

Abstract
Antagonists of VEGF-mediated angiogenesis are of great interest clinically for the treatment of solid tumors and certain forms of macular degeneration. We recently described a novel peptoid antagonist of VEGF Receptor 2 (VEGFR2) that binds to the extracellular domain of the receptor and inhibits VEGF-mediated autophosphorylation and subsequent downstream signaling. Given the structural similarities between peptides and peptoids, an obvious model for the mode of action of the peptoid is that it competes with VEGF for binding to VEGFR2. However, we present evidence here that this is not the case and that VEGF and the peptoid antagonist recognize non-overlapping surfaces located within the first three immunoglobulin-like sub-domains of the receptor. These data argue that the peptoid inhibits receptor-mediated autophosphorylation by a novel allosteric mechanism that may prevent the receptor from acquiring the conformation necessary to propagate downstream signals.
1. Introduction
Vascular endothelial growth factor receptor −1 and −2 (VEGFR1 & VEGFR2) are members of the Type III receptor tyrosine kinase family of proteins and both have seven Ig-like domains in the extracellular domain (ECD) with 32 % sequence identity in the ECD 1. VEGFR1 and VEGFR2 are required for embryonic development and they both bind VEGF with high affinity. The association of VEGF with VEGFR2 is a critical event in angiogenesis2 while the function of VEGFR1 is less clear 3. Antagonists of hormone-mediated receptor activation are of keen interest in the treatment of solid tumors since they require the formation of new vasculature to survive and grow4. These include monoclonal antibodies5–8, other protein based molecules9 or peptides10 targeting VEGF or the ECD of VEGFR2, as well as low molecular weight tyrosine kinase inhibitors11. Almost all of the molecules that target the hormone or the ECD of the receptor function by binding to the VEGFR2•VEGF interaction region of either the hormone or receptor. One well-studied exception is the monoclonal antibody Bevacizumab (Avastin), which binds to a site on VEGF neighboring the hormone-binding site12. Even in this case however, the antibody functions by blocking hormone binding to the receptor, presumably via steric interference. To the best of our knowledge, none of the known inhibitors targeted to VEGF or the VEGFR2 ECD operates via a non-competitive mechanism.
We have recently reported a relatively potent anti-angiogenic peptoid agent GU40C4 (Figure 1A) that disrupts VEGF-induced receptor autophosphorylation and, therefore, downstream events leading to angiogenesis13. This compound has also been shown to be effective in inhibiting tumor growth in an animal model. GU40C4 is a derivative of a compound isolated from a screening experiment that registered binding of VEGFR2-expressing cells to peptoids immobilized on TentaGel beads. As expected, the peptoids were found to bind to the ECD of the receptor13.
Figure 1.
Chemical structures of the peptoids employed in this study. (A) The high-affinity, VEGFR2-binding dimeric peptoid GU40C4 and its fluorescently labeled derivative. (B) Peptoids GU40A-E that were initially identified from the on-bead cell-based screen for VEGFR2 ligands13 and the control peptoid used in all assays.
In this report, we probe the mechanism of action of this novel inhibitor. Somewhat surprisingly, we find that GU40C4 and VEGF can bind simultaneously to the receptor. Moreover, we demonstrate that all of the peptoids isolated in the initial screen bind to the same or closely overlapping sites on the receptor. These and other data described below argue that the screen has identified a previously unrecognized “hotspot” for ligand binding on the receptor surface and that interaction of the peptoid with this receptor surface apparently antagonizes VEGF-triggered receptor activation via a novel allosteric mechanism.
2. Results
2.1. The five VEGF Receptor Ligands Have Similar Binding Properties
The screen mentioned above identified five VEGFR2-binding peptoids (Fig. 1B) from a library of approximately 300,000 compounds with three fixed C-terminal residues and six randomized residues. Two of these peptoids (GU40C and GU40E) were shown to bind to the ECD of VEGFR2 with dissociation constants (KD) in the range of 1–3 micromolar. We further demonstrated that these peptoids bound to the ECD of VEGFR1 with similar affinities13. We initiated this study by first asking if any of the other three peptoids exhibited significant specificity for VEGFR1 or VEGFR2. This was done using an ELISA-like binding assay in which increasing concentrations of fluoresceinated peptoid was introduced into 96-well plates coated with the receptor ECD. The amount of fluorescence retained after washing was measured. All of the peptoids bound the ECDs of both VEGFR1 and VEGR2 with KD values in the low to sub micromolar range (Fig. 2A).
Figure 2.
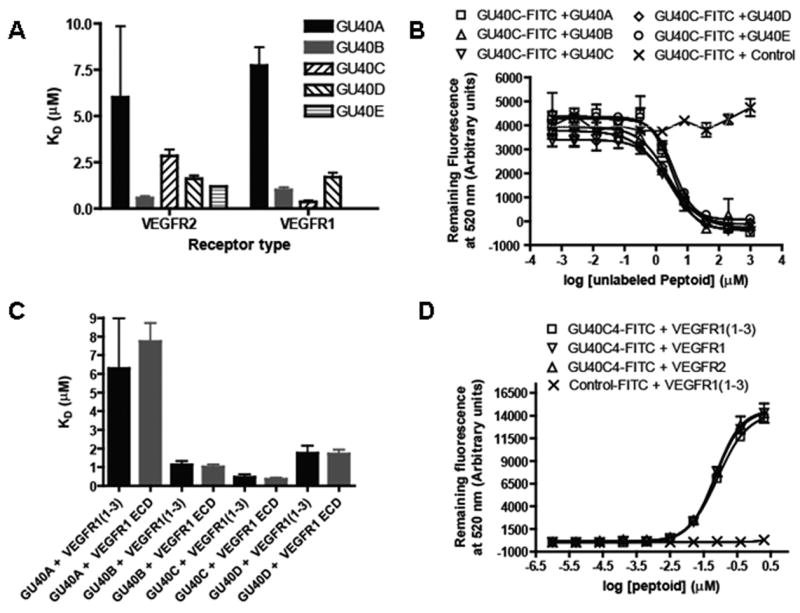
Binding of the peptoids to the extracellular domains (ECDs) of VEGFR1 and VEGFR2 in an ELISA-like assay. (A) KD values of the complexes between GU40A-E and the VEGFR1 or VEGFR2 ECD as measured by the titration of immobilized ECD with fluorescein-labeled peptoid and monitoring the amount of fluorescence retained. All of the peptoids bound both the VEGFR1 and VEGFR2 ECDs with KDs in the low μM range. (B) Competitive binding experiments that monitor the retention of fluorescein-labeled GU40C by immobilized VEGFR2 ECD in the presence of the indicated amounts of other unlabeled GU40A-E or an unlabeled control peptoid not selected to bind VEGFR2. All of the VEGFR2-binding peptoids competed with labeled GU40C. (C) Bar graph showing the KD values of peptoids GU40A-D binding to a recombinant protein representing the first three IgG-like domains of VEGFR1 (called VEGFR1(1–3)) (black bars) compared to the corresponding binding to the complete ECD of VEGFR1 (grey bars). (D) Binding isotherms of fluorescein-labeled GU40C4 dimer interaction with the VEGFR1 ECD, VEGFR2 ECD and VEGFR1(1–3). GU40C4-FITC denotes the conjugation product obtained by treating the thiol-containing GU40C4 derivative with maleimide-fluorescein isothiocyanate (FITC).
We next evaluated whether these five peptoids recognize different surfaces of the receptor. Fluoresceinated GU40C (GU40C-FITC) binding to the immobilized receptor was challenged with increasing concentrations of unlabeled GU40A-E. All five peptoids competed with labeled GU40C for limiting receptor (Fig. 2B). Furthermore, all of the peptoids evinced similar IC50s in this competition experiment. These data suggest that all five peptoids bind to the same, or closely overlapping, sites on the protein.
The ECDs of VEGFR1 and VEGFR2 contain seven IgG-like sub-domains. In order to begin to map the particular region recognized by the peptoids, we conducted the same ELISA-like binding assay using a construct containing only the first three N-terminal sub-domains of VEGFR1 (VEGFR1(1–3)), which includes the region of the receptor to which VEGF itself binds12, 15, 16. The KD values obtained from these data again fall within the low to sub micromolar range for all of the peptoids, showing that they all bind to a surface located within these three sub-domains (Fig. 2C).
2.2. The GU40C4 peptoid and VEGF do not compete for binding to the VEGFR2 ECD
The three N-terminal domains of VEGFR1 also encompass the VEGF binding region of the receptor, consistent with the idea that the peptoid antagonists function by competing with the hormone for binding to the receptor. To probe more directly for peptoid-hormone competition, we examined binding of fluorescein-labeled GU40C4 dimer which displayed improved binding properties compared to the GU40C monomer, to the receptor constructs in the presence of VEGF. First, we demonstrated that, as expected, the dimeric GU40C4 behaved similarly to the monomeric GU40C in that it bound to the ECDs of VEGFR1, VEGFR2 and VEGFR1(1–3) with similar affinities (Fig. 2D). For the competitive binding assay, fluoresceinated-GU40C4 was kept constant (100 nM) and unlabeled VEGF, GU40C4 or a control peptoid that does not bind to the VEGFRs were introduced at the concentrations indicated in Fig. 3. As expected, unlabeled GU40C4 displaced the fluorescein-conjugated GU40C4, but VEGF did not, even at hormone concentrations well above that necessary to saturate the receptor, indicating these two ligands have separate binding sites. The control peptoid had no effect on GU40C4•receptor interaction.
Figure 3.
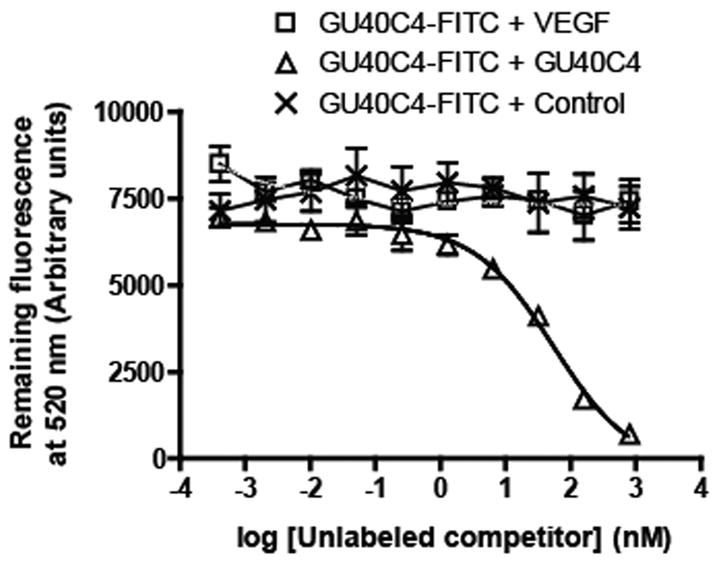
Competitive binding experiments that monitor the retention of fluorescein-labeled GU40C4 by immobilized VEGFR2 ECD in the presence of the indicated concentrations of unlabeled competitor (VEGF, unlabeled GU40C4 or a control peptoid not selected to bind VEGFR2). Only unlabeled GU40C4 competed the GU40C4-FITC•VEGFR2 ECD complex.
If it is indeed the case that GU40C4 and VEGF have different binding sites on VEGFR2, then it should be possible to observe simultaneous binding of both the peptoid and the hormone on the receptor. To address this point, GU40C4 was fluoresceinated and VEGF was biotinylated, allowing both to be visualized independently. The fluorescence emission of GU404-FITC can be detected at 520 nm (green channel) and, by using a streptavidin-coated quantum dot (Qdot655), the hormone can be detected at 655 nm (red channel). Eight wells of 96-well clear bottom plates were coated with the ECD of VEGFR2 and four different binding experiments were carried out in duplicate. In the first two, either GU40C4-FITC (100 nM) or biotin-VEGF (100 nM) alone was incubated with the immobilized receptor. In the third, equimolar amounts (100 nM each) of GU40C-FITC and biotin-VEGF were added. Finally, the fourth experiment was the same as third, except 100-fold excess of unlabeled GU40C was also included in the solution. After allowing the solutions to equilibrate for one hour and thorough washing, Streptavidin-Qdot655 was introduced to each well followed by another thorough wash. The amount of fluorescence remaining in each well was measured at 520 nm and 655 nm separately (Fig. 4). The level of fluorescence in each of the first two experiments served as a measure of the unimpeded binding of the peptoid and VEGF, respectively, to the immobilized receptor. As seen in Fig. 4, when both the peptoid and VEGF were present at 100 nM, strong signals in both the red and green channels were observed that were similar in intensity to those observed in the wells containing only one of the ligands. Addition of a large excess of unlabeled GU40C4 to both of the labeled ligands and the immobilized receptor competed binding of FITC-GU40C4, but not biotin-VEGF, to the immobilized receptor. Taken together, these data argue strongly that VEGF and the GU40C4 peptoid can simultaneously co-occupy the receptor.
Figure 4.
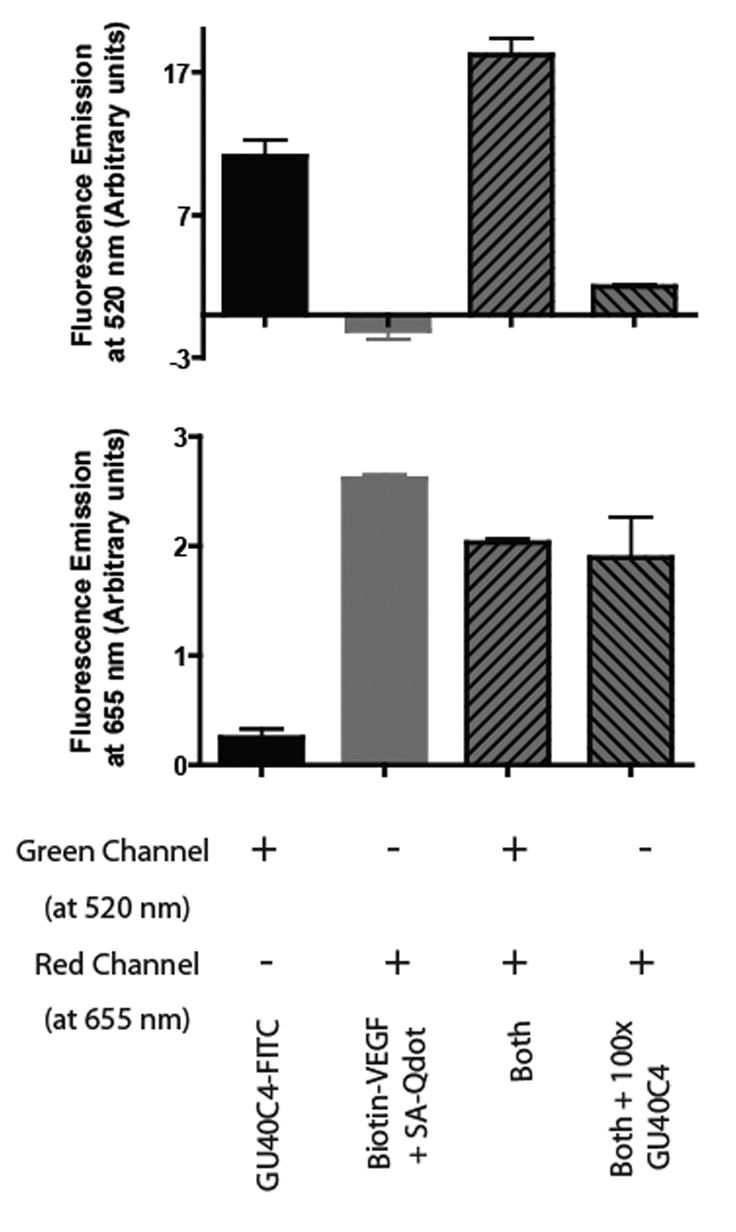
Retention of GU40C4-FITC and biotin-VEGF by immobilized VEGFR2 ECD. Retention of GU40C4-FITC was monitored at 520 nM (fluorescein emission) while retention of biotin-VEGF was monitored at 655 nm after treatment with red-emitting streptavidin-Qdot655. The top graph shows the retained fluorescence in the 520 nm channel (green-FITC detection) and the bottom graph shows the retained fluorescence at 655 nm (red-Qdot655 detection). The first and second bars in each graph represent the intensity of the retained fluorescence when the immobilized VEGFR2 ECD was treated with GU40C4-FITC alone or biotin-VEGF alone, respectively. The third bar represents the intensity of the retained fluorescence when both GU40C4-FITC and biotin-VEGF were added to the immobilized receptor. Note that the amounts of hormone and peptoid were in large excess over that of the immobilized receptor ECD. The fourth bar represents the same experiment as the third, except that 100-fold excess of unlabeled GU40C4 was also included. In all cases, streptavidin-Qdot655 was added to each well, followed by washing, prior to reading the fluorescence intensity in each channel.
To confirm that this surprising result was not some artifact of the particular assay employed, we conducted similar experiments, but immobilized biotin-VEGF on streptavidin-coated plates. As shown in Fig. 5A, a low level of FITC-GU40C4 is retained by the streptavidin-coated plates, presumably representing non-specific binding of the labeled peptoid to the avidin or the plate. This is supported by the fact that other labeled control peptoids provided essentially the same result in this assay (not shown). A similar level of non-specific binding was observed when the labeled peptoid was incubated with the biotin-VEGF-coated plates, showing that the peptoid does not bind directly to the hormone. When the soluble ECD of VEGFR2 was also added however, significant additional retention of labeled GU40C4 was observed. These results are consistent with binding of the GU40C4•VEGFR2 ECD complex to the immobilized VEGF. This interpretation is supported by the fact that this VEGFR2-dependent retention of the labeled peptoid could be competed by excess unlabeled VEGF, showing that it represents specific binding of the peptoid•receptor complex to the VEGF-coated plate.
Figure 5.
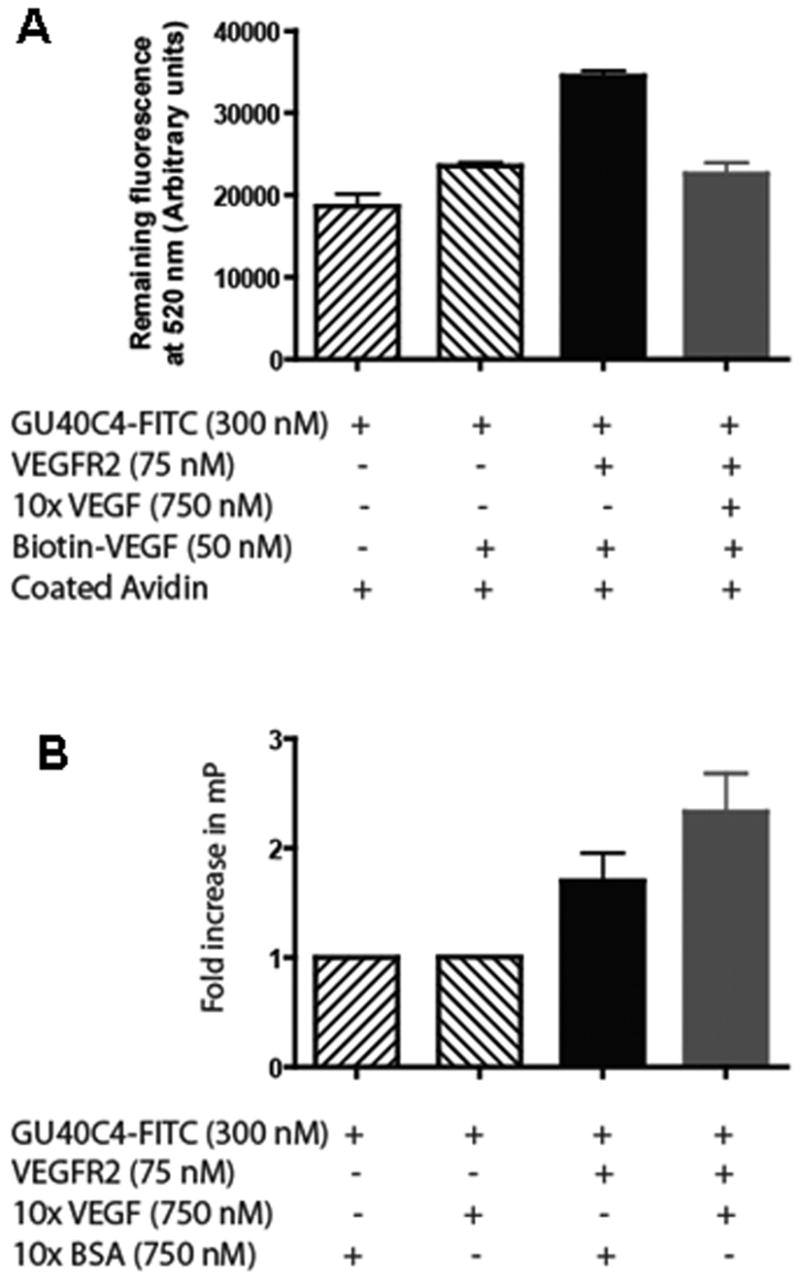
Formation of a peptoid•VEGFR2 ECD•VEGF complex. (A) The reagents indicated in the figure were incubated in the wells of biotin-VEGF captured avidin-coated plates. After washing, the intensity of the retained fluorescence at 520 nm was monitored to score retention of GU40C4-FITC. A significant increase in GU40C4-FITC retention above background (first bar) was observed only in the presence of VEGFR2 ECD (third bar). This retention was abolished by excess soluble VEGF. (B) Fluorescence anisotropy experiment monitoring the change in anisotropy evinced by GU40C4-FITC in the presence of the indicated reagents.
We also wished to validate this surprising result using a solution assay to ensure that the results observed when one of the ligands is immobilized on a surface reflects the behavior of the molecules accurately when they are free in solution. Therefore, we examined the effect of VEGF and the ECD of VEGFR2 on the anisotropy of FITC-GU40C4. The level of fluorescence anisotropy observed when 300 nM FITC-GU40C4 was incubated with 750 nM BSA as a control protein was set to one (Fig. 5B). Incubation of the labeled peptoid with 750 nM VEGF did not alter the anisotropy, again showing that GU40C4 does not bind to the hormone directly. However, addition of 75 nM VEGFR2 ECD resulted in a pronounced increase in the anisotropy value, consistent with binding of the peptoid to the receptor. Interestingly, when both the VEGFR2 ECD and the hormone were incubated with FITC-GU40C4, a further increase in anisotropy was observed, consistent with the formation of a FITC-GU40C4•VEGFR2•VEGF co-complex that tumbles even more slowly than the peptoid-receptor complex.
Finally, to probe the issue of how VEGF might affect VEGFR2 ECD•GU40C4 binding in a more quantitative fashion, a titration experiment was conducted in which the quantitative affinity for the fluorescein-labeled GU40C4 peptoid for immobilized VEGFR2 ECD was measured in the presence or absence of saturating amounts of VEGF. As shown in Fig. 6, virtually identical results were obtained in each titration demonstrating that the KD of the GU40C4•VEGFR2 ECD complex is essentially unaffected by binding of VEGF to the VEGFR2 ECD.
Figure 6.
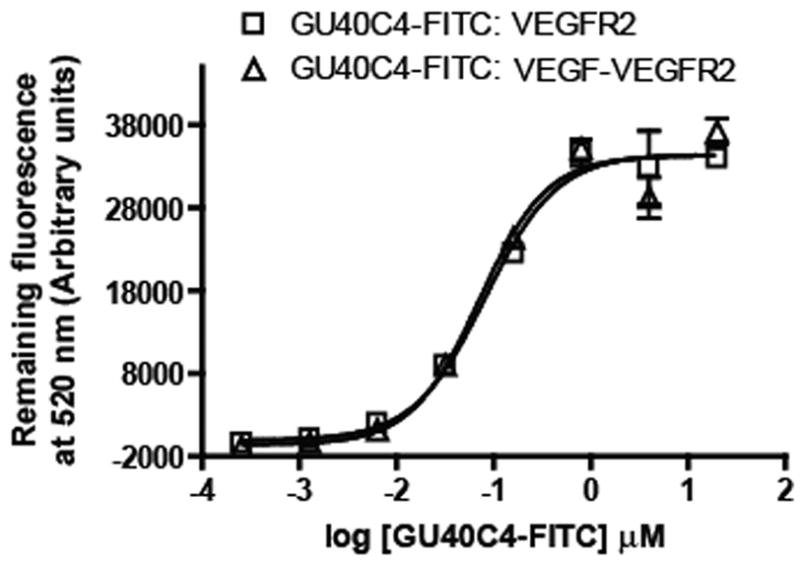
Quantitative determination of the affinity of GU40C4-FITC for immobilized VEGFR2 ECD in the presence or absence of saturating amounts of VEGF, which was added to the immobilized receptor prior to introduction of the peptoid. VEGF did not alter the affinity of the peptoid for the receptor.
3. Discussion
In a recent report we identified five peptoids in a library of ≈300,000 molecules that were capable of binding to cells expressing human VEGFR2 but not to cells lacking VEGFR213. Biochemical experiments confirmed that two of these five peptoids recognized the ECD of VEGFR2 with KDs in the low μM range. Since VEGFR2 operates as a homodimer, we hypothesized that joining together two molecules of one of the VEGFR2-binding peptoids with a suitable linker would result in a much higher affinity compound. Indeed, GU40C4 was shown to bind to the ECD of VEGFR2 with a KD of 20–40 nM. This compound was an antagonist of VEGF-dependent VEGFR2 autophosphorylation in cultured cells and also exhibited anti-angiogenic activity in vivo13.
Given precedents from previous studies of VEGF- or VEGFR2-targeted anti-angiogenic agents, we anticipated that the mechanism of action of the peptoid would be to interfere with hormone•receptor binding. Surprisingly, several different assays that monitored binding of the hormone and/or the peptoid to the ECD of VEGFR2 or VEGFR1 (Figs. 3–6) failed to show evidence of this anticipated competition. Indeed, the data shown in Fig. 5 provide direct evidence for a VEGF•VEGFR2•GU40C4 ternary complex. Specifically, we demonstrated that immobilized VEGF is able to retain GU40C4 in the presence, but not in the absence, of the ECD of VEGFR2 (Fig. 5A). Furthermore, the addition of VEGF to a FITC-GU40C4•VEGFR2 ECD complex resulted in a marked increase in fluorescence anisotropy, consistent with formation of a ternary complex, whereas mixing VEGF and FITC-GU40C4 in the absence of the receptor did not result in a change in anisotropy (Fig. 5B).
Thus, we conclude that GU40C (one “arm” of the dimeric peptoid (Fig. 1)) binds to a site on the VEGFR2 that is distinct from that of the hormone-binding site and that, furthermore, binding of the peptoid does not sterically inhibit subsequent receptor association. Moreover, the data shown in Fig. 2 demonstrate that all five of the peptoids isolated in the original screen compete with one another for binding to the VEGFR2 ECD, strongly suggesting that they recognize the same surface, although allosteric effects cannot be completely ruled out. Therefore, we suggest that the original screen and this study have revealed a previously unrecognized “hotspot” 17 18 for ligand binding on the VEGFR2 ECD that is distinct from the hormone binding site. Interestingly, binding assays using a construct containing only the first three immunoglobulin-like domains of the ECD (Fig. 2) reveal that this hotspot is in the same general region of the protein as the VEGF binding site12. Finally, this hotspot must exist on both VEGFR2 and VEGFR1 since the peptoids fail to discriminate between these two closely related receptors.
The finding that the peptoids do not recognize the hormone binding site is somewhat surprising in that many VEGFR1- and VEGFR2-binding peptides have been identified through phage display or other means and all of these molecules compete with VEGF for binding to the receptor19–24. Peptoids are peptide-like molecules and we initiated this project anticipating that we would isolate peptoid mimics of VEGF, at least with regard to binding to the receptor. Indeed, the first three C-terminal residues of all of the peptoids in the library were held constant in hopes of mimicking key residues in VEGF that contact the receptor12. Clearly, this design did not achieve its intended goal. Moreover, as will be reported elsewhere, studies of the structure/activity relationships evinced by various derivatives of GU40C show clearly that these three C-terminal side chains are dispensable for binding to the receptor (Udugamasooriya, et al., unpublished results).
While the data clearly rule out direct competition of the peptoid and the hormone for receptor binding, they do not clearly define what the mechanism of antagonism might be. In general, one is driven to the conclusion that the peptoid must interfere with some event downstream of formation of the hormone•receptor complex. While the structural details are unknown, binding of VEGF to VEGFR2 must drive a conformational change in the receptor that is transmitted across the membrane25, resulting in the activation of the intracellular tyrosine kinase activity. Therefore, one model is that the peptoid interferes with this conformational transition through binding to the receptor. Unfortunately, it is very difficult to probe such events directly in the context of the intact receptor and thus a mechanism based on peptoid-antagonized conformational changes must be treated as speculative.
4. Experimental
4.1. Synthesis of peptoids
Syntheses of peptoids GU40A-E was performed on Knorr Amide MBHA resin (substitution: 0.78 mmol/g resin; Novabiochem). First, Fmoc-Cys(Trt)-OH was loaded onto the bead (HOBt, HBTU, DIPEA, overnight) in order to facilitate subsequent fluorescein coupling to the thiol via maleimide chemistry. After removal of Fmoc group (20% piperidine in DMF), 2.0 M bromoacetic acid and 3.2 M diisopropylcarbodiimide (DIC) was added, and the reaction was completed using a microwave-assisted protocol (1000W microwave oven, and 10% power was delivered for 2 ×15 seconds)14. After washing with DMF, the beads were treated with 2 M primary amine and microwaved as described above. These two steps were repeated until the 9-mer peptoid was complete. After the final washing with 10 × DMF and 3 × dichloromethane (DCM), peptoids were cleaved off the resin by treatment with 95% TFA, 2.5% TIS and H2O. TFA was evaporated and the product was purified by HPLC and confirmed by MALD-TOF analysis.
To attach fluorescein, pure peptoid was dissolved in 1×PBS, 5 mM EDTA, pH=6.9 buffer and mixed with fluorescein-maleimide at a 1:1 ratio. The solution was incubated overnight and the product was purified by HPLC.
Dimeric peptoid GU40C4 and its fluoresceinated derivative were synthesized as described in our previous report13.
4.2. ELISA-like binding assay
White, clear bottom 96-well plates (Corning Inc.) were coated with of 1 μg/mL recombinant human VEGFR protein (R&D Systems) in sensitizing buffer (0.621g NaHCO3 & 0.275g Na2CO3 dissolved in 100 mL of ddH2O, pH=9.5) overnight at 4 °C. Each well was washed with 3 × 200 μL of phosphate-buffered saline (PBS) and blocked with Startingblock buffer (Pierce) or AquaBlock™/EIA/WB buffer (EastCoast Bio). 50 μL of serial dilutions of FITC-labeled peptoids dissolved in Startingblock buffer or TBST were added to each well & allowed to incubate for 1 hour at room temperature. Wells were washed with 5 × 200 μL of PBS and the remaining fluorescence was measured at 520 nm using a plate reader (Fluostar Optima, BMG Laboratories, Durham, NC). For competition assays, serial dilutions of unlabeled peptoids were mixed with constant amounts of FITC-labeled peptoids. In the direct binding assay (Fig. 6), a set of VEGFR2-coated wells were saturated with 50 nM VEGF for 1h at room temperature and washed before introducing the serial dilutions of GU40C4-FITC.
4.3. GU40C4-FITC and biotin-VEGF simultaneous visualization assay (Fig. 4)
Eight wells of a white, clear bottom 96-well plate (Corning Inc.) were coated with 1 μg/mL recombinant human VEGFR2 protein (R&D Systems) in sensitizing buffer (0.621g NaHCO3 & 0.275g Na2CO3 dissolved on 100 mL of ddH2O, pH=9.5) overnight at 4 °C. The wells were washed with 3 × 200 μL of PBS and blocked with Startingblock buffer (Pierce). Four different experimental sets were designed each in duplicate. The first two wells were treated 2 × 50 μL with 100 nM GU40C4-FITC and the next two wells 2 × 50 μL with 100 nM biotin-VEGF, dissolved in Startingblock buffer. The fifth and sixth wells were treated 2 × 50 μL of a mixture of 100 nM each of GU40C4-FITC and biotin-VEGF. The last two wells were treated exactly as the fifth and sixth, except the mixture also contained 10 mM unlabeled GU40C4. All wells were allowed to equilibrate in the dark at room temperature for one hour. Wells were washed thoroughly with PBS three times. 10 nM streptavindin-Qdot655 was introduced in Startingblock buffer for 30 minutes in the dark to each well. The wells were washed thoroughly four times with PBS. The entire procedure was conducted with an additional four wells that lacked immobilized receptor (two for GU40C4-FITC treatment and two for biotin-VEGF) as controls. The remaining fluorescence was probed through two sets of excitation and emissions. First, excitation at 485 nm and emission at 520 nm (to probe FITC signal – green channel) and the second excitation at 355 nm and emission at 655 nm (to probe Qdot655 – red channel). The values of control wells were subtracted from each original value to obtain the final data.
4.4. GU40C4-FITC and VEGF simultaneous detection assay (Fig. 5)
The assay was composed of two parts: 1. confirmation of GU40C4-FITC binding to VEGFR2 by fluorescence anisotropy and, 2. subsequent capture of GU40C4-FITC-VEGFR2 complex on VEGF-coated ELISA plates. First, in four different vials GU40C4-FITC (300 nM) was incubated; (1) without VEGFR2 (2) with VEGF (750 nM) only (3) with VEGFR2 (75 nM) (4) VEGFR2 (75 nM) with ten fold excess of VEGF (750 nM). For the vials 1 and 3, BSA (750 nM) was introduced to equalize the amount of VEGF added to vials 2 and 4. After incubation for 6 h at 4 °C, fluorescence polarization change (mP) was measured using Panvera Beacon 2000 instrument (Invitrogen). Separately, eight wells of 96-well clear bottom plate were coated with 2 μg/mL avidin using the sensitizing buffer as described earlier. These wells were treated with biotinylated-VEGF (50 nM) for 1h at 4 °C. After washing with PBS above (1), (3) & (4) mixtures were introduced onto the biotinylated-VEGF captured avidin coated wells and equilibrate overnight at 4 °C. After thorough wash with PBS, fluorescein signal was measured at 520 nm using the plate reader.
Acknowledgments
This work was supported by a contract from the National Heart, Lung, and Blood Institute (NO1-HV-28185) for the UT Southwestern Center for Proteomics Research, a grant from the Welch Foundation (I-1299), and the Effie Marie Cain Scholarship for Angiogenesis Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Yamane A, Seetharam L, Yamaguchi S, Gotoh N, Takahashi T, Neufeld G, Shibuya M. Oncogene. 1994;9:2683. [PubMed] [Google Scholar]
- 2.Ferrara N. Endocrine reviews. 2004;25:581. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 3.Roskoski R., Jr Crit Rev Oncol Hematol. 2007;62:179. doi: 10.1016/j.critrevonc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23:1011. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 5.Brekken RA, Overholser JP, Stastny VA, Waltenberger J, Minna JD, Thorpe PE. Cancer Res. 2000;60:5117. [PubMed] [Google Scholar]
- 6.Gerber HP, Kowalski J, Sherman D, Eberhard DA, Ferrara N. Cancer Res. 2000;60:6253. [PubMed] [Google Scholar]
- 7.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Nature. 1993;362:841. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 8.Prewett M, Huber J, Li Y, Santiago A, O’Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin DJ. Cancer Res. 1999;59:5209. [PubMed] [Google Scholar]
- 9.Getmanova EV, Chen Y, Bloom L, Gokemeijer J, Shamah S, Warikoo V, Wang J, Ling V, Sun L. Chem Biol. 2006;13:549. doi: 10.1016/j.chembiol.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 10.D’Andrea LD, Del Gatto A, Pedone C, Benedetti E. Chem Biol Drug Des. 2006;67:115. doi: 10.1111/j.1747-0285.2006.00356.x. [DOI] [PubMed] [Google Scholar]
- 11.Klohs WD, Hamby JM. Curr Opin Biotechnol. 1999;10:544. doi: 10.1016/s0958-1669(99)00033-6. [DOI] [PubMed] [Google Scholar]
- 12.Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, de Vos AM. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:7192. doi: 10.1073/pnas.94.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Amer Chem Soc. 2008;130 doi: 10.1021/ja711193x. In press. [DOI] [PubMed] [Google Scholar]
- 14.Olivos HJPGA, Reddy MM, Saloney D, Kodadek T. Org Lett. 2002;4:4057. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 15.Shinkai A, Ito M, Anazawa H, Yamaguchi S, Shitara K, Shibuya M. The Journal of biological chemistry. 1998;273:31283. doi: 10.1074/jbc.273.47.31283. [DOI] [PubMed] [Google Scholar]
- 16.Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, de Vos AM. Cell. 1997;91:695. doi: 10.1016/s0092-8674(00)80456-0. [DOI] [PubMed] [Google Scholar]
- 17.Clackson T, Wells JA. Science (New York, NY) 1995;267:383. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 18.Thanos CD, DeLano WL, Wells JA. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15422. doi: 10.1073/pnas.0607058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An P, Lei H, Zhang J, Song S, He L, Jin G, Liu X, Wu J, Meng L, Liu M, Shou C. International journal of cancer. 2004;111:165. doi: 10.1002/ijc.20214. [DOI] [PubMed] [Google Scholar]
- 20.Binetruy-Tournaire R, Demangel C, Malavaud B, Vassy R, Rouyre S, Kraemer M, Plouet J, Derbin C, Perret G, Mazie JC. The EMBO journal. 2000;19:1525. doi: 10.1093/emboj/19.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Mousawi M, Tchistiakova L, Yurchenko L, Pietrzynski G, Moreno M, Stanimirovic D, Ahmad D, Alakhov V. The Journal of biological chemistry. 2003;278:46681. doi: 10.1074/jbc.M308681200. [DOI] [PubMed] [Google Scholar]
- 22.Hetian L, Ping A, Shumei S, Xiaoying L, Luowen H, Jian W, Lin M, Meisheng L, Junshan Y, Chengchao S. The Journal of biological chemistry. 2002;277:43137. doi: 10.1074/jbc.M203103200. [DOI] [PubMed] [Google Scholar]
- 23.Jia H, Jezequel S, Lohr M, Shaikh S, Davis D, Soker S, Selwood D, Zachary I. Biochemical and biophysical research communications. 2001;283:164. doi: 10.1006/bbrc.2001.4761. [DOI] [PubMed] [Google Scholar]
- 24.Zilberberg L, Shinkaruk S, Lequin O, Rousseau B, Hagedorn M, Costa F, Caronzolo D, Balke M, Canron X, Convert O, Lain G, Gionnet K, Goncalves M, Bayle M, Bello L, Chassaing G, Deleris G, Bikfalvi A. The Journal of biological chemistry. 2003;278:35564. doi: 10.1074/jbc.M304435200. [DOI] [PubMed] [Google Scholar]
- 25.Ruch C, Skiniotis G, Steinmetz MO, Walz T, Ballmer-Hofer K. Nature structural & molecular biology. 2007;14:249. doi: 10.1038/nsmb1202. [DOI] [PubMed] [Google Scholar]