

Abstract
The influenza A virus M2 integral membrane protein is an ion channel that permits protons to enter virus particles during uncoating of virions in endosomes and also modulates the pH of the trans-Golgi network in virus-infected cells. The M2 protein is a homo-oligomer of 97 residues, and analysis by chemical cross-linking and SDS/PAGE indicates M2 forms a tetramer. However, a higher order molecular form is sometimes observed and, thus, it is necessary to determine the active form of the molecule. This was done by studying the currents of oocytes that expressed mixtures of the wild-type M2 protein (epitope tagged) and the mutant protein M2-V27S, which is resistant to the inhibitor amantadine. The composition of mixed oligomers of the two proteins expressed at the plasma membrane of individual oocytes was quantified after antibody capture of the cell surface expressed molecules and it was found that the subunits mixed freely. When the ratio of wild-type to mutant protein subunits was 0.85:0.15, the amantadine sensitivity was reduced to 50% and for a ratio of 0.71:0.29 to 20%. These results are consistent with the amantadine-resistant mutant being dominant and the oligomeric state being a tetramer.
Keywords: influenza A virus, M2 protein, amantadine, channel oligomeric form
The influenza A virus M2 integral membrane protein is thought to function as an ion channel that permits protons to enter virus particles during uncoating of virions in endosomes and also to modulate the pH of the trans-Golgi network (for reviews see refs. 1 and 2). Direct electrophysiological evidence that the M2 protein has ion channel activity has been obtained by expressing the M2 protein in oocytes of Xenopus laevis (3–6) or in mammalian cells (7, 8). The M2 protein ion channel activity is specifically blocked by the anti-influenza virus drug amantadine and is activated at the lowered pH found intralumenally in endosomes and the trans-Golgi network (3, 4, 6, 8). In addition, reconstitution of purified M2 protein or introduction of the transmembrane domain peptide into planar lipid bilayers resulted in amantadine-sensitive ion channel activity that was activated by low pH (9–11).
The M2 protein is a homo-oligomer of 97 residues that is expressed at the plasma membrane of virus-infected cells and it is oriented in membranes such that it has 24 N-terminal extracellular (or lumenal) residues, a 19 residue transmembrane domain, and a 54 residue cytoplasmic tail (12). The native form of the M2 protein is minimally a homotetramer consisting of a pair of either disulfide-linked dimers or disulfide-linked tetramers (13–15). In studies with chemical cross-linking reagents (13), and when large amounts of M2 were purified on sucrose gradients (10), a small amount of a larger complex (150–180 kDa) has been identified that appears to contain only M2 molecules and could represent a higher-order structure of M2 oligomers.
The biologically active oligomer of the vast majority of cellular ion channel proteins spans the membrane many times (e.g., Ca2+ and Na+ channels have 24 transmembrane domains, K+ channels have 4 subunits each of 6 transmembrane domains). If the active M2 ion channel is the homotetramer, the M2 ion channel is one of the smallest ion channels discovered to date and it is an excellent molecule for understanding the mechanistic details of channel function. Thus, it is exceedingly important to determine, unambiguously, the subunit stoichiometry of the M2 ion channel. The subunit stoichiometry of a voltage-activated potassium channel was demonstrated by determining the fraction of toxin-resistant channels resulting from coexpression of toxin-sensitive (scorpion toxin charybdotoxin) and toxin-insensitive Shaker channel α-subunits (16). A related approach was taken to determine the subunit stoichiometry of the nicotinic acetylcholine receptor (17). The subunit stoichiometry of minK (a protein that underlies the activity of the cardiac delayed rectifier, ISK) was also determined by coexpressing wild-type (wt) minK and a dominant negative point mutant of minK, which reaches the plasma membrane but passes no current (18).
We have determined the active subunit stoichiometry of the influenza virus wt M2 ion channel by expressing mixed oligomers of M2 and amantadine-resistant M2 subunits of known subunit composition and then measuring whole cell surface currents before and after treatment with the inhibitor: the data indicate the active M2 oligomer is a tetramer.
MATERIALS AND METHODS
Cells and Viruses.
HeLa T4 cells (a stable line of HeLa cells that express the human CD4 molecule) (19) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum. Recombinant vaccinia virus vTF7.3, which expresses the bacteriophage T7 RNA polymerase gene (20), was provided by Bernard Moss (National Institutes of Health, Bethesda).
Plasmids and mRNA Synthesis.
cDNA to the M2 protein mRNA (wt M2) (21, 22) and the mutant M2-V27S (3, 4) were cloned into pTM3 between the NcoI site and HindIII such that mRNA sense transcripts could be generated using the bacteriophage T7 RNA polymerase promoter and T7 polymerase. M2tag cDNA was obtained by a PCR reaction such that it encoded nine extra C-terminal residues (YPYDVPDYA), which form the epitope for the mAb 12CA5 (23). For in vitro transcription, plasmid DNAs were linearized downstream of the T7 promoter and the M2 cDNA with HindIII. In vitro synthesis and quantitation of 7mG(5′)ppp(5′)G-capped mRNA was carried out as described (3)
Protein Expression in Mammalian Cells and Oocytes, Metabolic Labeling, Immunoprecipitation, and SDS/PAGE.
Proteins were expressed in HeLa T4 cells by using the vaccinia virus T7 RNA polymerase (vac/T7)-mediated transient expression system (20). Subconfluent monolayers of HeLa T4 cells in a 3.5-cm dish were infected with vaccinia virus vTF7.3 at an input multiplicity of infection of 10 plaque-forming units per cell for 30 min and transfected with plasmid DNA by using a cationic liposome reagent (24) synthesized in our laboratory. At 5 h posttransfection, the cells were incubated for 30 min in methionine- and cysteine-free DMEM. The cells were then labeled with [35S]Pro-mix (100 μCi/ml; 1 Ci = 37 GBq; Amersham) and incubated for various times in chase medium (DMEM) containing 2 mM methionine and 2 mM cysteine. The cells were lysed in RIPA buffer containing 50 mM iodoacetamide and 1% (vol/vol) aprotinin (Sigma) as described (25), and proteins were immunoprecipitated with M2-specific 14C2 mAb (26). Polypeptides were analyzed by SDS/PAGE using 17.5% polyacrylamide gels containing 4 M urea as described (27). Radioactivity was analyzed and quantified by using a Fujix BAS 1000 image analyzer and macbas software (Fuji Medical Systems, Stamford, CT).
Oocytes expressing the M2 proteins were incubated in ND96 supplemented with 200 μCi/ml [35S]methionine (Amersham) 24–48 h after injection. These oocytes were briefly washed and their currents measured as described below. After the recording procedure, which lasted 10–15 min, the oocyte-expressed surface M2 protein was quantified by an antibody capture procedure. Individual oocytes were incubated with 100 μl of ND96 containing 1:100 diluted mAb 14C2 ascites fluid at 4°C for 30 min, washed five times with ice-cold ND96, and oocytes homogenized in 100 μl of RIPA buffer containing 50 mM iodoacetamide and 1 mM phenylmethylsulfonyl fluoride, and lysates were extracted with 1,1,2-trichlorotrifluoroethane (Freon) to remove yolk and pigment proteins (4). Protein A-Sepharose beads were added to recover immune complexes and processed for SDS/PAGE as described above.
Culture and Microinjection of Oocytes.
X. laevis oocytes (stage V) prepared as described previously (28) were microinjected with 50 nl of mRNA (1 ng/nl) on the day after defolliculation, incubated for 24 h in ND96 (pH 7.6), and finally incubated for 24 h in ND96 (pH 8.5) at 19°C before use (3).
Measurement of Membrane Current.
Whole cell current was measured with a two-electrode voltage clamp (3). The electrodes were filled with 3 M KCl, and the oocytes were bathed in either Barth’s solution (88.0 mM NaCl/1.0 mM KCl/2.4 mM NaHCO3/0.3 mM NaNO3/0.71 mM CaCl2/0.82 mM MgSO4/15 mM Hepes, pH 7.5), or a modified solution during the recording.
RESULTS
It has been shown previously that it is possible to determine the subunit composition of several ion channels by measuring the activity of mixed oligomers of a wt and inhibitor-insensitive (or inactive) mutant subunit (16, 18). This approach requires that there is a random distribution of the various mixtures of the two types of subunit and that all forms of the oligomer have an equal chance of being expressed at the cell surface.
We investigated if it was possible to create mixed oligomers between wt M2 protein (sensitive to the M2 ion channel inhibitor, amantadine) and an amantadine-resistant M2 mutant. To facilitate monitoring the formation of mixed oligomers, an epitope tag was added to the C terminus of wt M2 (M2tag), since it was anticipated that the addition of nine extra residues would not affect ion channel activity or cell surface expression but would cause a readily detectable shift in electrophoretic mobility of M2tag in comparison to that of untagged M2. Such a mobility shift would permit the formation of mixed oligomers to be monitored biochemically (see below). The M2 mutant M2-V27S was selected for study, because it has been shown previously that expression of M2-V27S in oocytes leads to surface currents that are highly resistant to inhibition by amantadine (amanR). As shown in Fig. 1, when either M2tag or M2-V27S were expressed in oocytes and membrane currents measured, the amplitudes of the currents were found to be increased similarly by low pH, but were very different with respect to inhibition by amantadine: the current of M2tag was amantadine sensitive (amanS), whereas the current of M2-V27S was amanR. To examine the ability of M2tag and M2-V27S to form mixed oligomers, the proteins were expressed in HeLa-T4 cells using the vac/T7 expression system and transfecting differing amounts of the plasmid DNAs encoding M2tag and M2-V27S. For technical reasons we chose to perform this experiment in mammalian cells, but as will be shown below similar data can be obtained from expression of M2 protein in oocytes. As shown in Fig. 2, when the M2 species were immunoprecipitated with M2-specific mAb 14C2, M2tag exhibited a more distinct and slower electrophoretic mobility than M2-V27S (and wt M2; data not shown) under reducing (A) and nonreducing (B) conditions. All three species of disulfide-linked dimer (none, one, or two tags) and all five predicted species of disulfide-linked tetramer (none, one, two, three, or four tags) were found and the distribution of M2tag to M2-V27S in the disulfide-linked dimers and tetramers (Fig. 2B) followed a binomial distribution (determined on a Fuji Bio-Imager). Confirmation that the quantized mobility shifts were due to addition of the epitope tags was obtained by the finding that it was possible to immunoprecipitate the M2tag species but not the completely untagged M2-V27S species using the tag epitope-specific antibody, mAb 12CA5 (data not shown). Chemical cross-linking of the lysates with dithiobis(succinimidyl proprionate) was also done to show that the bulk of the oligomer formed a species with a mobility consistent with M2 being a homotetramer, but a small amount of a higher molecular weight (150–180 kDa) oligomer also formed as seen near the origin of the gel (Fig. 2C). Thus, the data shown in Fig. 2 demonstrates that the gel electrophoretic mobility difference method can be used to study the distribution of M2tag (amanS) to M2-V27S (amanR) subunits and that M2tag and M2-V27S form mixed oligomers randomly. When cells individually expressing M2tag or M2-V27S were lysed and mixed and the M2 oligomers immunoprecipitated with M2-specific mAb 14C2 or tag epitope-specific mAb 12CA5 no mixed oligomer species were observed, thus ruling out the possible dissociation and reforming of oligomers during the experimental procedures used (data not shown).
Figure 1.

M2tag and M2-V27S are similarly activated by low pH and yield similar whole cell surface currents, but their amantadine sensitivities differ. Whole cell membrane currents of oocytes expressing M2tag (open bars) or M2-V27S (hatched bars) were measured in Barth’s solution at pH 7.5, after 30 s of incubation in Barth’s solution at pH 6.2, and again after 2 min of incubation at pH 6.2 with 100 μM amantadine. Currents are plotted as a multiple of current at pH 7.5. Values are given as mean ± SEM, n ≥ 5. Note that M2tag was fully sensitive to amantadine and that M2-V27S was fully resistant to amantadine.
Figure 2.

M2tag and M2-V27S freely form-mixed oligomers. HeLa-T4 cells were infected with vaccinia virus vTF7.3 for 30 min and then transfected with various amount (0–2.5 μg) of pTM3-M2tag or pTM3-M2-V27S DNA, with the total amount of DNA adjusted to 5 μg per dish by adding pTM3 vector DNA. At 3 h posttransfection cells were labeled with [35S]-Pro-mix (100 μCi/ml) for 30 min (A and B). RIPA buffer lysates were prepared in the presence of 50 mM iodoacetamide and immunoprecipitated with M2-specific mAb 14C2. Polypeptides were analyzed on SDS/PAGE under (A) reducing or (B) nonreducing conditions. In A, the amount of M2tag as a percentage of total M2 species was: lane 1, 0%; lane 2, 15%; lane 3, 35%; lane 4, 50%; lane 5, 72%; lane 6, 87%, and lane 7, 100%. (C) Cells were lysed in 1% Nonidet P-40, 50 mM Tris⋅HCl (pH 8.0), 100 mM NaCl, and 50 mM iodoacetamide and treated with dithiobis(succinimidyl proprionate) prior to immunoprecipitation with mAb 14C2 and analysis by SDS/PAGE under nonreducing conditions. D, disulfide-linked dimers of M2; T, disulfide-linked tetramers of M2; M, multimer of M2.
A second requirement for using this strategy is that the current measured from oocytes expressing purely amanS, purely amanR, or mixtures of the two subunits differ only in amantadine sensitivity, i.e., that these mixtures have approximately equal amplitudes when measured in the absence of amantadine, that their current increases by the same amount when activated by decreased pH, and that their ion selectivity be the same. We measured the currents of oocytes expressing comparable total quantities of protein and found that the amplitude of the current measured with oocyte bathing solution (pH 6.2) fell within a narrow range from 1.1–1.4 μA at −120 mV, a 5-to 7-fold increase over the current measured at pH 7.5. We also determined that the current–voltage relationship for oocytes expressing purely amanS subunits, purely amanR subunits, and mixtures were similar when measured at pH 7.5 and when measured at pH 6.2, thus showing that these oocytes differ only in amantadine sensitivity.
An important advance over using the approach first described by MacKinnon (16) with the mixed oligomer strategy to determine subunit stoichiometry is the ability to measure the ratio of the surface-expressed mixed oligomers in the very same oocytes used for electrophysiological measurements without relying on the ratio of microinjected RNA and making the assumption that different RNAs are translated with the same efficiency. First, the amount of M2tag and M2-V27S expressed at the surface of oocytes was determined by using the M2 ectodomain-specific mAb 14C2 to capture metabolically labeled cell surface molecules and to compare this amount to total expressed and immunoprecipitated M2tag or M2-V27S protein. After a 24-h continuous metabolic label, 30% of total M2tag protein and 30% of M2-V27S protein could be captured at the cell surface, indicating that there was no difference in surface expression of the two proteins (data not shown). The less than 100% cell surface expression levels of the M2 species reflects the slow intracellular transport rate in oocytes incubated at 18°C. The cell surface antibody binding assay was used to quantify the proportion of M2tag to M2-V27S subunits expressed in mixed oligomers at the cell surface. Polypeptides were analyzed on SDS/PAGE under both reducing and nonreducing conditions. An example of the mixed oligomers (analyzed under nonreducing conditions), expressed at the cell surface after microinjection of an mRNA mixture heavily biased for M2tag over M2-V27S, is shown in Fig. 3. For quantification of the data the sum of the subunits containing the M2tag to those with M2-V27S (untagged) was determined by analysis of the polypeptides under reducing conditions (as shown in Fig. 2A) and analysis of the radioactivity was performed using a BioImager. In the example shown in Fig. 3, the ratio of M2tag to M2-V27S in the subunits expressed at the cell surface was 0.71:0.29.
Figure 3.

Cell surface expression of M2tag and M2-V27S oligomers in oocytes of X. laevis. Synthetic mRNAs encoding M2tag, M2-V27S, or a mixture of the two RNAs and using water as a control were microinjected (50 nl of RNA of 0.85 μg/μl for M2tag, 50 nl of 0.25 μg/μl for M2-V27S, and 50 nl of RNA containing 0.75 μg/μl M2tag mixed with 0.25 μg/μl M2-V27S) into oocytes of X. laevis. At 24 h after injection, oocytes were labeled with [35S]methionine for 18 h and surface-expressed M2 protein captured with M2-specific mAb (14C2). Oocytes were washed, homogenized, and detergent lysates prepared in the presence of 50 mM iodoacetamide as described. Antibody was recovered by adding protein A-Sepharose beads and immune complexes were analyzed by SDS/PAGE under nonreducing conditions.
The principle for determining the stoichiometry of the active M2 oligomer is shown in Fig. 4 with the assumption being made that the oligomer is a tetramer. If the M2 ion channel has n subunits then on making mixed oligomers some channels will have n amanS subunits, some will have n amanR subunits, and many will have both subunit types. Among the different subunit types there will presumably be a range of amantadine sensitivity. The overall fraction of current inhibited by amantadine, Inhmix, at amantadine concentration [A] is given by the sum of the blocked current fraction contributed by each of the i channel species i:
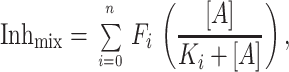 |
1 |
where Fi is the fraction of channels of species i, which contains i wt M2tag subunits in the entire population, Ki is the apparent inhibitory constant for that channel species, and n is the subunit stoichiometry. As we have shown (Fig. 2), if the various mixed oligomers form randomly then the fraction of the channels of each of the i species (Fi) will be determined by the law of mass action and the relative abundance of the M2tag (wt) (amanS) and M2-V27S mutant (amanR) subunits:
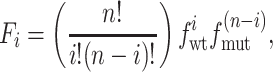 |
2 |
where fwt and fmut are the fractions of wt M2tag and mutant M2-V27S subunits, respectively. Thus, for example, for a tetrameric channel complex, the fraction of channels with one amanS and three amanR subunits will be directly proportional to the concentration of wt M2tag monomers, but proportional to the cube of the fraction of amanR monomers.
Figure 4.

Principle for determining active M2 protein oligomers. The cartoon depicts possible oligomers resulting from coexpression of amantadine-sensitive and epitope-tagged M2 subunits (○) with an amantadine-insensitive (V27S) mutant subunit ( ) assuming that the active oligomer is a tetramer. All possible combinations of the two subunits in a tetrameric channel are shown. As shown in Fig. 2, all five species can be identified.
To evaluate the value of n we needed to determine whether the amanS subunit or the amanR subunit is dominant. Mixtures of mRNAs encoding both amanS wt M2tag and amanR mutant M2-V27S protein were injected into oocytes and cell surface expression quantified as shown in Fig. 3. It was found that oocytes that expressed only a small fraction of the amanR subunit (15% or 21%; Fig. 5) had currents that were quite resistant to amantadine. These results suggest that the amanR form is dominant. For several of the cells that expressed a small fraction of amanR subunits, we confirmed that the amanR form was dominant by comparing the fraction of tetramers containing purely amanS subunits with Inhmix. If amanR is dominant, these values should be equal. For oocytes having a total of about 85% amanS subunits, about one-half of the tetramers consisted of purely amanS subunits (Fig. 2B, lane 6). When the currents of these oocytes were measured, the amantadine-sensitive component was about 50% of the total current, consistent with the amanR subunit being dominant. Thus, having demonstrated that the analytical requirements were satisfied, Eqs. 1 and 2 can be combined to provide an expression that relates the experimentally determined Inhmix to the subunit stoichiometry, n:
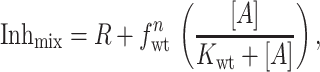 |
3 |
where R is the residual blocked current fraction due to all channel species having at least one amanR subunit, and fwt represents the fraction of amanS M2tag wt subunits. The second term is the blocked current fraction contributed by the fully amanS form (in this case, having only M2tag subunits that were shown above to be fully amanS). The apparent inhibitory constant for the fully wt M2 channel, Kwt, has been determined to be 0.3 μM amantadine (5). Thus, it is possible to calculate the blocked fraction for channels comprised of only the amanS subunits, Inhwt, from:
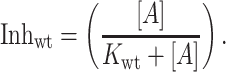 |
4 |
In these experiments [A] always exceeded Kwt, and thus Inhwt ≈1.
Figure 5.

Amantadine sensitivity of ion channel activities of mixed oligomers of known stoichiometry of M2tag and M2-V27S. Oocytes were injected with mRNA encoding M2tag, M2-V27S, or mixtures of these two mRNAs. After incubation for 2 days, the membrane currents of oocytes were measured in Barth’s solution at pH 6.2 with or without 20 μM of amantadine. The time course of the fraction of M2 ion channel current that was inhibited by amantadine, Inhmix, is plotted after addition of amantadine for M2tag protein (•), M2-V27S (♦), and for mixtures of M2tag protein and M2-V27S of defined stoichiometry (open symbols). The fraction of M2tag expressed in each batch of cells is given by the number above the curves and was measured by metabolically labeling a subset of the oocytes as described in Fig. 3.
Combining Eqs. 3 and 4 with Inhwt = 1, yields:
 |
5 |
Inhmix and fwt in Eq. 5 can be determined for individual cells from electrical measurements and the gel electrophoretic mobility difference method (Fig. 5), respectively. R represents the contribution to blocking by hybrids containing at least one amanR subunit. As suggested by the high degree of blocking found for small fractions of the amanR M2-V27S subunits (Fig. 5) and the correlation between the Inhmix and the fraction of tetramers comprised of purely amanS subunits (see above), these hybrids are significantly more resistant to amantadine than the fully amanS form. To simplify the analysis further and guard against the possibility of residual currents influencing the measurements, we performed experiments under conditions that minimize the value of R. These conditions are (i) low enough amantadine concentration to avoid nonspecific inhibition of the amantadine insensitive subunits and (ii) high fraction of amanS subunits, fwt, to shift the channel population toward the fully amanS form. When the value of R is minimized, the stoichiometry n can be estimated from Eq. 5 by neglecting R:
 |
6 |
Taking logarithms yields:
 |
7 |
We measured the Inhmix for eight oocytes that expressed mixtures of amanS and amanR subunits with a majority (71–91%) of the subunits being amanS. Because the inhibition by amantadine is only slowly reversible, the most appropriate means to quantify the inhibited current is to determine the time course of inhibition and to choose one time point for all measurements (isochronic condition) (5). As discussed above, the value of the current recorded from these oocytes was in the same range as the currents recorded from oocytes expressing purely amanS or amanR subunits. The determination of Inhmix was made with two concentrations of amantadine, 20 and 100 μM. For 100 μM of amantadine, Inhmix was determined 2 min after application, but for 20 μM amantadine, the steady state of inhibition was not reached until about 6 min, at which time Inhmix was determined. The subunit stoichiometry, n, determined from these measurements was 4.16 ± 0.17 (mean ± SEM).
DISCUSSION
The data presented here, using the methodology developed by MacKinnon (16), indicate that the active form of the influenza virus M2 ion channel is a homotetramer. We have extended the original protocol of microinjecting known mixtures of mRNAs by making a careful quantification of the amount of the M2 mixed oligomers expressed at the surface of the oocyte. However, a caveat has to be added that the unitary conductance of the M2 ion channel in living cells has not been measured, and if this conductance differed greatly for ion channels containing mixtures of amanS and amanR subunits from that of ion channels containing solely one type of subunit, the analysis would not be valid. However, we do not think this is the case, because the levels of protein expression and the current amplitude were nearly equal among the oocytes expressing mixtures, pure amanS, and pure amanR subunits.
Are there other oligomeric forms of the M2 ion channel that would have yielded results similar to those that we obtained? For example, is it possible that the active form is an octamer able to be inhibited by one amanR subunit? We think that this is unlikely, as can be shown from the following calculation of Inhmix. For fwt = 0.91 (which is the weakest case argument; see Table 1, line 2), if the active channel is an octamer, the calculated value of Inhmix = (0.91)8 = 0.47. This value is lower than the experimentally determined value of 0.69. If two amanR subunits are required to confer resistance to inhibition upon an octamer, the fraction of octamers with this composition of subunits will be the product of the sixth power of the fraction of wt subunits and the second power of the fraction of amantadine resistant subunits or [(fwt)6 × (1 − fwt)2]. The following calculation suggests it would be unlikely that we would have failed to detect this case that an octamer contains two amanR subunits: if fwt = 0.91, the calculated value of Inhmix = [1 − (0.91)6 × (0.09)2] = 0.996, a value sufficiently greater than the experimentally observed value of 0.69 to have been detected. The only molecular conformation that we could not have distinguished from a tetramer is one we consider to be only remotely possible: an active oligomeric form consisting of four tetramers, each of which contributes one transmembrane helix to the pore region, but for which resistance to inhibition can only be conferred by an amanR subunit in one of the four pore helices, i.e., there is quasi-equivalence and not functional equivalence of tetramers. In this model, the inhibition properties of the remaining 12 helices are irrelevant, and the mathematical analysis reduces to that of a simple tetramer. Although the present analysis cannot rule out a model with 16 subunits, only 4 of which contribute to the pore and determine sensitivity to inhibition, we believe such a model to be highly unlikely because it lacks a mechanism for linking the 4 tetramers. Models involving four tetramers, in which all of the subunits can contribute to resistance to inhibition, can be shown to be unlikely by the above analysis used for an octamer. Thus, the analytical method used firmly supports the conclusion that the active oligomeric form is a tetramer.
Table 1.
Prediction of the subunit stoichiometry of the active form of the M2 ion channel protein
| fM2tag (fwt) | Inhmix | In
(Inhmix)
|
|---|---|---|
| In (fwt) | ||
| 0.84 | 0.50 | 3.98 |
| 0.91 | 0.69 | 3.93 |
| 0.85 | 0.51 | 4.14 |
| 0.79 | 0.38 | 4.10 |
| 0.77 | 0.34* | 4.12 |
| 0.80 | 0.38* | 4.34 |
| 0.85 | 0.50* | 4.27 |
| 0.71 | 0.24* | 4.17 |
| 0.85 | 0.49* | 4.38 |
| mean n = 4.16 ± 0.17 | ||
The fraction of blocked currents at equilibrium, Inhmix, was determined using 100 μM of amantadine and 20 μM of amantadine (∗). Equilibrium inhibition is shown (2 min for 100 μM of amantadine and 6 min for 20 μM of amantadine). The inhibited fraction for the M2tag protein, Inhwt, at both 100 μM and 20 μM of amantadine was 100% at equilibrium (10 determinations). When these values for fraction-blocked current were substituted into Eq. 7, a value of n close to four was obtained. In this experiment, a high ratio of fwt (70–90%) was used. This condition favors a small R term (Eq. 6) by shifting the channel population towards the fully amantadine-sensitive form, allowing an accurate estimate of n to be made.
The M2 ion channel tetramer is made up of four 97-residue polypeptide chains containing a single 19-residue hydrophobic domain, which both anchors the protein in membranes and contributes in whole or in part to the pore-forming region of the channel (3, 4, 9, 29). There is no evidence to suggest that the M2 active complex is associated with unknown cellular proteins (13). Thus, the influenza virus M2 ion channel protein is truly a minimalistic or primitive ion channel by comparison to the multi-membrane spanning domain structure of the majority of eukaryotic ion channels. The significance of the 150–180 kDa multimer of M2 (13) (see also Fig. 2) is not known but may arise through extensive cross-linking or aggregation of multiple M2 tetramers. However, as discussed above, it is unlikely that the active form of the M2 protein is associated with this high molecular weight multimer.
The influenza virus M2 ion channel is an essential component of the virion and has to function after the entry of the virus into cells during virus uncoating in endosomes to allow protons to enter the virion to weaken protein–protein interactions. Nonetheless, the M2 protein is only found in virions in small amounts; it is estimated that there are on average 14–68 M2 subunits per virion (30). Thus, the finding that the active form of the M2 ion channel is a homotetramer suggests there are only ≈3–17 channel complexes per virion.
Acknowledgments
We are very grateful to Diana Brassard for constructing the M2tag DNA molecule. This research was supported by Public Health Service Research Grants AI-20201 (R.A.L.) and AI-31882 (L.H.P.) from the National Institute of Allergy and Infectious Diseases and Merck, Sharp and Dohme, Inc. for providing to L.H.P. a postdoctoral fellowship for Q.T. R.A.L. is an Investigator of the Howard Hughes Medical Institute.
ABBREVIATION
- wt
wild type
References
- 1.Hay A J. Semin Virol. 1992;3:21–30. [Google Scholar]
- 2.Lamb R A, Holsinger L J, Pinto L H. In: The Influenza A Virus M2 Ion Channel Protein and its Role in the Influenza Virus Life Cycle. Wimmer E, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 303–321. [Google Scholar]
- 3.Pinto L H, Holsinger L J, Lamb R A. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 4.Holsinger L J, Nichani D, Pinto L H, Lamb R A. J Virol. 1994;68:1551–1563. doi: 10.1128/jvi.68.3.1551-1563.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C, Takeuchi K, Pinto L H, Lamb R A. J Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimbo K, Brassard D L, Lamb R A, Pinto L H. Biophys J. 1996;70:1335–1346. doi: 10.1016/S0006-3495(96)79690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang C, Lamb R A, Pinto L H. Virology. 1994;205:133–140. doi: 10.1006/viro.1994.1628. [DOI] [PubMed] [Google Scholar]
- 8.Chizhmakov I V, Geraghty F M, Ogden D C, Hayhurst A, Antoniou M, Hay A J. J Physiol (London) 1996;494:329–336. doi: 10.1113/jphysiol.1996.sp021495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duff K C, Ashley R H. Virology. 1992;190:485–489. doi: 10.1016/0042-6822(92)91239-q. [DOI] [PubMed] [Google Scholar]
- 10.Schroeder C, Ford C M, Wharton S A, Hay A J. J Gen Virol. 1994;75:3477–3484. doi: 10.1099/0022-1317-75-12-3477. [DOI] [PubMed] [Google Scholar]
- 11.Tosteson M T, Pinto L H, Holsinger L J, Lamb R A. J Membr Biol. 1994;142:117–126. doi: 10.1007/BF00233389. [DOI] [PubMed] [Google Scholar]
- 12.Lamb R A, Zebedee S L, Richardson C D. Cell. 1985;40:627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- 13.Holsinger L J, Lamb R A. Virology. 1991;183:32–43. doi: 10.1016/0042-6822(91)90115-r. [DOI] [PubMed] [Google Scholar]
- 14.Sugrue R J, Hay A J. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panayotov P P, Schlesinger R W. Virology. 1992;186:352–355. doi: 10.1016/0042-6822(92)90096-8. [DOI] [PubMed] [Google Scholar]
- 16.MacKinnon R. Nature (London) 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- 17.Cooper E, Couturier S, Ballivet M. Nature (London) 1991;350:235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- 18.Wang K-W, Goldstein S A N. Neuron. 1995;14:1303–1309. doi: 10.1016/0896-6273(95)90277-5. [DOI] [PubMed] [Google Scholar]
- 19.Maddon P J, Dalgleish J S, McDougal P R, Clapham P R, Weiss R A, Axel R. Cell. 1986;47:333–342. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- 20.Fuerst T R, Niles E G, Studier F W, Moss B. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hull J D, Gilmore R, Lamb R A. J Cell Biol. 1988;106:1489–1498. doi: 10.1083/jcb.106.5.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zebedee S L, Richardson C D, Lamb R A. J Virol. 1985;56:502–511. doi: 10.1128/jvi.56.2.502-511.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green N, Alexander H, Olson A, Alexander S, Shinnick T M, Sutcliffe J G, Lerner R A. Cell. 1982;28:477–487. doi: 10.1016/0092-8674(82)90202-1. [DOI] [PubMed] [Google Scholar]
- 24.Rose J K, Bonagurio B, Whitt M A. BioTechniques. 1991;10:520–525. [PubMed] [Google Scholar]
- 25.Paterson R G, Lamb R A. In: The Molecular Biology of Influenza Viruses and Paramyxoviruses. Davidson A, Elliott R M, editors. Oxford: IRL/Oxford Univ. Press; 1993. pp. 35–73. [Google Scholar]
- 26.Zebedee S L, Lamb R A. J Virol. 1988;62:2762–2772. doi: 10.1128/jvi.62.8.2762-2772.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamb R A, Choppin P W. Virology. 1976;74:504–519. doi: 10.1016/0042-6822(76)90356-1. [DOI] [PubMed] [Google Scholar]
- 28.Shimbo K, Brassard D L, Lamb R A, Pinto L H. Biophys J. 1995;69:1819–1829. doi: 10.1016/S0006-3495(95)80052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C, Lamb R A, Pinto L H. Biophys J. 1995;69:1363–1371. doi: 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zebedee S L, Lamb R A. Proc Natl Acad Sci USA. 1989;86:1061–1065. doi: 10.1073/pnas.86.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]