

Abstract
Two factors that contribute to the progression of Parkinson disease are a brain defect in mitochondrial respiration and the generation of hydrogen peroxide (H2O2) by monoamine oxidase (MAO). Here we show that the two are linked. Metabolism of the neurotransmitter dopamine, or other monoamines (benzylamine, tyramine), by intact rat brain mitochondria suppresses pyruvate- and succinate-dependent electron transport. MAO inhibitors prevent this action. Mitochondrial damage is also reversed during electron flow. A probable explanation is that MAO-generated H2O2 oxidizes glutathione to glutathione disulfide (GSSG), which undergoes thiol-disulfide interchange to form protein mixed disulfides, thereby interfering reversibly with thiol-dependent enzymatic function. In agreement with this premise, direct addition of GSSG to mitochondria resulted in similar reversible inhibition of electron transport. In addition, the monoamines induced an elevation in protein mixed disulfides within mitochondria. These observations imply that (i) heightened activity and metabolism of neurotransmitter by monoamine neurons may affect neuronal function, and (ii) apparent defects in mitochondrial respiration associated with Parkinson disease may reflect, in part, an established increase in dopamine turnover. The experimental results also target mitochondrial repair mechanisms for further investigation and may, in time, lead to newer forms of therapy.
Parkinson disease is a progressive neurodegenerative disorder that affects primarily the dopamine (DA) neurons projecting from the substantia nigra pars compacta to the putamen and caudate regions of the brain (1). A genetically based or environmentally produced defect in mitochondrial respiration (2–4) and a natural metabolic pathway [i.e., monoamine oxidase (MAO)] that clears neurotransmitter from the cytosol of DA neurons (5–7) are believed to contribute to the development or progression of the disease. Because DA turnover is elevated in the parkinsonian brain (1) [as it is in animal models (8)], it follows that the surviving DA neurons are exposed to an increased flux of hydrogen peroxide (H2O2), derived as a consequence of MAO activity (Eq. 1):
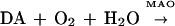 |
1 |
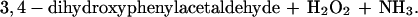 |
Therefore, H2O2-mediated damage is a prominent possibility.
The respiratory chain is associated with the inner mitochondrial membrane and is separated by an intermembrane space from MAO, which is localized to the outer membrane. Therefore, it is not immediately evident that metabolism of DA by MAO should affect the respiratory chain. However, H2O2 generated by MAO at the outer membrane has the ability to evoke changes, either directly or indirectly, at the distant inner membrane. In addition, contact sites exist between the inner and outer membranes (9).
Normally H2O2 is detoxified by the enzyme glutathione peroxidase (GSH-Px), resulting in the formation of glutathione disulfide (GSSG):
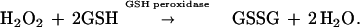 |
2 |
This is particularly true for mitochondria, which have no catalase, but do possess an abundant supply of both GSH and GSH-Px (9–11). Previous studies have demonstrated either loss of GSH (12) or accumulation of GSSG (13) within mitochondria during the metabolism of monoamines, including DA, by MAO. Although removal of H2O2 by GSH-Px extinguishes a direct oxidative threat, formation of GSSG can have deleterious consequences. GSSG reacts spontaneously (Eq. 3) with thiol groups in proteins (Pr-SH) to form protein mixed disulfides (Pr-SSG); this reaction is catalyzed by thioltransferases or thioredoxins (14, 15):
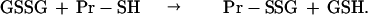 |
3 |
When the affected protein thiols are essential for biologic activity, that function is suppressed (e.g., succinate dehydrogenase, NADH dehydrogenase, ATPase, isocitrate dehydrogenase, and succinate-supported mitochondrial electron transport) (16, 17). Therefore, the thiol redox state is an important determinant of mitochondrial function, which, in turn, affects cellular viability (10, 18).
METHODS
Mitochondria were freshly isolated from whole brain (less the cerebellum) of male Sprague–Dawley rats (250–275 g; Taconic Farms) by the procedure of Clark and Nicklas (19). In a few experiments, the brains of pregnant female rats were used when they became available from unrelated experiments dealing with embryonic tissue. Isolation of mitochondria was carried out in the cold with the use of a refrigerated Sorvall RC2 centrifuge, equipped with an SS 34 rotor. The mitochondria were suspended in the isolation buffer, which consisted of 5 mM Mops containing 0.225 M mannitol, 0.075 M sucrose, and 1.0 mM EGTA, adjusted to pH 7.4 with KOH. After a 1-hr incubation period with and without MAO substrates and/or inhibitors, mitochondrial electron transport, supported by either pyruvate or succinate, was measured by the method of Berridge and Tan (17). Experiments were typically carried out at 37°C; additional experiments were conducted at ambient temperature, where indicated in the text. The measurement of electron transport in isolated mitochondria is based on the reduction of the dye (3-[4,5-dimethylthiozol-2-yl]-2,5-diphenyltetrazolium bromide; MTT). The same assay, applied to whole cells with glucose serving as substrate, is widely employed as an index of cell survival or proliferation (20, 21).
Experiments were generally constructed with 5 replicate samples consisting of 300 μl mitochondrial suspension (2 mg protein/ml) to which was added the MAO substrates (30 μl, 500 μM final concentration) and/or MAO inhibitors (30 μl, 2 μM deprenyl, and 2 μM clorgyline, preincubated with the mitochondria for 5 min). After a 1-hr incubation period with gentle shaking, 750 μl of a mixture of MTT (0.42 mg/ml) and pyruvate or succinate (15 mM) was added. Samples were quenched with 750 μl of lysing buffer (17) after 5 min at 37°C (or 15 min for experiments conducted at room temperature). The lysing buffer consisted of 10% (wt/vol) SDS and 45% (vol/vol) dimethylformamide, adjusted to pH 4.7 with glacial acetic acid. Samples were read after 5 min or longer. Absorbance readings were taken in duplicate with a plate reader (ATTC model 340; SLT Laboratory Instruments, Hillsborough, NC) and were reported as the difference between 550 nm and 620 nm. Spectrophotometry was preceded by a shaking period of 99 sec on the plate reader. Results for individual samples were expressed as a percent of the mean control value in the experiment. In several experiments (including the data presented in Fig. 2), an alternate analytic procedure was used (assay method B). Samples from the incubation procedure were chilled on ice, MTT and succinate were added, and then 150 μl aliquots were transferred to a microtiter plate, which was incubated in a 37°C oven for 15–30 min before quenching. The latter procedure was not preferred because it encompassed a lag phase as samples warmed from 0°C and, therefore, it was replaced by the primary procedure described above. Mitochondrial protein was determined by the method of Lowry et al. (22), with bovine serum albumin as standard. Protein mixed-disulfides were measured with a modification of the procedure described by Akerboom and Sies (23); the GSH released by reduction of disulfides in a protein pellet was measured with a modification (24) of the Tietze recycling assay (25).
Figure 2.

Partial reversal of inhibition with time during succinate-supported MTT reduction. Pooled results from three to four experiments (n = 15–18 per group) show a comparison of the time periods 0–15 min (15′) and 15–30 min (30′). Inhibition decreased significantly (P < 0.01) with time for both DA and benzylamine (two-tailed Student’s t test).
Chemicals were obtained from the following sources: benzylamine⋅HCl, dopamine⋅HCl, MTT, Mops, and succinic acid from Sigma; tyramine⋅HCl from Aldrich; clorgyline from Research Biochemicals International (Natick, MA); deprenyl from Midepex (Budapest, Hungary), and SDS from Pierce. The succinic acid was neutralized by titration with KOH. All other chemicals were the highest available grade. For statistical assessment, multiple comparisons were conducted by ANOVA, followed by the Tukey–Kramer test. Where appropriate, a two-tailed Student’s t test was used.
RESULTS AND DISCUSSION
These studies were conducted to evaluate the effect of MAO activity on mitochondrial electron transport. For this purpose, the MTT assay described by Berridge and Tan (17) was used. The latter investigators studied MTT reduction by isolated mitochondria with succinate as substrate and found that MTT reduction occurs at two sites (17): The major site (70–80% of the total) lies between cytochrome c and cytochrome a with a lesser contribution (20–30%) from a site between the S3 iron-sulfur center of complex II and the points of inhibition by antimycin A or chlorpromazine. Mitochondrial MTT reduction is sensitive to inhibition by the sulfhydryl reagent p-chloromercuribenzoate and, therefore, it is suitable for assessing protein–thiol susceptibility to MAO-generated H2O2. We initiated studies with succinate as electron donor, but also studied pyruvate-supported MTT reduction, which focuses on complex I of the electron transport chain.
Three MAO substrates were studied: DA, benzylamine, and tyramine. DA and tyramine are mixed MAO-A/MAO-B substrates, while benzylamine is an MAO-B substrate. In initial experiments, succinate-supported MTT reduction was suppressed after 1 hr to 72.1 ± 4.5% (SEM), 77.4 ± 3.9%, and 81.0 ± 3.1% of control, respectively, by 500 μM DA, benzylamine, and tyramine (P < 0.01, ANOVA followed by the Tukey–Kramer test, n = 9–15). With a lower concentration of 100 μM DA, MTT reduction was 90.1 ± 3.1% of control (P < 0.01). No inhibition was observed when 500 μM DA was added to control samples after the incubation period was complete (101.4 ± 0.9% of control, n = 10), indicating that the amines did not interfere with the MTT assay per se. In addition, incubation without added substrates did not impair the ability of mitochondria to carry out the reduction of MTT.
If MAO activity is required for the inhibition of mitochondrial electron transport, MAO inhibitors should block the effect. Fig. 1 illustrates the effects of a combination of 2 μM deprenyl (selective MAO-B inhibitor) and 2 μM clorgyline (selective MAO-A inhibitor). Suppression of mitochondrial reduction of MTT by DA, benzylamine, or tyramine was fully prevented by this combination (P < 0.001). These results show a direct dependence upon MAO activity for suppression of electron transport by monoamines.
Figure 1.

Inhibition of mitochondrial electron transport by MAO substrates (500 μM for 1 hr) and protection by MAO inhibitors (2 μM deprenyl plus 2 μM clorgyline). Results are pooled from three independent experiments (n = 15 per group). The mean control absorbance at 550 nm was 0.34 absorbance units. ∗, P < 0.001 vs. control; ∗∗, P < 0.001 vs. MAO substrate alone (ANOVA followed by Tukey–Kramer multiple comparison test).
During initial experiments, inhibition was less when the MTT assay was extended to a longer time span (assay method B; see Methods). This is illustrated in Fig. 2, which shows that the inhibition decreased by more than half when the 15- to 30-min time period was compared to the 0- to 15-min time period. To test directly the possibility that reversal of inhibition took place as a result of succinate-supported electron flow, we incubated mitochondria with 500 μM DA for 1 hr as usual, and then added succinate for an additional 10 min before adding MTT. Pre-incubation with succinate in three experiments restored MTT reduction to 97.4 ± 1.8% of corresponding control samples (P < 0.001, n = 14) compared with 75.3 ± 1.8% of untreated control without pre-incubation with succinate. This observation, in the absence of MAO inhibitors, confirms that damage to electron transport can be repaired during electron flow.
In Parkinson disease, the major mitochondrial defect is associated with complex I (NADH dehydrogenase activity) (2). Therefore, we performed additional experiments with pyruvate (an NAD-linked substrate) used in place of succinate to evaluate effects encompassing NADH dehydrogenase. Pyruvate was half as effective as succinate in supporting MTT reduction by rat brain mitochondria. However, as with succinate, pyruvate-supported MTT reduction was significantly suppressed by exposure to DA. Indeed pyruvate-based activity was more severely compromised by DA (44.0 ± 0.6% of control, P < 0.001, n = 10 per group, two-tailed Student’s t test).
Although MTT reduction is normally studied at 37°C (17, 26), experiments with mitochondria are frequently conducted at lower temperatures (12, 19). Therefore, additional experiments with pyruvate were conducted at ambient temperature. As previously observed with succinate, inhibition of pyruvate-supported electron transport by DA (50.2 ± 0.9% of control, P < 0.001) was completely prevented by MAO inhibitors (103.1 ± 1.5% of control, P < 0.001 compared with DA alone, n = 9–10/group). As with succinate, reversal of mitochondrial damage was observed when pyruvate was added 15 min before MTT: 38.4 ± 2.0% vs. 17.7% ± 1.4% inhibition before and after reversal with pyruvate, respectively (P < 0.001, n = 13–14/group). The apparently lesser reversal by pyruvate at room temperature compared with succinate at 37°C (see above) was due to the temperature used, since similar results were obtained with succinate at room temperature.
An explanation of the experimental results is provided in Fig. 3, which illustrates the linkage between MAO activity (outer membrane) and electron transport (inner membrane). The mechanism of inhibition of electron transport is presumed to be as follows: MAO-generated H2O2 is detoxified within the mitochondria by GSH-Px, producing GSSG. A portion of the GSSG undergoes thiol-disulfide exchange with free sulfhydryl (-SH) groups of proteins to form Pr-SSG, resulting in suppression of SH-dependent electron transport.
Figure 3.

Schematic representation of the MAO-dependent pathway for suppression of mitochondrial electron flow and subsequent recovery. MAO activity (outer membrane) leads to formation of GSSG, which forms disulfide linkages (Pr-SSG) with cysteine resides of proteins associated with the inner membrane, resulting in suppression of electron transport. The inhibition is relieved by reduction of GSSG by GSSG reductase, leading to reversal of the reaction that forms Pr-SSG. The NADPH-dependent protein-disulfide reductase activity of thioredoxin may also participate.
To test for the presumptive role of GSSG in mediating inhibition of electron transport, we added GSSG directly to mitochondria. It is known that GSSG can be taken up by mitochondria (27). Incubation of mitochondria with GSSG for 1 hr suppressed succinate-supported electron transport: 500 μM GSSG = 77.9% ± 0.6% of control and 200 μM GSSG = 89.7 ± 1.5% (P < 0.001, n = 14–19). Therefore, GSSG generated by GSH-Px within mitochondria can inhibit electron transport. Like the inhibition by DA, that evoked by GSSG was reversed by pre-incubation with succinate (P < 0.001): 500 μM GSSG = 92.5 ± 1.4% and 200 μM GSSG = 99.7 ± 0.7% (n = 9–14). Levels of Pr-SSG were elevated by 64.8 ± 8.2% after exposure to 500 μM GSSG in five independent experiments (P < 0.005, paired two-tailed t test).
Reversal of damage during electron transport can be initiated by reduction of flavin adenine dinucleotide in succinate dehydrogenase, leading to the chemical reduction of NAD+ via the “back-reaction” and, subsequently, NADP+ via transhydrogenase. Pyruvate dehydrogenase reduces NAD+ directly. NADPH is required by GSSG reductase for conversion of GSSG to GSH, which would facilitate the reduction of Pr-SSG and the reversal of damage. NADPH is also a cofactor for the protein disulfide oxidoreductase activity of thioredoxin (15). It is well established that metabolism of NAD-linked substrates by brain mitochondria leads to a major shift in the redox ratios for pyridine nucleotide cofactors in favor of their reduced forms. In this regard, the NADPH/NADP+ ratio is increased more readily than the NADH/NAD+ ratio (19).
The results and interpretations described here are supported by the following observations: Incubation of rat brain mitochondria with MAO substrates leads to loss of GSH (12) and accumulation of GSSG (28), which is retained within the mitochondria (13, 29). We observed elevated mitochondrial Pr-SSG (P < 0.001, Tukey–Kramer test) after incubation with monoamines for 1 hr at 37°C (pmols Pr-SSG/mg mitochondrial protein): 112 ± 7 (DA), 105 ± 10 (benzylamine), and 124 ± 8 (tyramine), compared with 15 ± 2 (control, mean ± SEM, n = 4–5 independent experiments). Increased Pr-SSG after incubation with monoamines has been observed previously (P. Werner and G.C., unpublished data). Increased Pr-SSG was also observed in the current experiments when DA was incubated with mitochondria at ambient temperature (120 ± 8 pmols/mg protein). These observations, as a whole, support the view that protein S-thiolation can form the basis for inhibition of mitochondrial electron transport. In addition, inhibition of succinate dehydrogenase, isocitrate dehydrogenase, and NADH dehydrogenase has been observed in primate intestinal mitochondria exposed to GSSG (16), in concordance with our observations concerning suppressed electron transport. In vivo, treatment of rats with haloperidol, which increases DA turnover, elevates Pr-SSG in both the midbrain and striatum (30), while inhibition of complex I has been observed after chronic administration of l-dopa (31).
DA can autoxidize to generate quinones that form adducts with protein thiols. In this way, the quinones can suppress thiol-dependent enzymatic function, such as electron transport. However, the similar effects (Fig. 1) of tyramine and benzylamine, which lack the autoxidizable catechol grouping, exclude quinones as causative agents. In addition, the ability of MAO inhibitors to completely prevent the action of DA indicate that autoxidation is not the route for DA toxicity to mitochondria.
In contrast to the MAO-dependent effects reported here with intact mitochondria, two reports (32, 33) described related experiments with disrupted mitochondrial preparations in which complex I activity was inhibited by catecholamines, but MAO inhibitors did not protect. In another report (31), inhibition of MAO protected complex I in sonicated mitochondrial preparations from damage by either 10 mM DA or 10 mM 6-hydroxydopamine. However, the freeze–thaw or sonication procedures used in the latter studies rupture the mitochondria and release both GSH and soluble GSH-Px, sensitizing the submitochondrial particles and membrane fragments to direct damage by H2O2 or quinones. Therefore, mechanisms of damage in the latter experiments are quite different from that indicated for intact mitochondria (Fig. 3).
The average concentration of DA in rat striatum is in the range of 65 μM (10 μg/g). This represents a lower concentration limit because DA is localized to dopaminergic nerve terminals, representing a much smaller mass of tissue. Estimates of catecholamine concentrations in peripheral sympathetic neurons range from 600 μM (34) to as high as 50 mM when the vesicular storage pool is included (35). Therefore, concentrations of 500 μM monoamine, used in the current experiments, are appropriate to investigate the effect of MAO activity on mitochondrial function. The mean concentration of GSH in fresh rodent brain is in the range 1.5–3 mM (24). However, because GSH is heterogeneously distributed (36, 37), local concentrations may be much higher in axons, nerve terminals, and glia. The concentration 500 μM GSSG, used in the current experiments, is representative of a partial oxidation of cellular GSH.
Our results demonstrate that mitochondrial electron transport is reversibly suppressed by MAO activity. Damage was greatest when pyruvate was the electron donor. Therefore, observations on diminished mitochondrial complex I activity in autopsy specimens from parkinsonian brain (2) can reflect, in part, the increased turnover of DA that characterizes surviving nigrostriatal DA neurons (1, 8); this may be particularly relevant during treatment with l-dopa, which provides DA to the brain. Because the extent of complex I deficiency (30–40%) seems too large to be accounted for solely by surviving DA neurons, it would appear that other cell types contribute to the observations made with autopsy specimens. Glia, which possess uptake mechanisms for DA, represent a cell type that may be at risk for suppression of mitochondrial respiration by monoamines. A genetic or environmentally based mitochondrial defect, amplified by DA turnover, can make neurons vulnerable to activation of the N-methyl-d-aspartate (NMDA) subclass of glutamate receptors (38, 39). In addition, diminished cellular respiration can increase cellular vulnerability by limiting the synthesis of GSH (40), a process that requires ATP.
The observation that mitochondrial repair was facilitated by electron transport is of particular interest. It means that susceptibility to mitochondrial damage will be controlled by mechanisms that either prevent GSSG accumulation or reverse protein S-thiolation. In vivo, a slow reversal over time (7 days) of mitochondrial complex I suppression induced by chronic l-dopa treatment of rats has been reported (31). Mitochondrial protection and repair mechanisms deserve serious assessment in model experiments and autopsy specimens of brain, as they may play a significant role in preventing or reversing mitochondrial defects in Parkinson disease.
Acknowledgments
This work was supported in part by a grant (to G.C.) from the U.S. Public Health Service (National Institutes of Health Grant NS-23017) and by a Fellowship (to R.F.) from the Parkinson’s Disease Foundation (New York).
ABBREVIATIONS
- MAO
monoamine oxidase
- DA
dopamine
- Pr-SH
protein thiol
- Pr-SSG
protein-glutathione mixed disulfide
- MTT
(3-[4,5-dimethylthiozol-2-yl]-2,5-diphenyltetrazolium bromide
- GSH
glutathione
- GSH-Px
glutathione peroxidase
- GSSG
glutathione disulfide
References
- 1.Hornykiewicz O, Kish S J. Adv Neurol. 1986;45:19–34. [PubMed] [Google Scholar]
- 2.Schapira A H V, Cooper J M, Dexter D, Clark J B, Genera P, Marsden C D. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 3.Mizuno Y, Ikebe S, Hattori N, Nakagawa-Hattori Y, Mochizuki H, Tanaka M, Ozawa T. Biochim Biophys Acta. 1995;1271:265–274. doi: 10.1016/0925-4439(95)00038-6. [DOI] [PubMed] [Google Scholar]
- 4.Gorrell J M, DiMonte D, Graham D. Environ Health Perspect. 1996;104:652–654. doi: 10.1289/ehp.96104652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen G. Adv Neurol. 1986;45:119–125. [PubMed] [Google Scholar]
- 6.Parkinson Study Group. Arch Neurol. 1989;46:1052–1060. doi: 10.1001/archneur.1989.00520460028009. [DOI] [PubMed] [Google Scholar]
- 7.Spina M B, Cohen G. Proc Natl Acad Sci USA. 1989;86:1398–1400. doi: 10.1073/pnas.86.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hefti F, Melamed E, Wurtman R J. Brain Res. 1980;195:123–137. doi: 10.1016/0006-8993(80)90871-9. [DOI] [PubMed] [Google Scholar]
- 9.Panfili E, Sandri G, Ernster L. FEBS Lett. 1991;290:35–37. doi: 10.1016/0014-5793(91)81219-x. [DOI] [PubMed] [Google Scholar]
- 10.Reed D J. Annu Rev Pharmacol Toxicol. 1990;30:603–631. doi: 10.1146/annurev.pa.30.040190.003131. [DOI] [PubMed] [Google Scholar]
- 11.Huang J, Philbert M A. Brain Res. 1995;680:16–22. doi: 10.1016/0006-8993(95)00209-9. [DOI] [PubMed] [Google Scholar]
- 12.Sandri G, Panfili E, Ernster L. Biochim Biophys Acta. 1990;1035:300–305. doi: 10.1016/0304-4165(90)90092-b. [DOI] [PubMed] [Google Scholar]
- 13.Werner P, Cohen G. FEBS Lett. 1990;280:44–46. doi: 10.1016/0014-5793(91)80200-m. [DOI] [PubMed] [Google Scholar]
- 14.Rabenstein D L, Millis K K. Biochim Biophys Acta. 1995;1249:29–36. doi: 10.1016/0167-4838(95)00067-5. [DOI] [PubMed] [Google Scholar]
- 15.Holmgren A. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 16.Benard O, Balasubramanian K A. Int J Biochem Cell Biol. 1995;27:589–595. doi: 10.1016/1357-2725(95)00019-L. [DOI] [PubMed] [Google Scholar]
- 17.Berridge M V, Tan A S. Arch Biochem Biophys. 1993;303:474–482. doi: 10.1006/abbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- 18.Meister A. Biochim Biophys Acta. 1995;1271:35–42. doi: 10.1016/0925-4439(95)00007-q. [DOI] [PubMed] [Google Scholar]
- 19.Clark J, Nicklas W B. J Biol Chem. 1970;245:4724–4731. [PubMed] [Google Scholar]
- 20.Mossman T. J Immunol Methods. 1983;65:55–63. [Google Scholar]
- 21.Hansen M B, Nielsen S E, Berg K. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 22.Lowry O, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 23.Akerboom T P M, Sies H. Methods Enzymol. 1981;77:373–382. doi: 10.1016/s0076-6879(81)77050-2. [DOI] [PubMed] [Google Scholar]
- 24.Slivka A, Spina M B, Cohen G. Neurosci Lett. 1987;74:112–118. doi: 10.1016/0304-3940(87)90061-9. [DOI] [PubMed] [Google Scholar]
- 25.Tietze F. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 26.Slater T F, Sawyer B, Sträuli U. Biochim Biophys Acta. 1963;77:383–393. doi: 10.1016/0006-3002(63)90513-4. [DOI] [PubMed] [Google Scholar]
- 27.McKernan T B, Woods E B, Lash L H. Arch Biochem Biophys. 1991;288:653–663. doi: 10.1016/0003-9861(91)90248-h. [DOI] [PubMed] [Google Scholar]
- 28.Werner P, Cohen G. Ann NY Acad Sci. 1993;679:364–369. doi: 10.1111/j.1749-6632.1993.tb18323.x. [DOI] [PubMed] [Google Scholar]
- 29.Olafsdottir K, Reed D J. Biochim Biophys Acta. 1988;964:377–382. doi: 10.1016/0304-4165(88)90038-4. [DOI] [PubMed] [Google Scholar]
- 30.Shivakumar B R, Ravindranath V. Brain Res. 1992;595:256–262. doi: 10.1016/0006-8993(92)91058-m. [DOI] [PubMed] [Google Scholar]
- 31.Przedborski S, Jackson-Lewis V, Muthane U, Jiang H, Ferreira M, Naini A B, Fahn S. Ann Neurol. 1993;34:715–723. doi: 10.1002/ana.410340515. [DOI] [PubMed] [Google Scholar]
- 32.Ben-Shachar D, Zuk R, Glinka Y. J Neurochem. 1995;64:718–723. doi: 10.1046/j.1471-4159.1995.64020718.x. [DOI] [PubMed] [Google Scholar]
- 33.Glinka Y, Tipton K F, Youdim M B H. J Neurochem. 1996;66:2004–2010. doi: 10.1046/j.1471-4159.1996.66052004.x. [DOI] [PubMed] [Google Scholar]
- 34.Corrodi H, Jonsson G. J Histochem Cytochem. 1967;15:65–78. [Google Scholar]
- 35.Anden N E, Fuxe K, Hamberger B, Hokfelt T. Acta Physiol Scand. 1966;67:306–312. doi: 10.1111/j.1748-1716.1966.tb03317.x. [DOI] [PubMed] [Google Scholar]
- 36.Slivka A, Mytilineou C, Cohen G. Brain Res. 1987;409:275–284. doi: 10.1016/0006-8993(87)90712-8. [DOI] [PubMed] [Google Scholar]
- 37.Philbert M A, Beiswanger C M, Waters K D, Reuhl K R, Lowndes H E. Toxicol Appl Pharmacol. 1991;107:215–227. doi: 10.1016/0041-008x(91)90204-r. [DOI] [PubMed] [Google Scholar]
- 38.Zeevalk G D, Nicklas W J. J Pharmacol Exp Ther. 1991;257:870–878. [PubMed] [Google Scholar]
- 39.Marey-Semper I, Gelman M, Levi-Strauss M. J Neurosci. 1995;15:5912–5918. doi: 10.1523/JNEUROSCI.15-09-05912.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Monte D A, Chan P, Sandy M S. Ann Neurol. 1992;32:S111–S115. doi: 10.1002/ana.410320719. [DOI] [PubMed] [Google Scholar]