Twenty-five of these homozygous lethal excision lines (xl lines) were used to make mosaic flies that are homozygous for xl only in the eye using the EGUP/hid method.
Generation of mosaic flies
Generation of mosaic flies followed the EGUF/hid method of Stowers and Schwarz (1999). In this method, a combination of the FRT/FLP and GAL4/UAS systems is used to induce, only in the eye, mitotic recombination between the chromosome carrying a lethal mutation of interest (xl in this case) and the GMR-hid-carrying chromosome. Mitotic eye clones that are not homozygous for xl are killed off by the dominant photoreceptor lethal transgene GRM-hid, resulting in adult eyes homozygous only for xl. Site specific recombination is achieved by recombining the flipase recognition site FRT in each of the xl- and GMR-hid-carrying chromosomes and providing the site specific recombinase FLP activity in precursor eye cells (driven by the eyeless promoter) through the use of the EGUF (eyeless Gal4 UAS FLP) stock available from the Stock Center. The mosaic flies had the genotype, xl FRT/GMR-hid FRT;;EGUF/EGUF. The GMR-hid FRT;;EGUF/EGUF stock obtained from the Bloomington Stock Center was used to recombine FRT into the xl-carrying chromosome and to construct the mosaic flies.
Sequencing xl embryos
Since xls are homozygous lethal in the adult and xl stocks are kept over the FM7a balancer, one cannot sequence the inaE gene in the adult. Sequencing was carried out in xl embryos, which were viable, using a modification of the procedure described by Hagedorn et al. (2006). The FM7a balancer which was normally used to balance the xl stocks (xl/FM7a × FM7a/⇁) was replaced with the FM7c-Kr-GFP balancer, which had the GFP-tagged Krüppl gene inserted into the balancer chromosome. Thus, the stock was kept as (xl/FM7c Kr-GFP × FM7c Kr-GFP/⇁). Krüppl-GFP is expressed in >3 hr old embryos, and all embryos carrying the balancer fluoresce at >3 hr. The only embryos that do not fluoresce are males hemizygous for xl. These were identified under a fluorescence microscope (Olympus SZX7) at 10 hr and used in sequencing.
Quantitative RT PCR of xl mutants
For the same reason given above, Quantitative RT PCR also had to be carried out on embryos. Total RNA was extracted from ~5 embryos using Qiagen RNeasy mini kit, and VersoTM SYBR Green 1-Step QRT-PCR ROX kit was used for the preparation of the reaction mixture. Measurements were carried out on the Applied Biosystems 7300 real-time PCR unit, and the ABI7300 system software was used for data analysis. The following primer pairs were used. Primers 1 and 2 (inaE-D specific): (5′TCGTGTCTTGTAGAGATCGA3′) and (5′TTATATAGAAGGCGGGCGGC3′); primer pairs 3/4 (inaE-A specific): (5′TCATCCCAAGCCCGACGAGCA3′) and (5′ATGCGCTTTGTAGGTGTGTG3′); primer pairs 5/6 (both inaE-A& -D): (5′TTGTTAGCGTCTCGTTGGTT3′) and (5′AGGTGCACCATATCGTGATT3′); RP49: (5′CGGTTACGGATCGAACAAGCG3′) and (5′TTGGCGCGCTCGACAATCT).
Supplementary Material
Table 1.
Quantification of the data in Fig. 1.
| Data summary ofFig. 1 B | |||||||
|---|---|---|---|---|---|---|---|
| Peak amplitude (mV) | % Recovery of Peak amplitude at | ||||||
| 10 s | 30s | 70 s | 110 s | 150 s | 180 s | ||
| wt (n = 6) | 28.8 ± 4.8 | 91 ± 4 | 103 ± 4 | ||||
| N125 (n = 9) | 24.7 ± 3.6 | 27 ± 11 | 55 ± 16 | 85 ± 11 | 93 ± 8 | 96 ± 4 | |
| P343 (n = 6) | 18.5 ± 3.9 | 2 ± 2 | 22 ± 14 | 78 ± 12 | 85 ± 17 | 95 ± 4 | 96 ± 4 |
|
| |||||||
| Data summary of Fig. 1 C | |||||||
| genotype (mean peak amplitude ± S.D. in mV) (n = 7 for all except #) wt (28.2 ± 3.4); P365/+ (14.0 ± 2.3); P365/P365 (0.5 ± 0.3); N125;;P365/+ (0.4 ± 0.3); N125/P-line;;P365/+ (3.0 ± 0.3); P-line/P-line;;P365/+ (4.8 ± 0.7); N125;;P365/P[inaE-RD] (14.4 ± 2.4); N125;P[inaE-RA];P365/+ (1.2 ± 0.6) (n = 5)# | |||||||
|
| |||||||
| Data summary of Fig. 1D | |||||||
| (n = 7 for all) | Peak amplitude (mV) | % peak amplitude at 30 s white light | |||||
|
| |||||||
| wt | 30.5 ± 1.5 | 56 ± 4 | |||||
| N125 | 26.8 ± 2.9 | 21 ± 7 | |||||
| N125;;P[inaE-RD] | 30.9 ± 2.5 | 60 ± 4 | |||||
| N125;P[inaE-RA] | 28.9 ± 2.9 | 13 ± 9 | |||||
Acknowledgments
We thank Kim Gilbert for help with the manuscript preparation, Chaoxian Geng and Guohua Li for their contributions in the early phases of the work, and Eric Harness, Junko Kitamoto, and Chia-Ping Huang for their technical assistance. The inaEN125 mutant was a generous gift from Martin Heisenberg in the 70’s. We gratefully acknowledge the receipt of Df(1)benC02, Df(1)AR10, FM7c Kr-GFP, and Yp3 stocks from Howard Nash, John Thomas, Henry Chang, and Mary Bownes, respectively. Other stocks were obtained from the Bloomington Stock Center. The microarray work was carried out in the Center for Medical Genomics, Indiana University School of Medicine (Director: Howard Edenberg). Confocal microscopy was carried out in the departmental confocal facility (Director: Don Ready), the sorting of xl mutant embryos were carried out in Henry Chang’s laboratory, electron microscopy was carried out in Purdue University Life Science Microscope Facility (Director: Debra Sherman), and lipase assays were performed in Purdue Metabolomics Profiling Facility. We gratefully acknowledge the help of Don Ready, Henry Chang, and Bruce Cooper. This work was supported by NEI grant EY00033 to WLP.
References
- Acharya JK, Jalink K, Hardy RW, Hartenstein V, Zuker CS. InsP3 receptor is essential for growth and differentiation but not for vision in Drosophila. Neuron. 1997;18(6):881–887. doi: 10.1016/s0896-6273(00)80328-1. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Stat Soc B. 1995;57:289–300. [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams E-J, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163(3):463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomquist BT, Shortridge RD, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak WL. Isolation of a putative phospholipase C gene of Drosophila, norpA, and its role in phototransduction. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- Bonini NM, Bui QT, Gray-Board GL, Warrick JM. The Drosophila eyes absent gene directs ectopic eye formation in a pathway conserved between flies and vertebrates. Development. 1997;124(23):4819–4826. doi: 10.1242/dev.124.23.4819. [DOI] [PubMed] [Google Scholar]
- Chyb S, Raghu P, Hardie RC. Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature. 1999;397:255–259. doi: 10.1038/16703. [DOI] [PubMed] [Google Scholar]
- Craig BA, Black MA, Doerge RW. Gene expression data: The technology and statistical analysis. J Agric Biol and Environ Stat. 2003;8(1):1–28. [Google Scholar]
- Hagedorn EJ, Bayrakta JL, Kandachar VR, Bai T, Englert DM, Chang HC. Drosophila melanogaster auxilin regulates the internalization of Delta to control activity of the Notch signaling pathway. J Cell Biol. 2006;173(3):443–452. doi: 10.1083/jcb.200602054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie RC. Regulation of TRP Channels via Lipid Second Messengers. Annu Rev Physiol. 2003;65:735–759. doi: 10.1146/annurev.physiol.65.092101.142505. [DOI] [PubMed] [Google Scholar]
- Hardie RC. TRP channels and lipids: from Drosophila to mammalian physiology. J Physiol. 2007;578:9–24. doi: 10.1113/jphysiol.2006.118372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie RC, Martin F, Cochrane GW, Juusola M, Georgiev P, Raghu P. Molecular basis of amplification in Drosophila phototransduction: roles for G protein, phospholipase C, and diacylglycerol kinase. Neuron. 2002;36:689–701. doi: 10.1016/s0896-6273(02)01048-6. [DOI] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- Huang FD, Matthies HJ, Speese SD, Smith MA, Broadie K. Rolling blackout, a newly identified PIP2-DAG pathway lipase required for Drosophila phototransduction. Nat Neurosci. 2004;7:1070–1078. doi: 10.1038/nn1313. [DOI] [PubMed] [Google Scholar]
- Johnson EC, Pak WL. Electrophysiological study of Drosophila rhodopsin mutants. J Gen Physiol. 1986;88:651–673. doi: 10.1085/jgp.88.5.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung H-T, An L, Tseng-Crank J, Kim E, Harness EL, Zhou Y, Kitamoto J, Li G, Doerge RW, Pak WL. Phototransduction in Drosophila: Use of microarrays in cloning genes identified by chemically induced mutations causing ERG defects. In: Fliesler SJ, Kisselev OG, editors. Signal Transduction in the Retina. Boca Raton, FL: CRC Press; 2007. pp. 195–217. [Google Scholar]
- Masai I, Okazaki A, Hosoya T, Hotta Y. Drosophila retinal degeneration A gene encodes an eye-specific diacylglycerol kinase with cysteine-rich zinc-finger motifs and ankyrin repeats. Proc Natl Acad Sci USA. 1993;90:11157–11161. doi: 10.1073/pnas.90.23.11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minke B. Cell Calcium. 3. Vol. 40. 2006. TRP channels and Ca2+ signaling; pp. 261–275. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C. The TRP Superfamily of Cation Channels. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- Raghu P, Colley NJ, Webel R, James T, Hasan G, Danin M, Selinger Z, Hardie RC. Normal phototransduction in Drosophila photoreceptors lacking an InsP3 receptor gene. Mol Cell Neurosci. 2000;15:429–445. doi: 10.1006/mcne.2000.0846. [DOI] [PubMed] [Google Scholar]
- Raghu P, Usher K, Jonas S, Chyb S, Polyanovsky A, Hardie RC. Constitutive activity of the light-sensitive channels TRP and TRPL in the Drosophila diacylglycerol kinase mutant, rdgA. Neuron. 2000;26:169–179. doi: 10.1016/s0896-6273(00)81147-2. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1639. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sved J. Eyes absent (eya) Drosophila Inform Serv. 1986;63:169. [Google Scholar]
- Yoon J, Ben-Ami HC, Hong YS, Park S, Strong LLR, Bowman J, Geng C, Baek K, Minke B, Pak WL. Novel mechanism of massive photoreceptor degeneration caused by mutations in the trp gene of Drosophila. J Neurosci. 2000;20(2):649–659. doi: 10.1523/JNEUROSCI.20-02-00649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J, Leung H-T, Lee S, Geng C, Kim Y, Baek K, Pak WL. Specific molecular alterations in the norpA-encoded phospholipase C of Drosophila and their effects on electrophysiological responses in vivo. J Neurochem. 2004;89:998–1008. doi: 10.1111/j.1471-4159.2004.02384.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.