

Abstract
Cell–substratum adhesion is an essential requirement for survival of human neonatal keratinocytes in vitro. Similarly, activation of the epidermal growth factor receptor (EGF-R) has recently been implicated not only in cell cycle progression but also in survival of normal keratinocytes. The mechanisms by which either cell–substratum adhesion or EGF-R activation protect keratinocytes from programmed cell death are poorly understood. Here we describe that blockade of the EGF-R and inhibition of substratum adhesion share a common downstream event, the down-regulation of the cell death protector Bcl-xL. Expression of Bcl-xL protein was down-regulated during forced suspension culture of keratinocytes, concurrent with large-scale apoptosis. Similarly, EGF-R blockade was accompanied by down-regulation of Bcl-xL steady-state mRNA and protein levels to an extent comparable to that observed in forced suspension culture. However, down-regulation of Bcl-xL expression by EGF-R blockade was not accompanied by apoptosis; in this case, a second signal, generated by passaging, was required to induce rapid and large-scale apoptosis. These findings are consistent with the conclusions that (i) Bcl-xL represents a shared molecular target for signaling through cell-substrate adhesion receptors and the EGF-R, and (ii) reduced levels of Bcl-xL expression through EGF-R blockade lower the tolerance of keratinocytes for cell death signals generated by cellular stress.
Survival of normal, untransformed cells depends on the presence of environmental stimuli. If such survival signals are not provided, cells will undergo a process termed programmed cell death (PCD) or apoptosis. Both cell adhesion receptors and growth factor/cytokine receptors appear to play prominent roles in mediating survival of normal epithelial cells. For example, preventing cell–substrate interactions induces PCD in the immortalized keratinocyte line HaCaT (1). Similarly, normal anchorage-dependent mammary epithelial cells require activation of adhesion molecules of the integrin class to survive in vitro (2). The importance of growth factor/cytokine receptors to cell survival has been demonstrated in various cell systems. For example, activation of the interleukin 3 receptor supports survival of hemopoietic cells (3–6); the nerve growth factor receptor, the survival of neurons (7, 8); and the insulin like growth factor 1 receptor, the survival of fibroblasts (9, 10). Removal of the respective growth factor from the medium or receptor blockade has been shown to induce PCD.
We reported recently that normal human keratinocytes depend on activation of the epidermal growth factor receptor (EGF-R) for survival (11). Specifically, blocking EGF-R activation by exposure to either a ligand-antagonistic monoclonal antibody (mAb 425) or an EGF-R-selective tyrosine kinase inhibitor of the tyrphostin class (AG1478) significantly shortens the life span of cultured normal human keratinocytes from 5–7 weeks to ≈2 weeks. Keratinocyte PCD induced by EGF-R blockade occurs slowly and incrementally, being first detected after 5 days of EGF-R blockade. The induction of apoptotic keratinocyte death by EGF-R blockade can be greatly accelerated if these cells are passaged after at least 2 days of EGF-R blockade. The combination of EGF-R blockade and passaging induces large-scale PCD (75–95% of the total cell population) within 24 h. Taken together, these earlier results indicate that EGF-R blockade increases susceptibility of normal human keratinocytes to PCD induced by cellular stress such as passaging.
Little is known about adhesion- or cytokine-dependent regulation of molecular mechanisms controlling entry into PCD. In the present study we have begun to examine molecular targets downstream of either substratum adhesion or of the EGF-R that may be relevant to the control of keratinocyte survival and death. This investigation focused on known components of the cell death machinery that have been identified in recent years. The best studied death regulatory molecules are the Bcl-2 family of proteins (for a review, see ref. 12). Different members of this molecular family either protect cells from cell death (for example Bcl-2 and Bcl-xL) or promote cell death (for example Bcl-xS, Bad, Bak, Bax, and Bik). In hemopoietic cells it has been demonstrated that stimulation of cell surface receptors including CD40 (13), CD28 (14), and the interleukin 2 receptor (15, 16) not only can effect rescue from apoptosis but also up-regulate Bcl-xL expression. We therefore asked the question whether EGF-R stimulation and/or substratum adhesion similarly affect the expression of Bcl-2 family members. We demonstrate that EGF-R blockade and forced suspension share a common downstream event, the down-regulation of Bcl-xL expression. As Bcl-xL is a protector from apoptosis, our results indicate that engagement of adhesion receptors and of the EGF-R regulates at least one common intracellular mediator that maintains cell survival.
MATERIALS AND METHODS
Cell Culture and Reagents.
Human neonatal foreskin keratinocyte cultures were initiated and propagated in MCDB153 complete medium, as described (17). Complete MCDB153 (Sigma) contained 30 μM Ca2+ and was supplemented with amino acids, ethanolamine, phosphorylethanolamine, hydrocortisone, and insulin (all from Sigma); EGF (10 ng/ml, Collaborative Research); and bovine pituitary extract (prepared from pituitaries from Pel-Freez). All assays were performed using MCDB153 complete medium free of exogenous EGF and of bovine pituitary extract. Antigen specificity and EGF-R antagonistic properties of mAb 425, which recognizes a protein epitope on the human EGF-R, have been described in detail (18–21). Control mAb 15-6A, which is directed to a carbohydrate epitope, binds to but does not interfere with the activity of the EGF-R (22). In previous studies we have shown that the mAb 425 preparation used here is nontoxic, as high concentrations of EGF neutralize the effect of mAb 425 on keratinocyte proliferation (23). Poly(2)-hydroxy-ethyl methylacrylate [poly(HEMA)] was purchased from Sigma.
Analysis of DNA Integrity.
DNA integrity was determined by agarose gel electrophoresis according to Eastman (24).
Immunoblot Analysis.
Cells were harvested by trypsinization and immediately lysed in 1× Laemmli buffer (62.5 mM Tris⋅HCl, pH 6.8/2% SDS/10% glycerol) supplemented with protease inhibitors (2 μg/ml aprotinin, 2 μg/ml leupeptin, and 0.5 mM phenylmethylsulfonyl fluoride) and boiled for 3–5 min. Equal amounts of protein, normalized to cell number were separated on 12% SDS/polyacrylamide gels followed by blotting to polyvinylidene difluoride membranes (Millipore). Membranes were blocked for 1 h at room temperature using 5 vol % dry milk in PBS. After incubation with antibodies binding specifically to Bcl-2 (Dako, Glostrup, Denmark; #M 887; diluted to 5 μg/ml), Bak (Calbiochem; diluted to 2.5 μg/ml), or Bcl-x (mAb 2A1.24; 5 μg/ml; kindly provided by L. H. Boise, University of Chicago, Chicago), immune complexes were detected using secondary anti-rabbit/anti-mouse antibodies coupled to biotin followed by incubation with the ABC detection system according to the manufacturer’s instructions (Vector Laboratories) and visualized by enhanced chemiluminescence (Renaissance; DuPont/NEN). To assess involucrin expression, either the upper portion of the polyvinylidene difluoride membrane was used or duplicate aliquots of samples were separated on 9.5% polyacrylamide gels followed by detection using a rabbit anti-involucrin antibody (BT-601; Biomedical Technologies, Stoughton, MA) diluted 1:12. Protein extracts from chicken DT-40 cells transfected with human Bcl-xL (D.L.E., unpublished results) served as positive controls for Bcl-xL. In one lane of each gel, Prestained protein molecular weight standards were applied (Life Technologies, Gaithersburg, MD).
Reverse Transcription–PCR (RT-PCR).
RT-PCR analysis of bcl-x transcripts was performed as described (25). Control β-actin sequences were amplified as described (26).
RNase Protection Assay.
RNA was resuspended in diethyl pyrocarbonate-treated water and stored at −20°C. The RNase protection assay was carried out described (27). Briefly, antisense RNA probes were prepared using the T3 promoter in Bluescript (Stratagene), labeled with [32P]CTP, purified on a 6% acrylamide sequencing gel, and used the same day. Total RNA (25 μg) was hybridized with 2 × 105 cpm antisense probe in 80% formamide buffer at 60°C. The bcl-x probe was generated from a human cDNA clone and covers nucleotides 104–433 (GenBank accession no. L20121L20121), the human bcl-2 probe was generated from a human cDNA clone and covers the N-terminal 214 nucleotides of the bcl-2 coding region (GenBank accession no. M13994M13994), and mouse cyclophilin antisense probe (Ambion, Austin, TX) was used as an internal control for standardization of expression levels between samples. RNA from fibroblasts transfected with human bcl-x and human bcl-2, served as positive controls, respectively. Samples were fractionated on a 6% sequencing gel; the gel was dried, exposed to a PhosphorImager screen, and the relative signal intensities calculated using the image quant program (Molecular Dynamics).
RESULTS
Keratinocyte Apoptosis Induced by Forced Suspension Culture.
A previous study has shown that apoptosis was induced in the immortalized keratinocyte cell line HaCaT by forced suspension culture (1). To determine if normal keratinocytes respond similarly to the HaCaT line, we seeded early passage human neonatal keratinocytes onto plastic pretreated with poly(HEMA), a substrate known to prevent adhesion. After 24 h of culture on poly(HEMA) in complete MCDB153 medium, a pattern of DNA degradation consistent with apoptosis was evident in normal early passage keratinocytes (Fig. 1). In agreement with earlier observations (24) in other epithelial cells, we found DNA degradation in the form of higher molecular weight smears rather than the nucleosome ladders that are commonly observed in hemopoietic cells. To assess quantitatively the loss of cell viability caused by forced suspension culture, we determined with time the fraction of cells that could be rescued by transfer into tissue culture-treated plates. Rescued cells were scored by their ability to reattach and, upon further culture, commence cell cycle progression as determined by [3H]thymidine uptake. As shown in Table 1, the percentage of normal keratinocytes able to reattach to tissue culture-treated plastic decreased steadily during the first 12 h of culture on poly(HEMA). By 24 h, almost all cells (>98%) had initiated an irreversible cell death program as determined by their inability to reattach. Similarly, as described earlier (1), many HaCaT cells underwent PCD. However, as compared with normal keratinocytes, a significant fraction (40–60%) of HaCaT cells could be rescued 24 h or even 4 days after forced suspension culture, indicative of a subpopulation that is resistant to apoptosis induced by this means (Table 1). These results indicate that, compared with the HaCaT line, early passage normal keratinocytes are more dependent upon substratum adhesion for survival.
Figure 1.
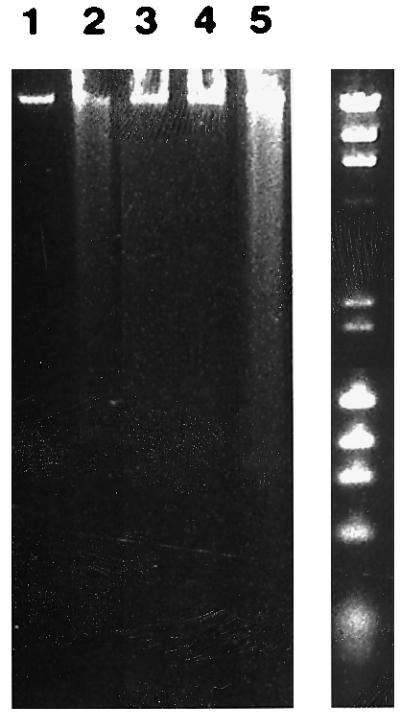
DNA degradation upon forced suspension culture or EGF-R blockade of normal human neonatal keratinocytes. High molecular weight DNA derived from keratinocytes cultured for 24 h on tissue culture-treated plastic (lane 1) or poly(HEMA) (lane 2) was analyzed. Lane 3 contains DNA from control cells cultured 3 days in MCDB153 medium, passaged, and harvested 24 h after passaging. Lane 4 contains DNA of keratinocytes cultured for 3 days in the presence of EGF-R antagonistic mAb 425 harvested before passaging. Lane 5 shows DNA degradation in keratinocytes cultured for 3 days in the presence of mAb 425, passaged, and harvested 3 days after passaging. Molecular weight standards (λHindIII and φX174HaeIII) are shown to the right.
Table 1.
Survival of normal human keratinocytes and immortalized keratinocytes (HaCaT) after various time of forced suspension culture
| Time,* h | Cell number†
|
|
|---|---|---|
| Normal keratinoytes | HaCat | |
| 0 | 1.86 ± 0.2 | 2.4 ± 0.15 |
| 1 | 1.62 ± 0.1 | |
| 2 | 1.25 ± 0.09 | |
| 4 | 0.93 ± 0.08 | |
| 6 | 0.79 ± 0.09 | |
| 8 | 0.54 ± 0.08 | |
| 10 | 0.35 ± 0.03 | |
| 12 | 0.25 ± 0.01 | |
| 24 | <0.01 | 1.1 ± 0.1 |
| 48 | <0.01 | 1.1 ± 0.1 |
| 72 | <0.01 | 0.7 ± 0.1 |
| 96 | <0.01 | 1.2 ± 0.1 |
Indicates time after seeding cells on poly(HEMA); actual cell counts were obtained 24 h after transfer of cells to tissue culture-treated plastic. Cell number at time point 0 was obtained by seeding cells on tissue culture-treated plastic and determination of attached cells 8 h after seeding.
Total cell number (×104) per well; results represent mean ± SD of triplicate determinations. Representative results of one of three experiments are shown.
Keratinocyte Apoptosis Following EGF-R Blockade.
EGF-R blockade for 3–5 days followed by passaging induced rapid, large-scale PCD in normal human keratinocytes (11). In this earlier study, EGF-R activation by endogenous or exogenous ligands was blocked using either an EGF-R antagonistic antibody (mAb 425) or the EGF-R-selective tyrphostin AG1478. It must be stressed that, as determined by morphological criteria or TUNEL (terminal deonucleotidyltransferase-mediated UTP end labeling) staining, large-scale keratinocyte apoptosis did not occur if the cultures were simply incubated with the EGF-R antagonists for 3–5 days; however, routine passaging at this point, followed by reseeding onto tissue culture-treated plastic, led to rapid induction of apoptosis (11). Routine passaging consisted of trypsinization using a 0.5% trypsin/0.01% EDTA solution in Hanks’ balanced salt solution followed by washing cells one time in a 10% fetal calf serum solution in Hanks’ balanced salt solution and reseeding in complete MCDB153 medium. Consistent with our earlier results, no evidence of DNA degradation was detectable in adherent keratinocytes after 3 days of mAb 425 treatment and cultured in MCDB153 medium free of exogenous EGF and bovine pituitary extract (Fig. 1, lane 4); however, passaging of mAb 425-treated cells (Fig. 1, lane 5) but not control cells at this time followed by replating in compete MCDB153 medium led to significant DNA degradation within 24 h. The pattern of DNA degradation in the mAb 425-pretreated and passaged cells was similar to that observed in keratinocytes cultured for 24 h on poly(HEMA) (Fig. 1, lane 2). These results are consistent with the conclusion that EGF-R blockade primes keratinocytes for cell death, which can be precipitated by passaging. The temporal separation between the priming event (i.e., EGF-R blockade) and the triggering of large-scale apoptosis by passaging afforded us the opportunity to study the regulation of molecules relevant to apoptosis that occur prior to its acute onset. We therefore investigated EGF-R-dependent expression patterns of the Bcl family of proteins.
Expression of Bcl-2-Like Regulators of Cell Death by Cultured Keratinocytes.
As determined by immunoblot analyses, Bcl-2 was weakly expressed or undetectable in early-passage normal keratinocyte cultures initiated from different donors (data not shown) whereas Bcl-x was expressed in each of the three keratinocyte cultures analyzed (Fig. 2). Due to differential splicing, the bcl-x gene is known to encode two different Bcl-x proteins, a long (Bcl-xL; 26 kDa) and a short (Bcl-xS; 21 kDa) form, which serve opposite functions in control of cell survival (25); whereas Bcl-xL promotes survival, Bcl-xS promotes cell death. Consistent with an earlier report (28) the immunoreactive Bcl-x species detected in keratinocytes by immunoblot analysis corresponded to the larger (i.e., protective) Bcl-xL protein whereas no evidence of the smaller Bcl-xS isoform was detected. Analysis of bcl-x transcripts in keratinocytes using RT-PCR followed by ethidium bromide gel electrophoresis of the amplification products confirmed this result. Using primers that span the bcl-x cDNA region containing the spliced exon, a single amplimer was detected corresponding to bcl-xL (Fig. 2B).
Figure 2.

Down-regulation of Bcl-xL expression in keratinocytes by EGF-R blockade. (A) Immunoblot analysis of Bcl-xL protein expression in three different keratinocyte cultures maintained in the presence (+) of EGF-R antagonistic mAb 425 (10 μg/ml) (+) or in the presence of control mAb 15-6A (−) for 3 days. As a control, cell extracts of chicken bursal lymphocytes transduced with human Bcl-xL were included. To assess effects of EGF-R blockade on keratinocyte differentiation expression of involucrin was determined for all experimental conditions. (B) RT-PCR products generated using keratinocyte RNA and Bcl-xL-specific primers. Two keratinocyte cultures were maintained in the absence (lanes 1 and 2) and in the presence (lanes 3 and 4) of EGF-R antagonistic mAb 425 (10 μg/ml) for 3 days. Controls include amplification products derived from chicken bursal cells transduced with human bcl-xL and bcl-xS cDNAs. (Lower) To control for RNA integrity and quantity amplification products using primers specific for human β-actin are shown.
Regulation of Bcl-xL Expression by Substrate Engagement and the EGF-R.
We next examined whether substratum adhesion and/or EGF-R activation regulate Bcl-xL expression in keratinocytes. As shown in Fig. 3A, forced suspension culture of normal keratinocytes led within 24 h to significant loss of Bcl-xL protein expression, compared with control cells that were allowed to adhere. Down-regulation of Bcl-xL expression became evident between 4 and 6 h after seeding on poly(HEMA) and remained at low levels throughout the observation period of 24 h. Similarly, blockade of EGF-R activation by treatment with mAb 425 was associated with significant down-regulation of Bcl-xL protein when compared with controls treated with mAb 15-6A (Fig. 2A). Down-regulation of Bcl-xL protein expression by EGF-R blockade occurred between 8 and 24 h after addition of mAb 425 to cultures (Fig. 3B). In an earlier study, expression of Bcl-xL by keratinocytes has been related to the differentiation state of these cells (29). Specifically, Bcl-x immunostaining was strongest in the upper, differentiated layers of the epidermis but comparatively weak in basal cells. To determine whether changes of Bcl-xL expression subsequent to forced suspension culture or EGF-R blockade were related to accelerated keratinocyte differentiation we assessed expression of the early differentiation marker, involucrin in keratinocyte cultures treated with mAb 425 or seeded on poly(HEMA), respectively. As shown in Figs. 2, 3, and 5, no significant effects of either experimental treatments on involucrin expression were observed. Northern blot analysis demonstrated that, like Bcl-xL protein levels, steady-state bcl-x mRNA levels were also reduced in mAb 425-treated keratinocyte cultures, consistent with an effect of EGF-R on bcl-x transcription and/or RNA stability (Fig. 4A). RNase protection analysis of time-dependent effects of EGF-R blockade on steady-state bcl-x mRNA levels showed that significant down-regulation occurred between 1 and 6 h after addition of mAb 425 (Fig. 4B). Down-regulation of Bcl-xL expression by mAb 425 treatment was reversible; within 24 h after removal of the antibody, there was complete recovery of Bcl-xL expression (Fig. 5). These cultures were also able to re-adhere and proliferate upon passaging, as shown (11).
Figure 3.

Time-dependent down-regulation of Bcl-xL expression upon forced suspension culture or EGF-R blockade of normal keratinocytes. Bcl-xL protein expression was compared by immunoblot analysis of cells seeded for various times on plastic (P) or poly(HEMA) (A) or in the absence (−) and presence (+) of mAb 425 (B). Representative results of one of three experiments are shown.
Figure 5.

Reversible down-regulation of Bcl-xL expression upon lifting of EGF-R blockade. Bcl-xL protein expression of keratinocytes treated with control mAb 15-6A (−) and EGF-R antagonistic mAb 425 (+) for 3 days is shown in the first two lanes. Lanes 3–7 show time-dependent recovery of Bcl-xL expression levels in parallel cultures after removal of mAb 425 and addition of complete MCDB153 medium supplemented with EGF at 10 ng/ml. Representative results of one of two experiments are shown.
Figure 4.

Down-regulation of Bcl-xL steady-state mRNA expression by EGF-R blockade. (A) Northern blot analysis for Bcl-xL was performed on mRNA from keratinocytes incubated in the presence or absence of mAb 425 for 3 days. The results of one of two experiments are shown. (B) RNase protection assay was performed after incubation with and without mAb 425 for 0, 1, 6, and 24 h. No evidence of bcl-2 mRNA expression was found in RNase protection assay. Cyclophilin A served as an internal standard to ensure comparable RNA quantities. Positive controls for RNase protection assays included RNA isolated from avian fibroblasts transduced with human bcl-xL and bcl-2 cDNAs as indicated in B.
EGF-R-Dependent Expression of Bak by Keratinocytes.
At least two other members of the Bcl-2 family (Bax and Bak) are known to promote cell death. In immunoblot analysis of cultured keratinocyte extracts from two donors, immunoreactive protein species corresponding to Bak (Fig. 6) but not Bax (data not shown) were found. Consistent with an earlier study (30), expression of Bak was observed in A431 cells which served as a positive control in these experiments. In contrast to Bcl-xL, Bak expression did not change upon EGF-R blockade with mAb 425 (Fig. 6).
Figure 6.

Immunoblot analysis of Bak protein expression upon EGF-R blockade in normal keratinocytes. Bak expression in two keratinocyte cultures after EGF-R blockade for 3 days using mAb 425 as compared to cultures maintained in the presence of control mAb 15-6A are shown. In both experiments no significant modulation of Bak expression was observed whereas Bcl-xL was completely (culture 1) or partially (culture 2) down-regulated by EGF-R blockade. Positive controls include extracts of squamous carcinoma cells A431 (Bak) and of chicken bursal lymphocytes transduced with human bcl-xL.
DISCUSSION
Apoptotic death of normal keratinocytes can be induced by two distinct mechanisms: (i) prevention of substratum adhesion—e.g., by plating onto dishes coated with poly(HEMA)—which prevents cell attachment, or (ii) EGF-R blockade followed by routine passaging and replating onto tissue culture plastic. In this study we provide evidence that both methods of inducing keratinocyte death are associated with down-regulation of a protector from apoptotic cell death, Bcl-xL but not of another Bcl-2 family member that promotes apoptosis, Bak.
The balance of cell death protectors and inducers in the Bcl-2 family of proteins has been proposed as a common determinant of the susceptibility of cells to apoptotic cell death induced by various stimuli (12, 31). The Bcl-2 family members Bcl-2, Bcl-xL, and Bcl-w have been shown to protect cells from apoptosis, whereas Bad, Bak, Bax, and Bik induce apoptosis. The different members of the Bcl-2 protein family undergo homo- and heterodimerization, depending on their expression levels. The relative amounts of specific dimers are believed to determine susceptibility to apoptosis. For example, Bax homodimers promote PCD (32); however, Bcl-2 or Bcl-xL, when present at sufficiently high levels, can heterodimerize with Bax, thereby preventing the formation of Bax homodimers and protecting the cell from apoptotic death (32–34). Based on this model, reduced expression of Bcl-2 or Bcl-xL is predicted to allow the predominance of death promoters such as Bax or Bak to be manifest (30, 35, 36), thereby increasing cell susceptibility to PCD. Consistent with this model we found that EGF-R blockade is associated with down-regulation of the cell death protector Bcl-xL but does not affect expression of the cell death inducer Bak as detected in Western blot analyses. It should be noted that the balance of homo- versus heterodimers may be just one way in which this family of proteins can act, as Bcl-xL has recently been shown to protect cells from apoptosis independently of its ability to heterodimerize with Bax (37). That expression of Bcl-xL is subject to regulation by the EGF-R was suggested by a recent study of mammary epithelial cell lines established from transgenic mice that overexpress c-myc (38). These cell lines are prone to die by PCD due to overexpression of the c-myc transgene. Addition of the EGF-R ligands EGF and transforming growth factor α to these cell lines not only enhances their survival but also induces up-regulation of Bcl-xL mRNA and protein. These data suggest that EGF-R-dependent Bcl-xL expression protects epithelial cells that over-express c-myc from PCD. Our findings are in agreement with and extend these observations in several important ways. By using normal early-passage keratinocytes, we demonstrate that EGF-R activation supports survival not only of Myc-overexpressing but also of normal epithelial cells with unmodified levels of c-Myc expression. Thus, EGF-R activation provides a physiologically relevant survival signal for at least some normal epithelial cells.
Several lines of evidence presented in this study indicate that down-regulation of Bcl-xL expression is not sufficient to induce the keratinocyte death program, at least within the time frame of the experiments here described. First, although Bcl-xL is strongly down-regulated simply by blockade of the EGF-R, such treatment is not in itself sufficient to induce large-scale apoptosis, as measured by morphological criteria, TUNEL assays (11), and gel electrophoretic analysis of DNA degradation (Fig. 1). Second, even after EGF-R blockade has induced Bcl-xL down-regulation, its effects on keratinocyte survival remain reversible (11). Lifting the EGF-R blockade is accompanied by up-regulation of Bcl-xL expression within 24–48 h and resistance to cell death upon passaging. Taken together, these findings support the view that reduced Bcl-xL expression lowers the resistance of keratinocytes to acute cell death signals generated by passaging.
The importance of Bcl-xL to normal keratinocyte survival is further supported by our findings that two distinct mechanisms for inducing PCD (EGF-R blockade and lack of substratum adhesion) are associated with down-regulation of Bcl-xL expression. These results suggest that signaling pathways from the EGF-R and from adhesion receptors converge on Bcl-xL as a shared target relevant to epithelial cell survival. It is highly likely that cell–substratum adhesion molecules of the integrin class are implicated in the induction of PCD. Integrins comprise a family of heterodimeric transmembrane glycoproteins, each consisting of an α- and a β-chain that bind to extracellular matrix molecules. Keratinocytes express numerous integrins, including α6β4, α2β1, α3β1, and α5β1 (39). That integrins can regulate PCD in epithelial cells has been suggested by recent studies of Boudreau et al. (2) who showed that extracellular matrix engagement of β1 integrin delays PCD of normal mammary epithelial cells. Several studies have demonstrated that integrins, like growth factor receptors, possess signal transduction capacity, thereby providing a mechanism through which the extracellular matrix can influence cell phenotype and, possibly, survival. Notably, activation of integrins by ligand binding leads to the formation of a multimolecular complex at the inner cell membrane, called a focal adhesion plaque, that contains among other proteins a tyrosine kinase known as focal adhesion kinase (FAK) (40). Constitutively activated forms of FAK have been shown to rescue epithelial cell lines from apoptosis induced by forced suspension cultures consistent with an important contribution of the tyrosine kinase moiety of FAK to epithelial cell survival (41). By using the EGF-R selective tyrphostin AG1478 we have demonstrated earlier that, in the case of the EGF-R, the tyrosine kinase moiety is necessary to maintain keratinocyte viability (11). Taken together, these observations raise the intriguing possibility that activation of tyrosine kinases involved in integrin signaling (such as FAK) and receptor tyrosine kinases such as the EGF-R coordinately regulate Bcl-xL expression in epithelial cells. Little is known about the signal transduction pathways linking tyrosine kinases to regulation of cell survival except that inhibitors of phosphatidylinositol-3 kinase such as wortmannin can induce PCD in PC12 pheochromocytoma, RAT-1 fibroblasts, and hemopoietic cells of various lineages (42–44). Phosphatidylinositol-3 kinase is phosphorylated upon activation of tyrosine kinase receptors including the nerve growth factor receptor (45) and the EGF-R (46) and has also been found to be associated with FAK upon integrin engagement by substrate (47). It is thus possible that phosphatidylinositol-3 kinase is a shared target for both, receptor tyrosine kinase- and integrin-dependent signaling relevant to cell survival and Bcl-xL expression in keratinocytes.
Our results are consistent with the view that adhesion/tyrosine kinase-dependent mechanisms may regulate apoptosis susceptibility of epithelial cells on several, distinct levels. In support of this view, detachment from substrate may not only affect expression of Bcl-2 like apoptosis regulators in keratinocytes as shown in this study, but is also followed by induction of the expression of proteolytic enzymes of the Caspase family, particularly Caspase-1 (ICE) in mammary epithelial cells (2); Caspases have been implicated in the execution of cell death programs in diverse cell systems (48).
The findings that we have reported here for normal keratinocytes provide a possible explanation for the recently published observation that hyperplastic epidermis as observed in psoriatic plaques expresses higher Bcl-xL levels than normal epidermis (28). Psoriatic skin is characterized by up-regulated expression of the EGF-R (49–51) and its ligands transforming growth factor α (51–53) and amphiregulin (54), thus constituting a hyperactive EGF-R-dependent autocrine loop. In addition, aberrant expression of the α2, α3, α6, and β1 integrins has been observed in the suprabasal layers of hyperproliferative epidermis (55). Thus, the elevated levels of Bcl-xL in lesional psoriatic epidermis may very well be secondary to supra-physiological activation of the EGF-R and/or to enhanced engagement of integrin receptors.
Acknowledgments
We are grateful to Dr. L. Showe for help in performing RNase protection assays. We thank Dr. L. H. Boise for the Bcl-x antibody. This work was supported by National Institutes of Health Grants AR42998 and AR42682 (P.J.J.) and a grant from the Gustavus and Louise Pfeiffer Foundation (U.R.). B.R. was a fellow of the Deutsche Forschungsgemeinschaft (Ri 716/1-1).
ABBREVIATIONS
- EGF-R
epidermal growth factor receptor
- PCD
programmed cell death
- poly(HEMA)
poly(2)-hydroxy-ethyl methylacrylate
- TUNEL
terminal deonucleotidyltransferase-mediated UTP end labeling
- RT-PCR
reverse transcription–PCR
References
- 1.Frisch S M, Francis H. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boudreau N, Sympson C J, Werb Z, Bissel M J. Science. 1996;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pierce J H, Ruggiero M, Fleming T P, Di Fiore P P, Greenberger J S, Varticovski L, Schlessinger J, Rovera G, Aaronson S A. Science. 1988;239:628–631. doi: 10.1126/science.3257584. [DOI] [PubMed] [Google Scholar]
- 4.Collins M K, Marvel J, Malde P, Lopez-Rivas A. J Exp Med. 1992;176:1043–1051. doi: 10.1084/jem.176.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mekori Y A, Oh C K, Metcalfe D D. J Immunol. 1993;151:3775–3784. [PubMed] [Google Scholar]
- 6.Baffy G, Miyashita T, Williamson J R, Reed J C. J Biol Chem. 1993;268:6511–6519. [PubMed] [Google Scholar]
- 7.Martin D P, Ito A, Horigome K, Lampe P A, Johnson E., Jr J Neurobiol. 1992;23:1205–1220. doi: 10.1002/neu.480230911. [DOI] [PubMed] [Google Scholar]
- 8.Garcia I, Martinou I, Tsujimoto Y, Martinou J C. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 9.Rubin R, Baserga R. Lab Invest. 1995;73:311–331. [PubMed] [Google Scholar]
- 10.Sell C, Baserga R, Rubin R. Cancer Res. 1995;55:303–306. [PubMed] [Google Scholar]
- 11.Rodeck U, Jost M, Kari C, Shih D-T, Lavker R M, Ewert D L, Jensen P J. J Cell Sci. 1997;110:113–121. doi: 10.1242/jcs.110.2.113. [DOI] [PubMed] [Google Scholar]
- 12.White E. Genes Dev. 1996;10:1–16. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, Karras J G, Howard R G, Rothstein T L. J Immunol. 1995;155:3722–3725. [PubMed] [Google Scholar]
- 14.Boise L H, Minn A J, Noel P J, June C H, Accavitti M A, Lindsten T, Thompson C B. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 15.Broome H E, Dargan C M, Krajewski S, Reed J C. J Immunol. 1995;155:2311–2317. [PubMed] [Google Scholar]
- 16.Mueller D L, Seiffert S, Fang W, Behrens T W. J Immunol. 1996;156:1764–1771. [PubMed] [Google Scholar]
- 17.McNeill H, Jensen P J. Cell Regul. 1990;1:843–852. doi: 10.1091/mbc.1.11.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murthy U, Basu A, Rodeck U, Herlyn M, Ross A H, Das M. Arch Biochem Biophys. 1987;252:549–560. doi: 10.1016/0003-9861(87)90062-2. [DOI] [PubMed] [Google Scholar]
- 19.Rodeck U, Herlyn M, Herlyn D, Molthoff C, Atkinson B, Varello M, Steplewski Z, Koprowski H. Cancer Res. 1987;47:3692–3696. [PubMed] [Google Scholar]
- 20.Murthy U, Rieman D J, Rodeck U. Biochem Biophys Res Commun. 1990;172:471–476. doi: 10.1016/0006-291x(90)90696-k. [DOI] [PubMed] [Google Scholar]
- 21.Rodeck U, Williams N, Murthy U, Herlyn M. J Cell Biochem. 1990;44:69–79. doi: 10.1002/jcb.240440202. [DOI] [PubMed] [Google Scholar]
- 22.Basu A, Murthy U, Rodeck U, Herlyn M, Mattes L, Das M. Cancer Res. 1987;47:2531–2536. [PubMed] [Google Scholar]
- 23.Vardy D A, Kari C, Lazarus G S, Jensen P J, Zilberstein A, Plowman G D, Rodeck U. J Cell Physiol. 1995;163:257–265. doi: 10.1002/jcp.1041630206. [DOI] [PubMed] [Google Scholar]
- 24.Eastman A. Methods Cell Biol. 1995;46:41–55. doi: 10.1016/s0091-679x(08)61923-8. [DOI] [PubMed] [Google Scholar]
- 25.Boise L H, Gonzalez-Garcia M, Postema C E, Ding L, Lindsten T, Turka L A, Mao X, Nunez G, Thompson C B. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 26.Rodeck U, Bossler A, Kari C, Humphreys C W, Gyorfi T, Maurer J, Thiel E, Menssen H D. Int J Cancer. 1994;59:78–82. doi: 10.1002/ijc.2910590116. [DOI] [PubMed] [Google Scholar]
- 27.Gilman M. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore R E, Seidman J G, Smith J A, Struhl K, editors. New York: Wiley; 1989. pp. 4.7.1–4.7.8. [Google Scholar]
- 28.Wrone-Smith T, Johnson T, Nelson B, Boise L H, Thompson C B, Nunez G, Nickoloff B J. Am J Pathol. 1995;146:1079–1088. [PMC free article] [PubMed] [Google Scholar]
- 29.Krajewski S, Krajewska M, Shabaik A, Wang H G, Irie S, Fong L, Reed J C. Cancer Res. 1994;54:5501–5507. [PubMed] [Google Scholar]
- 30.Farrow S N, White J H, Martinou I, Raven T, Pun K T, Grinham C J, Martinou J C, Brown R. Nature (London) 1995;374:731–733. doi: 10.1038/374731a0. [DOI] [PubMed] [Google Scholar]
- 31.Oltvai Z N, Korsmeyer S J. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- 32.Oltvai Z N, Milliman C L, Korsmeyer S J. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 33.Yin X M, Oltvai Z N, Korsmeyer S J. Curr Top Microbiol Immunol. 1995;194:331–338. doi: 10.1007/978-3-642-79275-5_38. [DOI] [PubMed] [Google Scholar]
- 34.Chao D T, Linette G P, Boise L H, White L S, Thompson C B, Korsmeyer S J. J Exp Med. 1995;182:821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chittenden T, Harrington E A, O’Connor R, Flemington C, Lutz R J, Evan G I, Guild B C. Nature (London) 1995;374:733–736. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- 36.Kiefer M C, Brauer M J, Powers V C, Wu J J, Umansky S R, Tomei L D, Barr P J. Nature (London) 1995;374:736–739. doi: 10.1038/374736a0. [DOI] [PubMed] [Google Scholar]
- 37.Cheng E H, Levine B, Boise L H, Thompson C B, Hardwick J M. Nature (London) 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- 38.Nass S J, Li M, Amundadottir L T, Furth P A, Dickson R B. Biochem Biophys Res Commun. 1996;227:248–256. doi: 10.1006/bbrc.1996.1497. [DOI] [PubMed] [Google Scholar]
- 39.Watt, F. M. & Jones, P. H. (1993) Development (Cambridge, U.K.) Supplement, 185–192.
- 40.Lewis J M, Schwartz M A. Mol Biol Cell. 1995;6:151–160. doi: 10.1091/mbc.6.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frisch S M, Vuori K, Ruoslahti E, Chan-Hui P Y. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao R, Cooper G M. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 43.Minshall C, Arkins S, Freund G G, Kelley K W. J Immunol. 1996;156:939–947. [PubMed] [Google Scholar]
- 44.Scheid M P, Lauener R W, Duronio V. Biochem J. 1995;312:159–162. doi: 10.1042/bj3120159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pang L, Sawada T, Decker S J, Saltiel A R. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 46.Hu P, Margolis B, Skolnik E Y, Lammers R, Ullrich A, Schlessinger J. Mol Cell Biol. 1992;12:981–990. doi: 10.1128/mcb.12.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen H C, Guan J L. J Biol Chem. 1994;269:31229–31233. [PubMed] [Google Scholar]
- 48.Patel T, Gores G J, Kaufman S H. FASEB J. 1996;10:587–597. doi: 10.1096/fasebj.10.5.8621058. [DOI] [PubMed] [Google Scholar]
- 49.Nanney L B, Stoscheck C M, Magid M, King L., Jr J Invest Dermatol. 1986;86:260–265. doi: 10.1111/1523-1747.ep12285389. [DOI] [PubMed] [Google Scholar]
- 50.King L, Jr, Gates R E, Stoscheck C M, Nanney L B. J Invest Dermatol. 1990;95:10S–12S. doi: 10.1111/1523-1747.ep12505661. [DOI] [PubMed] [Google Scholar]
- 51.Valyi-Nagy I, Jensen P J, Albelda S M, Rodeck U. J Invest Dermatol. 1992;99:350–356. doi: 10.1111/1523-1747.ep12616672. [DOI] [PubMed] [Google Scholar]
- 52.Gottlieb A B, Chang C K, Posnett D N, Fanelli B, Tam J P. J Exp Med. 1988;167:670–675. doi: 10.1084/jem.167.2.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finzi E, Harkins R, Horn T. J Invest Dermatol. 1991;96:328–332. doi: 10.1111/1523-1747.ep12465223. [DOI] [PubMed] [Google Scholar]
- 54.Cook P W, Pittelkow M R, Keeble W W, Graves-Deal R, Coffey R, Jr, Shipley G D. Cancer Res. 1992;52:3224–3227. [PubMed] [Google Scholar]
- 55.Hertle M D, Kubler M D, Leigh I M, Watt F M. J Clin Invest. 1992;89:1892–1901. doi: 10.1172/JCI115794. [DOI] [PMC free article] [PubMed] [Google Scholar]