

Abstract
σ32, the product of the rpoH gene in Escherichia coli, provides promoter specificity by interacting with core RNAP. Amino acid sequence alignment of σ32 with other sigma factors in the σ70 family has revealed regions of sequence homology. We have investigated the function of the most highly conserved region, 2.2, using purified products of various rpoH alleles. Core RNAP binding analysis by glycerol gradient sedimentation has revealed reduced core RNAP affinity for one of the mutant σ32 proteins, Q80R. This reduced core interaction is exacerbated in the presence of σ70, which competes with σ32 for binding of core RNAP. When a different but more conserved amino acid was introduced at this position by site-directed mutagenesis (Q80N), this mutant sigma factor still displayed a significant reduction in its core RNAP affinity. Based on these results, we conclude that at least one specific amino acid in region 2.2 is involved in core RNAP interaction.
Keywords: protein–protein interaction, sigma factor, transcription initiation
In eubacteria there exist a number of different sigma factors that are involved in the expression of specific sets of genes. The primary sigma factor controls the expression of primary housekeeping genes, and a number of alternative sigma factors regulate gene expression at specific stages of growth, or as a response to outside stimuli. In spite of the diversity of sigma factors, they all have similar activities during transcription initiation (reviewed in refs. 1 and 2). They direct transcription initiation by interacting with core subunits of RNA polymerase to form a holoenzyme, by recognizing the −10 and −35 regions of DNA promoters, and by stabilizing the separation of DNA strands. Ultimately, the sigma factor, at least in the case of σ70, dissociates from the rest of the RNAP, and the core RNAP subunit enters the elongation phase of transcription.
As expected of a group of proteins with similar functions, a sequence alignment of various sigma factors has revealed four regions of homology (2, 3). Region 2 is further divided into four subregions, and these regions have been implicated in various functions during transcription initiation. Genetic studies by a number of groups have indicated that region 2.4 is involved in the recognition of the −10 region of the promoter (4–7). Based on DNA footprinting studies, conserved aromatic amino acids in region 2.3 have been shown to promote the separation of DNA strands (8, 9). Mutation and deletion analyses have implicated region 2.1 in core RNAP binding (10, 11). However, the role of the most highly conserved region, 2.2, during transcription remains unknown.
Several groups have speculated that the most conserved region on sigma factors may be the core RNAP binding region (2, 12, 13). Their assumption was based on the premise that all sigma factors bind on the same region of core RNAP. If this idea were true, then region 2.2 should form the primary core RNAP binding region. Contrary to this assumption, a deletion analysis of σ70, the primary sigma factor in Escherichia coli, has shown that a deletion of parts of regions 1.2 and 2.1 reduces binding to core RNAP (10). However, deletions can profoundly affect the structure of a protein and do not indicate the specific contact site on an intact protein.
In an attempt to elucidate the function of region 2.2 during transcription, we have analyzed four mutants of σ32, an alternative sigma factor that confers heat shock promoter specificity (14). All four mutants contain a single amino acid change in region 2.2. Because the mutations occurred in the highly conserved region and because most of the affected amino acids themselves were conserved, we explored the possibility that one or more of these σ32 mutants might be defective in their ability to interact with core RNAP. Using glycerol gradient sedimentation to examine protein–protein interaction, we have shown that a conserved amino acid in region 2.2 is critical for efficient core RNAP interaction.
MATERIALS AND METHODS
Bacterial Strains and Plasmids.
Bacterial strains and plasmids are listed in Table 1. Isolation of rpoH alleles has been described (15).
Table 1.
Bacterial strains and plasmids used in this study
| Strain | Plasmid | Relevant genotype | Characteristics | Source |
|---|---|---|---|---|
| 285c | rpoD285 | (15) | ||
| BB1554 | ΔdnaK52 sidB2 | (16) | ||
| TC28-2 | rpoH28-2(ts) | (17) | ||
| CG410 | dnaK756 | (18) | ||
| RL721 | rpoC3531(His6) zja∷kan | Gift of R. Landick (University of Wisconsin, Madison) | ||
| NUT21 | dnaK756/pUHE211-1 pDMI,1 | This lab | ||
| NUT25 | dnaK756/phis173 pDMI,1 | This lab; pUHE211-1 derivative | ||
| NUT81 | dnaK756/phisB2 pDMI,1 | This lab; pUHE211-1 derivative | ||
| NUT84 | dnaK756/phis28-2 pDMI,1 | This lab; pUHE211-1 derivative | ||
| NUT95 | dnaK756/phis185 pDMI,1 | This lab; pUHE211-1 derivative | ||
| pUHE211-1 | σ32-c-his, Apr | (19) | ||
| pDMI,1 | lacIq, Kmr | (19) | ||
| pJet40 | dnaK-P1 promoter | Gift of J. Erickson (Columbia University, New York) |
ts, temperature sensitive.
Cloning of rpoH Alleles.
Cloning of rpoH173, rpoH181, sidB2, and rpoH28–2(ts) was essentially the same as previously described (20). After the site of mutation in each allele was determined by DNA sequencing, these alleles were subcloned into an rpoH histidine-tagged vector, pUHE211–1, by a fragment exchange of MluI and PstI segment of the rpoH gene. The plasmid-encoding σ32 mutant, rpoH185, was generated by amplifying pUHE211–1 with a primer that changed nucleotides 238–240 from CAG to ACC in the rpoH gene. The PCR product was then sequenced to check for any nucleotide misincorporations. None were found.
σ32 Purification.
Overproducers of his-tagged σ32 were grown at 30°C in 1 liter of 2 × YT medium with 100 μg/ml ampicillin and 50 μg/ml kanamycin. At A600 = 1, isopropyl B-d-thiogalactoside was added to 0.5 mM. Cells were grown for 20 min and poured into tubes with ice. All subsequent steps were performed at 4°C. After centrifugation at 5,000 rpm for 10 min in a Sorvall SLA-3000 rotor, the cell pellet was resuspended in 18 ml of ice-cold buffer X (50 mM KH2PO4, pH 7.9 at 4°C/300 mM KCl/50 mM Ile/50 mM Phe) and subjected to 10,000 lb/in2 in an ice-cold French press. The cell lysate was centrifuged for 30 min at 15,000 rpm in a Sorvall SS-34 rotor. The supernatant was loaded onto a 3-ml nickel-nitrilotriacetic acid agarose column at a rate of 0.4 ml/min. The column was subsequently washed with 40 ml of buffer X and then with 10 ml of buffer X + 15 mM imidazole. Nickel-bound proteins were eluted with 30 ml of 15–150 mM linear gradient of imidazole in buffer X. Purified σ32 proteins were dialyzed against two changes of 1 liter of 50 mM KH2PO4/300 mM KCl/50% glycerol. All mutant and wild-type σ32 protein concentrations were determined according to Pace et al. (21).
Core RNA Polymerase (RNAP) and σ70 Purification.
Proteins were purified according to the protocol received from R. Landick’s lab (personal communication) with the following modification. σ70 was stripped from RNAP using BioRex 70 (Bio-Rad) as described (22). Purified proteins were dialyzed with two changes of 1 liter of TGED (10 mM Tris·HCl, pH 7.9 at 20°C/5% glycerol/100 mM EDTA/100 mM DTT) + 50% glycerol. A molar extinction coefficient of 198,500 M−1 cm−1 and 41,745 M−1 cm−1 was used to calculate core RNAP and σ70 concentration, respectively (20, 23).
In Vitro Transcription Assay.
Holoenzyme containing different concentrations of σ32 proteins was reconstituted under the following conditions: buffer A (50 mM Hepes, pH 7.9 at 4°C/0.1 mM EDTA/1 mM DTT/100 mM NaCl/10 mM MgCl2), 0.5 mg/ml BSA, 150 μM ATP, 150 μM CTP, 1.5 μM UTP, 1.1 nM core RNAP, and 3.5 μg of DNA template containing the dnaK-P1 promoter. The size of the DNA template is 3.0 Kbp, and the molar ratio of promoter to core RNAP is 16:1. After incubating the mixture for 10 min at 30°C, 150 μM GTP and 33 nM [α-32P]UTP were added. The final reaction volume was 100 μl. Then, 5 μl of 2 mg/ml rifampicin was mixed into the reaction. After 10 min, the reaction was stopped by adding 100 mM EDTA, 2 M NH4OAc, and 0.4 μg/μl tRNA. Samples were precipitated in isopropanol and washed with 70% ethanol. RNA transcripts were then dissolved in 16 μl of loading dye (80% formamide/10 mM EDTA/1 mg/ml xylene cyanol FF/1 mg/ml bromophenol blue). Twenty-five percent of the transcripts were loaded on a 6% polyacrylamide gel. RNA transcripts were visualized using a PhosphorImager and quantitated under imagequant software (Molecular Dynamics).
Glycerol Gradient Sedimentation.
Holoenzyme was reconstituted in buffer A at 30°C for 15 min in a 200-μl volume. The concentration of sigma factors, σ70 and σ32, and core RNAP in each experiment was 100 nM. Samples were loaded on top of a 5-ml linear 15–35% (vol/vol) glycerol gradient and centrifuged at 4°C in a Beckman SW50.1 rotor for 24 hr at 48,000 rpm. Eighteen fractions were collected from the bottom of the tube and subjected to Western blot analysis with σ32 antiserum to detect the sedimentation pattern of σ32.
RESULTS
Mutants of σ32 in Region 2.2.
We have isolated and cloned 15 rpoH alleles by suppressing the temperature sensitivity of rpoD285 (15), which contains a 42-bp deletion mutation in σ70, the primary sigma factor in E. coli. DNA sequence analysis has revealed two alleles with a single amino acid change in region 2.2. These are rpoH173 (Q80R) and rpoH181 (P74R). Another allele sidB2 (E81G) was isolated by suppressing the growth deficiency of ΔdnaK52 (16). We have cloned and sequenced the fourth rpoH allele, rpoH28–2 (G82S), isolated by Waghorne and Fuerst (17). G82S fails to permit growth of lambda phage at high temperatures. Amino acid alignment of sigma factors has revealed that G82 is an absolutely conserved residue among all sigma factors, whereas Q80 and E81 are highly conserved residues (3). P74, on the other hand, may be conserved only among heat shock sigma factors (24).
Purified Proteins Exhibit Different Levels of Transcriptional Activity.
We were able to purify σ32 proteins to greater than 95% purity with the exception of G82S, which was unstable. The remaining purified proteins were assayed for their activity using an in vitro transcription assay. The sigma factors were preincubated with core RNAP to facilitate the reconstitution of Eσ32 holoenzyme. The reconstituted RNAP then recognized the DNA template, which contained the dnaK-P1 promoter and a terminator from an E. coli rRNA transcription unit, producing a transcript of 290 nucleotides. When only core RNAP or σ32 was present in the reaction, no transcripts were seen (data not shown). However, when both proteins were present, significant levels of transcripts were detected (Fig. 1).
Figure 1.

Graphical representation of transcripts from a dnaK-P1 containing promoter using holoenzyme reconstituted with wild-type or four σ32 mutants and core RNAP. Core RNAP (1.1 nM) was used in each reaction with increasing concentration of sigma factors. Data are expressed as means ± SD of at least four experiments in each curve.
Our analysis revealed a relatively high activity for wild-type σ32. When the ratio of σ32 to core RNAP was one to one, approximately 60 fmol of transcripts were produced from a possible maximum of 110 fmol. A Lineweaver–Burk plot was drawn to obtain a better estimate of the maximal level of transcripts and the dissociation equilibrium constant, Kd, which would be equivalent to the concentration of the sigma factors at the half-maximal level of transcription (graph not shown). Based on the points plotted on the graph, the following linear equation was obtained:
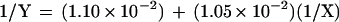 |
where Y is the concentration of the product and X is the concentration of σ32. Calculating the Y and the X intercepts provided the values for the maximal level of transcripts (91 fmol) and Kd (1 nM).
The results for the mutants were significantly different from those of the wild type. The level of transcripts for P74R at the equimolar concentration of core RNAP was only 20% of the wild type. E81G, a mutant that altered a conserved residue, was slightly lower in activity than P74R. Attempts to increase the concentration of mutant sigma factors to raise the maximal activity resulted in the precipitation of proteins. Q80R displayed the lowest level of activity. It was difficult to detect any transcriptional activity with equimolar amounts of sigma and core RNAP for this mutant sigma factor. Although it was not possible to delineate the maximal level of transcripts, it was clear from the titration curve that the Kd of Q80R–core RNAP complex would be considerably less than that of the wild type. Therefore, our findings indicate that although wild-type σ32 exhibits high activity, the three purified mutants are defective at some stage of transcription.
Binding to Core RNAP.
To substantiate further whether any of the mutants might be defective in core RNAP interaction, we used the glycerol gradient sedimentation technique. In the absence of core RNAP, σ32 sedimented near the top of the gradient (Fig. 2B). When an equimolar concentration of core RNAP was added into the reaction, σ32 sedimented to the bottom of the gradient, indicating that the sigma factor was binding to core RNAP (Fig. 2C). The sedimentation pattern of core RNAP with (Fig. 2A) or without (data not shown) σ32 was indistinguishable. The concentration of both σ32 and core RNAP used in this experiment was 100 nM. Even though this technique does not measure protein–protein interaction at equilibrium because of the changing conditions during sedimentation, a binding constant for a number of protein complexes has been estimated while taking into consideration the dilution effect during sedimentation (25–27). In our assay, an efficient interaction of the proteins suggests that the Kd of the complex is less than 100 nM. If the 2- to 3-fold dilution effect during glycerol gradient sedimentation is accounted for, then the Kd can be less than 33 nM. The above in vitro transcription analysis indicated that the Kd of σ32–core RNAP complex may be 1 nM.
Figure 2.

Western blot analysis of glycerol gradient sedimentation of sigma factors and core RNAP. The concentration of each protein in all experiments was 100 nM. The sedimentation pattern of both σ32 and core RNAP was determined using polyclonal antibodies [gift of C. Gross (University of California, San Francisco) and Y. N. Zhou (National Institutes of Health, Bethesda, MD)]. The left-most region of each panel represents the bottom of the tube (35% glycerol). A typical sedimentation pattern of β and β′ subunits of core RNAP with σ32 (A); σ32 only (B); σ32 with core RNAP (C); E81G with core RNAP (D); P74R with core RNAP (E); Q80R with core RNAP (F); σ32 + σ70 with core RNAP (G); Q80R + σ70 with core RNAP (H); P74R + σ70 with core RNAP (I); E81G + σ70 with core RNAP (J); and Q80N with core RNAP (K).
We examined the core RNAP binding affinities of E81G and P74R (Fig. 2 D and E, respectively). Although E81G appeared to trail the core RNAP by one-half, both mutants were almost identical to the sedimentation pattern of the wild type. These observations suggest that the cause of their decreased activity may lie elsewhere in the transcriptional process. However, another interpretation of these data is that the mutant proteins are indeed defective in core RNAP interaction, but the technique used in the experiment is not sensitive enough to detect the slight reduction in core RNAP affinity. Nevertheless, these two mutants did possess strong core RNAP affinity at the concentration used in the experiment. Q80R produced a strikingly different pattern of sedimentation (Fig. 2F). The majority of Q80R was unbound. The broad sedimentation behavior of Q80R in the presence of core RNAP also suggested that the complex is unstable, constantly associating and dissociating during sedimentation. To obtain an estimate of the binding constant of this complex, densitometry was used to calculate the percentage of the core RNAP-bound sigma. Our analysis indicated that only 20% was associated with core RNAP. Because one molecule of sigma interacts with one molecule of core RNAP, we estimated that the Kd for the mutant RNAP complex was approximately 100 nM, taking into consideration the dilution effect during sedimentation.
Competition for Core RNAP with σ70.
The Kd for the σ70–core RNAP interaction has been measured to be approximately 2 nM (23). If the Kd for the Q80R–core RNAP complex were 100 nM, then the addition of the core RNAP competitor σ70 in equimolar quantity would completely displace Q80R from the polymerase. To test this idea, we added σ70 into the reaction. In the following experiments, core RNAP was added into a mixture that already contained both types of sigma factors, either σ70 and σ32, or σ70 and Q80R. The results showed that even when σ70 is present, σ32 could effectively compete for core RNAP (Fig. 2G). Approximately 50% of σ32 was found in a complex with core RNAP. A similar percentage was observed in another report (28). However, this high affinity for core RNAP in σ32 was drastically reduced when a specific amino acid in region 2.2 was altered. As expected, Q80R was almost completely displaced in the presence of σ70 (Fig. 2H).
We then examined the effect of σ70 on the core RNAP binding affinity of E81G and P74R. In the absence of a core competitor, E81G and P74R were observed to interact with core RNAP quite efficiently (Fig. 2 D and E, respectively). When an equimolar amount of σ70 was introduced, P74R revealed a sedimentation pattern that was reminescent of the result obtained from σ32 (Fig. 2I). Slightly more than half of the sigma factor was found to be in complex with core RNAP. The other mutant, E81G, revealed a different result when σ70 was included in the reaction (Fig. 2J). Approximately 90% was found dissociated from core RNAP.
The Conserved Mutant Q80N Possesses Lower Transcriptional Activity.
To examine residue 80 of σ32 in greater detail, we generated the mutant Q80N (see Materials and Methods). This is a conserved change, as both are polar amino acids with an amide. The difference between the two residues is the shortening of the side chain by one angstrom. Although the change from a polar to a basic amino acid may potentially alter the stability of the structure, this is much less likely to occur with a conserved change, such as in glutamine to an asparagine. On the other hand, such a change may have a significant effect on protein–protein interaction.
Q80N was purified, and its activity was also determined using an in vitro transcription assay (Fig. 1). Surprisingly, the activity of this mutant was similar to the transcriptional activity seen in other mutants. In spite of the conserved change of amino acid, Q80N displayed a significantly lower activity than that of the wild type. In addition, the dose-response curve of Q80N was quite comparable to that of Q80R. This result suggests that Q80 is an essential residue for the function of the subunit of RNAP.
Q80N also Exhibits Reduced Core RNAP Affinity.
Encouraged by the above result, we determined the affinity of Q80N for core RNAP by glycerol gradient sedimentation and found that Q80N was as defective as Q80R in core RNAP binding (Fig. 2K). Approximately 25% of this mutant sigma factor was bound to core. Furthermore, the broad sedimentation behavior of Q80N indicated the unstable association with core RNAP. When the core RNAP competitor σ70 was included in the reaction, majority of Q80N failed to interact with core RNAP (data not shown). Therefore, we conclude that the length of the side chain is important for proper core RNAP association.
DISCUSSION
Mutations of Sigma that Affect Core RNAP Binding.
A few studies have been performed to identify the core RNAP binding region of sigma factors (10, 11, 29). Initially, a deletion analysis on σ70 identified a short peptide fragment containing sequences from region 1.2 and 2.1 that might be essential for core RNAP interaction (10). In support of this finding, a single amino acid substitution in region 2.1 of σE in Bacillus subtilis was shown to destabilize core RNAP interaction (11). The same study also reported that a number of single amino acid substitutions in region 2.2 had no effect on the function of the sigma factor, except for one residue that was believed to have destabilized the structure of the polypeptide. Unfortunately, the analogous residue corresponding to Q80 of σ32 was not investigated in this report.
Our study indicates that the most highly conserved region, 2.2, is involved in core RNAP binding. Using glycerol gradient sedimentation to observe holoenzyme formation with purified σ32 and core RNAP, we have shown that the mutation Q80R exhibits reduced core RNAP affinity. A similar result was obtained using a “small zone” gel filtration column (data not shown) (27). These results supported the initial observation of potential defects in core RNAP binding through in vitro transcription analysis. In addition, the use of purified proteins in our protein–protein interaction assay allowed us to estimate the Kd of the Q80R–core RNAP complex as approximately 100 nM. This is a 100-fold reduction in the core RNAP binding affinity, because the Kd of σ32–core RNAP complex was estimated to be 1 nM by our in vitro transcription experiment. Therefore, the reduction in the mutant’s Kd allowed us to predict that competition with equimolar σ70 would displace Q80R from core RNAP almost completely. Such displacement was observed.
The Structural Basis for the Effects of rpoH Mutations.
This report’s analysis of the σ32 mutants in region 2.2 is consistent with the recently determined crystal structure of a protease-resistant fragment of σ70, which is composed entirely of alpha helices and connecting loops (30). Q80, which we believe is critical for core RNAP interaction, has a corresponding amino acid, Q406, in σ70 (Fig. 3). The crystal structure reveals several aspects of Q406 that may support our observation with mutants at this position. Q406 is exposed to the solvent, which may promote favorable protein–protein interaction by being readily exposed on the surface of the protein. Structurally, it is located within the solvent-exposed hydrophobic patch, composed of highly conserved residues, and believed to be a critical region for core RNAP interaction (30). Finally, Q406 lies very close to the kink, centered about N383, which is thought to be an important structural motif for core RNAP interaction (30).
Figure 3.

Core RNAP binding region of the crystallized structure of σ70 (30). Residues His57, Pro74, Gln80, Glu81, and Gly82 of σ32 correspond to Asn383, Gln400, Gln406, Glu407, and Gly408 of σ70, respectively. (A) The frontal view of the region presumed to be important for core RNAP interaction is illustrated, where the kink is centered about Asn383. (B) The left-side view of A. [The figure was generated by using the program grasp (31).]
Glutamine is a polar amino acid with an amide group and has the potential to form hydrogen bonds with a residue or residues in core RNAP. Because the strength of hydrogen bonds is heavily dependent on the distance and the colinearity between hydrogen bond donors and acceptors, a substitution of glutamine with any other amino acid may have deleterious consequences to the polypeptide’s core RNAP affinity. When the substitution was a conserved amino acid, as it was the case with Q80N, a significant reduction in core RNAP interaction was noticed.
We were not able to purify G82S because of its extreme instability in vivo. This instability of G82S was first noticed by Yan Ning Zhou, using Western blot analysis to detect the level of proteins in vivo (personal communication). This residue is one of three residues in region 2.2 that are absolutely conserved among all sigma factors in the σ70 family (3). The other two are G85 and L86. G94C of σE in B. subtilis, which is analogous to G85 of σ32, failed to accumulate this sigma factor in vivo (11). We believe that this residue has also destabilized the alpha helix. The crystal structure of this region of σ70 has provided confirmation of our speculation (Fig. 3). In σ70, both G408 and G411, which correspond to G82 and G85 of σ32, lie in a region of closest proximity with neighboring alpha helices (30). Therefore, the introduction of amino acids bulkier than a glycine in the space-restricted hydrophobic environment should perturb the stability of the structure.
P74 is one of few residues in region 2.2 that are not conserved among all sigma factors. We thus were not surprised to discover that P74R was able to interact with core RNAP efficiently. Even when σ70 was added into the reaction, P74R was able to compete efficiently for core RNAP. The crystal structure of σ70 may provide an explanation for this mutant’s behavior (Fig. 3). The analogous residue on σ70 is Q400, an unconserved change. In addition, this residue is located somewhat removed from the hydrophobic patch consisting of solvent-exposed residues and is not part of an alpha helix but of a connecting loop. These features indicate that this residue may not be critical for core RNAP binding.
E81G appeared to associate with core RNAP almost as tightly as did wild type, even though its in vitro transcriptional activity was decreased. It is worth noting that E81 is a conserved residue, and the analogous residue of σ70, E407, is partially exposed to the solvent (Fig. 3B). There was the possibility that the core RNAP binding defect was subtle and undetectable by our assay. By introducing the core RNAP competitor, σ70, we were able to visualize the effect of the mutation on core RNAP affinity. Although there was a significant level of the sigma factor that interacted with core RNAP, most were found free of core RNAP. This evidence suggests that although Q80 has a profound effect, E81 has a subtle effect, at least in core RNAP interaction. However, we cannot rule out the possibility that there may be other defects for either of the mutants at this time.
Our work finally confirms the speculation that the most highly conserved region, 2.2, is involved in core RNAP binding. We conclude that mutations in Q80 lead to reduced core RNAP affinity, which directly contributes to its decreased activity during in vitro transcription. Because Q80 is a highly conserved residue, it would be interesting to determine whether mutations at this residue in other sigma factors can lead to the unstable Eσ complex. We are currently investigating this possibility.
Acknowledgments
We thank Audrey Nolte for isolating, cloning, and sequencing rpoH173. We are also indebted to Arun Malhotra for providing the figures for the crystal structure of σ70, and John Bertsch for assisting us in the illustration of the figures. We are indebted to Robert Landick and James W. Erickson for unpublished strains and to Yan Ning Zhou for showing us her unpublished data. Carol Gross and Richard Burgess provided helpful suggestions for improvement of the manuscript. This work was supported by Research Grants MV 484 from the American Cancer Society and AI-08722 from the National Institute of Allergy and Infectious Diseases.
ABBREVIATION
- RNAP
RNA polymerase
References
- 1.Gross C A, Lonetto M A, Losick R. In: Transcriptional Regulation. Yamamoto K, McKnight S, editors. New York: Cold Spring Harbor; 1992. pp. 129–176. [Google Scholar]
- 2.Helmann J D, Chamberlin M J. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- 3.Lonetto M, Gribskov M, Gross C A. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zuber P, Healy J, Carter H L, III, Cutting S, Moran C P, Losick R. J Mol Biol. 1989;206:605–614. doi: 10.1016/0022-2836(89)90569-x. [DOI] [PubMed] [Google Scholar]
- 5.Siegele D A, Hu J C, Walter W A, Gross C A. J Mol Biol. 1989;206:591–603. doi: 10.1016/0022-2836(89)90568-8. [DOI] [PubMed] [Google Scholar]
- 6.Waldburger C, Gardella T, Wong R, Susskind M M. J Mol Biol. 1990;215:267–276. doi: 10.1016/s0022-2836(05)80345-6. [DOI] [PubMed] [Google Scholar]
- 7.Daniels D, Zuber P, Losick R. Proc Natl Acad Sci USA. 1990;87:8075–8079. doi: 10.1073/pnas.87.20.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rong J C, Helmann J D. J Bacteriol. 1994;176:5218–5224. doi: 10.1128/jb.176.17.5218-5224.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juang Y L, Helmann J D. Biochemistry. 1995;34:8465–8473. doi: 10.1021/bi00026a030. [DOI] [PubMed] [Google Scholar]
- 10.Lesley S A, Burgess R R. Biochemistry. 1989;28:7728–7734. doi: 10.1021/bi00445a031. [DOI] [PubMed] [Google Scholar]
- 11.Shuler M F, Tatti K M, Wade K H, Moran C P. J Bacteriol. 1995;177:2887–2894. doi: 10.1128/jb.177.13.3687-3694.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neidhardt F C, Van-Bogelen R A, Lau E T. J Bacteriol. 1983;153:597–603. doi: 10.1128/jb.153.2.597-603.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gribskov M, Burgess R R. Nucleic Acids Res. 1986;14:6745–6763. doi: 10.1093/nar/14.16.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grossman A D, Erickson J W, Gross C A. Cell. 1984;38:383–390. doi: 10.1016/0092-8674(84)90493-8. [DOI] [PubMed] [Google Scholar]
- 15.Grossman A D, Zhou Y, Gross C, Heilig J, Christie G E, Calendar R. J Bacteriol. 1985;151:939–943. doi: 10.1128/jb.161.3.939-943.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bukau B, Walker G C. EMBO J. 1990;9:4027–4028. doi: 10.1002/j.1460-2075.1990.tb07624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waghorne C, Fuerst C R. J Bacteriol. 1985;164:960–963. doi: 10.1128/jb.164.2.960-963.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tilly K, McKittrick N, Zylicz M, Georgopoulos C. Cell. 1983;34:641–646. doi: 10.1016/0092-8674(83)90396-3. [DOI] [PubMed] [Google Scholar]
- 19.Gamer J, Bujard H, Bukau B. Cell. 1992;69:833–842. doi: 10.1016/0092-8674(92)90294-m. [DOI] [PubMed] [Google Scholar]
- 20.Calendar R, Erickson J W, Halling C, Nolte A. J Bacteriol. 1988;170:3479–3484. doi: 10.1128/jb.170.8.3479-3484.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pace C N, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe P A, Hager D A, Burgess R R. Biochemistry. 1979;18:1344–1352. doi: 10.1021/bi00574a034. [DOI] [PubMed] [Google Scholar]
- 23.Gill S C, Weitzel S E, von Hippel P H. J Mol Biol. 1991;220:307–324. doi: 10.1016/0022-2836(91)90015-x. [DOI] [PubMed] [Google Scholar]
- 24.Nakahigashi K, Yanagi H, Yura T. Nucleic Acids Res. 1995;23:4383–4390. [PMC free article] [PubMed] [Google Scholar]
- 25.McCracken S, Greenblatt J. Science. 1991;253:900–902. doi: 10.1126/science.1652156. [DOI] [PubMed] [Google Scholar]
- 26.Mason S W, Li J, Greenblatt J. J Mol Biol. 1992;223:55–66. doi: 10.1016/0022-2836(92)90715-v. [DOI] [PubMed] [Google Scholar]
- 27.Phizicky E M, Fields S. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberek K, Galitski T P, Zylicz M, Georgopoulos C. Proc Natl Acad Sci USA. 1992;89:3516–3520. doi: 10.1073/pnas.89.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y N, Walter W A, Gross C A. J Bacteriol. 1992;174:5005–5012. doi: 10.1128/jb.174.15.5005-5012.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malhotra A, Severinova E, Darst S A. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- 31.Nicholls A, Sharp K A, Honig B. Proteins Struct Funct Genet. 1991;11:282–296. [Google Scholar]